Introduction
Inflammation plays crucial roles in the development
of tumor. Tumor is regarded as a never healed injury (1). It has been proven that immune
inflammatory cell could enhance angiogenesis, accelerate cell
proliferation, invasion and metastasis in tumor development
(2). Tumor can recruit normal cells
to build tumor microenvironment. Inflammatory reaction stimulates
angiogenesis and tissue reconstitution to promote the progress of
cancer (3).
Angiogenesis is one of the crucial hallmarks of
tumor. Recent studies have indicated that multiple mechanisms were
involved in anti-angiogenesis therapy, such as immunity (4–6). It is
important to optimize tumor treatment strategies according to the
estimation of the change of tumor microenvironment after
anti-angiogenesis therapy. Several studies powerfully supported
that anti-angiogenesis therapy can overcome various suppressive
immunity network (7). Sunitinib can
reduce the number of MDSCs, Tregs and the expressions of IL-10,
TGF-β and PD-1 to relieve the immunity suppression in tumor
(8). In the nude mouse models,
neutralizing VEGFR can arrest VEGF signaling pathway, promote DCs
maturation and downregulate the number of Tregs. In view of
stablization of endothelial cells, targeting angiogenesis is one
prospective method to control process of tumor. Endostatin is
considered with be no side effects and less multidrug resistance,
which could efficiently inhibit the angiogenesis and the growth of
endothelial cells (9). However, there
is rare research of endostatin in the tumor microenvironment.
In our previous study, we had proven that endostatin
plays antitumor effect combined with DC-T cell therapy in lung
carcinoma. To investigate further mechanism of endostatin antitumor
effect, we explored the effect of endostatin on immune cells and
cytokines in tumor microenvironment, and the intervention of the
immune network, aiming to offer potential data for molecular
targeted therapy or adoptive cellular immunotherapy.
Materials and methods
Cells
Lewis lung cancer cell line (from lung
adenocarcinoma cell line of C57BL/6 mice) was purchased from
Shanghai Cell Bank of Chinese Academy of Sciences and was cultured
in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal
bovine serum. The cells (1×107) were resuspended in
RPMI-1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) and
mixed.
Animals
Male wild-type C57BL/6 mice (age, 6 weeks; weight,
18–22 g) were purchased from Beijing Laboratory Animal Center of
Chinese Academy of Sciences and fed in the specific-pathogen-free
animal laboratory. The feeding and use of laboratory animals
complied with Animal Experimentation Ethical Standards proposed by
Ethics Committee of Shandong University [SCXK (Lu) 2003–0003].
After Lewis lung carcinoma cells (LLCs) were
recovered and subcultured in complete medium, cells in log growth
phase were used and cell concentration was adjusted to
1×107/ml. Right rib skin of C57BL/6 mice was disinfected
with 75% alcohol and suspension of LLCs was collected with 1 ml
syringe (mixing upside down). Suspension (0.2 ml) was then given to
each mouse via subcutaneous injection, with 1×106 cells
being inoculated in each mouse.
C57BL/6 mice were divided into three groups (low
dose of endostatin, high dose of endostatin and PBS control) with 7
mice in each group. Intervention was given to tumor-bearing mice on
day 7. Tumor growth and diameter were measured every other day. The
mice were killed after 24 h with administration on day 14. For
control group, phosphate-buffered saline (0.2 ml) was given to each
C57BL/6 mouse by tail vein injection for a total of 14 days; for
low dose of endostatin group, was given to each mouse by tail vein
injection for a total of 14 days at a concentration of 7.5
mg/kg/day; for high dose of endostatin were given 15 mg/kg/day to
each mouse by tail vein injection on day 7 after the model of
tumor-bearing C57BL/6 mice was established.
To measure mouse weight and tumor inhibition rate,
electronic scale was used to measure the weight of mice in each
group every other day. After tumor-bearing C57BL/6 mice were
killed, tumor tissues were dissected and weighed. Tumor inhibition
rate = (1 - mean tumor weight of treatment groups/mean tumor weight
of control group) × 100%. For the measurement of tumor volume, the
maximum long and short diameters of tumors were measured using
vernier caliper. Then, mean tumor volume and tumor inhibition rate
in each group were calculated and growth curve was drawn. Tumor
volume = long diameter of tumor × short diameter of
tumor2/2.
Antibodies and reagents
Recombinant human endostatin (rhEndostatin; Simcere
Pharm, Nanjing, China); EZ-Sep™ mouse percollase (Amresco, Dallas,
TX, USA); RPMI-1640 medium, FBS (Gibco, Grand Island, NY, USA);
ConA, DMEM medium, phosphate-buffered saline (PBS) buffer (Sigma,
St. Louis, MO, USA); fluorescently-labeled antibody CD3, CD4, CD8,
CD11c, CD86, major histocompatibility complex (MHC) II, CD11b,
Gr-1, CD206, CD68 and NOS2 and their isotype controls (eBioscience,
Inc., San Diego, CA, USA); mouse lymphocyte factor ELISA kit
(Shanghai Enzyme-linked Biotechnology Co., Ltd., Shanghai, China);
BCA Protein Assay kit (Beyotime, Shanghai, China); anti-mouse
hypoxia-inducible factor-1α (HIF-1α), VEGF antibody (Abcam,
Cambridge, MA, USA); rmGM-CSF, rmIL-4 (Peprotech), rmIL-2, rmTNF-α
(Biological); anti-mouse CD31 nonlabeled immunohistochemical
monoclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA); MCO-15AC CO2 incubator (Sanyo, Osaka, Japan); sterile 1.5
laminar flow bechtop (Thermo Fisher Scientific); FACSCalibur flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA); NanoDrop
ND-1000 ultraviolet spectrophotometer (Agilent Technologies, Inc.,
Santa Clara, CA, USA); Model 680 Microplate Reader (Bio-Rad,
Hercules, CA, USA).
Immunohistochemistry
Tumor tissues were separated from tumor-bearing mice
and fixed with paraformaldehyde. The tissues were embedded with
paraffin and then sectioned. The paraffin sections were dewaxed to
water. The antigens were repaired by high pressure for 8 min using
streptavidin-peroxidase method. Subsequent procedures were
conducted according to instructions of secondary antibody kit and
DAB color development kit (Bio-Rad). Under a light microscope, mean
microvessel density (MVD) was counted under 6 high power fields
(×200) for each section.
Flow cytometry
Tumor tissues were dissected and cut into pieces.
After trypsinization, the tissues were filtered through 300 mm
stainless steel mesh to obtain monocytes. After cell density was
adjusted to 5×105/ml, 100 µl cell suspension was added
into each flow tube. FACS tubes contained phycoerythrin-labeled
anti-CD83 and anti-CD86 antibodies (BioLegend, San Diego, CA, USA),
allophycocyanin-labeled anti-CD68 antibody, phycoerythrin-labeled
anti-iNOS antiboody, fluorescein isothiocyanate-labeled anti-CD3,
anti-CD4, anti-IFN-γ, anti-CD206, anti-Gr-1 and anti-CD11c
antibodies (eBioscience, Inc.). Negative control, isotype control
and single line pipette groups were designed. Phycoerythrin-rat IgG
and fluorescein isothiocyanate-hamster IgG were added in control
groups. After being fully mixed with fluorescent-labeled antibody,
they were placed at room temperature away from light for 15 min.
FACS Aria II sorting flow cytometer (BD Biosciences, San Jose, CA,
USA) was used to detect molecule expressions on cell surface and
FlowJo software was used to analyze data.
Enzyme linked immunosorbent assay
(ELISA)
Antibodies were diluted with coating buffer to the
extent where protein level was 1–10 µg/ml. Then, 0.1 ml of
antibodies were taken and added to ELISA plate wells, which were
incubated at 4°C overnight. The required number of wells was
calculated according to the number of test samples, blank control
and standard samples. Distilled water (130 µl) was added into blank
and standard wells, while 100 µl cell supernatant was added into
sample wells for testing. Each sample group was made in triplicate.
The plate was covered and incubated in an incubator with 5%
CO2 at 37°C for 90 min. Afterwards, liquid in wells was
discarded and the wells were washed 3 times. After 100 µl biotin
antibody was added to each well, the plate was covered and
incubated in an incubator with 5% CO2 at 37°C for 1 h.
Then, the plate was washed with washing liquid for 3 times of 3
min. Enzyme conjugate (100 µl) was added into each well and the
plate was incubated in the incubator with 5% CO2 at 37°C
for 30 min. Afterwards, the plate was washed for 3 times. Color
developing reagent (100 µl) was added into each well and kept away
from light. The plate was incubated in the incubator with 5% CO2 at
37°C for 10–20 min. Finally, 100 µl stop buffer was added into each
well to terminate reaction and OD450 values were determined using a
microplate reader (Model 680; Bio-Rad, Hercules, CA, USA).
Western blotting
After tumor tissues were taken out from
tumor-bearing mice. They were chipped and then ground in a
homogenizer. Total protein was extracted through disruption on ice,
and 400 µl RIPA lysate was added to each group. After disruption,
cells were taken out and transferred to a centrifugal tube of 1.5
ml. Then, cells were centrifuged at 13,200 × g at 4°C for 20 min,
and the supernatant was collected. Bicinchoninic Acid Protein Assay
kit was used to determine the concentration of extracted protein
(Beyotime). Sodium dodecyl sulfate polyacrylamide gel
electrophoresis was used to transfer protein to polyvinylidene
fluoride film. The film was then covered with Tris-buffered saline
and Tween-20 (TBST) containing 5% skimmed milk for 2 h. After
primary antibodies (rabbit anti-mouse HIF-1α monoclonal antibody,
1:1,000; rabbit anti-mouse VEGF monoclonal antibody, 1:1,000;
Abcam) were added, the film was incubated at 4ºC overnight. The
film was washed five times (6 min each) with TBST. After secondary
antibody (horseradish peroxidase goat anti-mouse IgG antibody
conjugate, 1:2,000; Abcam) was added, the film was incubated at
37ºC for 1 h and washed with TBST. Electrochemiluminescence kit was
used to achieve chemiluminescence signals. SmartView
electrophoresis image analysis system (Smartview Enterprise Imaging
Solutions, Irvine, CA, USA) was used to obtain images. Quantity One
software (Bio-Rad) was applied to analyze gray values of each zone.
The ratio of each interested protein and gray values of β-actin was
calculated for statistical analysis.
Immunohistochemisty
Tumor tissues were separated from tumor-bearing mice
and fixed with formalin. The tissues were embedded with paraffin
and then sectioned. The paraffin sections were dewaxed to water.
Streptavidin-peroxidase method was used following kit instructions.
Primary antibodies were replaced with phosphate-buffered saline as
negative control. According to semi-quantitative integration, the
images were reviewed by two physicians from the Department of
Pathology and a conclusion was drawn. The results were scored
according to positive staining intensity and expression of positive
cells. Negative staining intensity: cells had no staining (score
0); light brown cells were weakly positive (score 1); brown cells
were moderately positive (score 2); brown cells without background
coloring were strongly positive (score 3). For expression of
positive cells, 5 different fields were chosen under ×400 light
microscope and 200 cells were counted for each field. The
percentage of positive cells was then calculated. If positive cells
≤5%, score was 0; if positive cells ≤25%, score was 1; if 25 %
<positive cells ≤50%, score was 2; if positive cells >50%,
score was 3. There were four grades of immunohistochemisty results
according the above scores: 0 was rated as negative (−), 1–4 were
rated as weakly positive (+), 5–8 were graded as moderately
positive (+), and 9–12 were rated as strongly positive (+++).
Statistical analysis
All data were analyzed using SPSS 18.0 software
(IBM, New York, NY, USA). Measurement data were expressed as means
± SD. Statistical differences between groups were determined using
one-way ANOVA, and LSD test was performed to make comparison
between any two groups. Ration comparison was conducted by
χ2 test and Fisher's exact test. P<0.05 was
considered to indicate a statistically significant difference.
Results
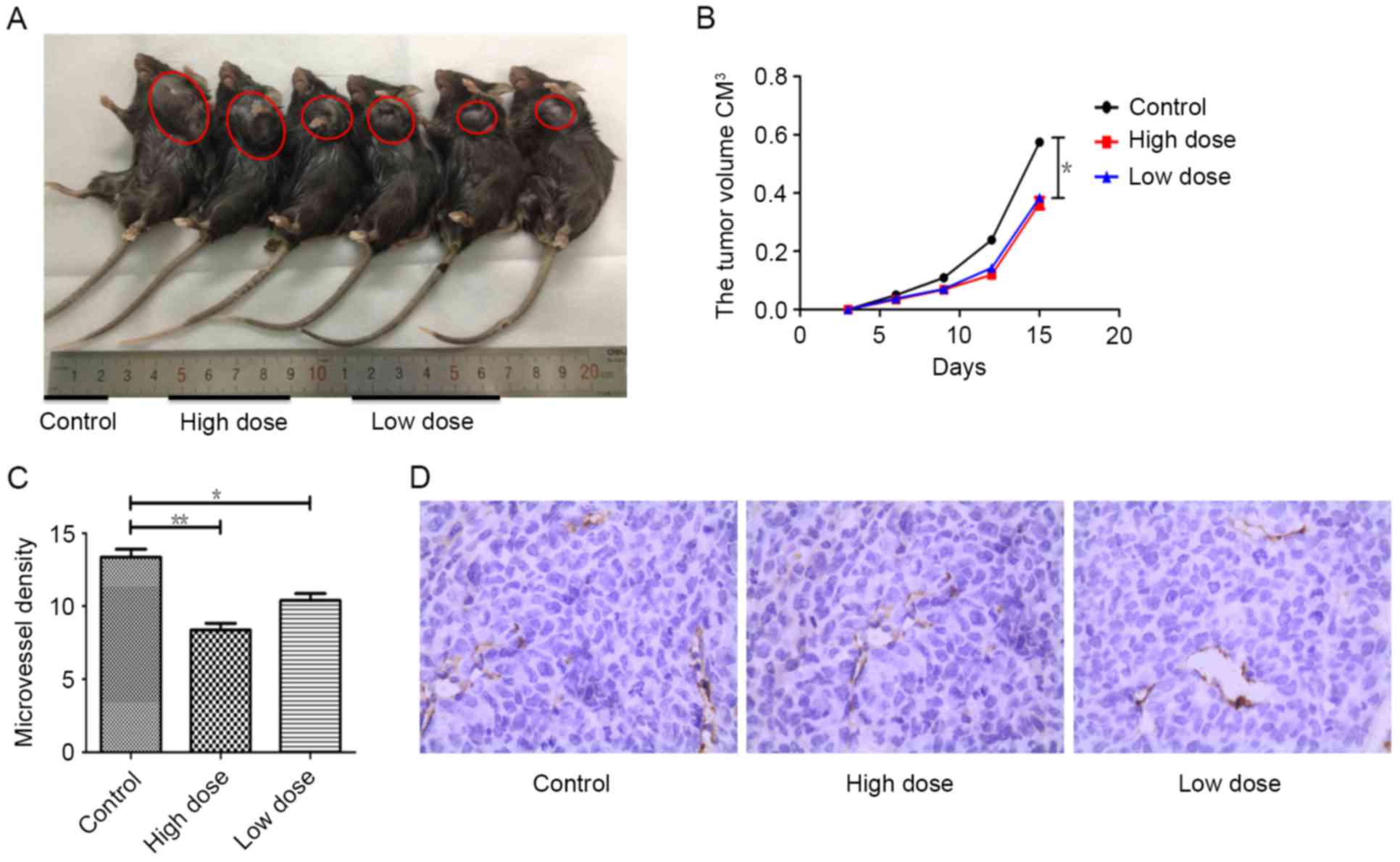
Tumor growth and angiogenesis were
strongly inhibited by endostatin therapy
To test the effect of endostatin therapy on tumor
growth and angiogenesis, tumor sizes were measured and
immunohistochemistry (IHC) were performed separately. Compared with
control group, tumor growth was actively suppressed by endostatin
therapy (P=0.013), while there was no difference of tumor growth
between low and high dose group of endostatin (P>0.05) (Fig. 1A and B). Using CD31 as the marker of
vascular endothelial cells, cytoplasm of endothelial cells was
stained with yellowish-brown. In contract to control group, tumor
MVD in mice receiving low dose of endostatin was fewer (P=0.014),
while MVD in mice receiving tumor high dose of endostatin was
significantly decreased (P=0.002), and the necrosis of tumor
tissues were also increased, which was different between these two
groups (P<0.05) (Fig. 1C and D).
These results indicate by dose dependent, endostatin markedly
inhibited tumor angiogenesis, promoted tumor necrosis and
significantly reduced tumor growth.
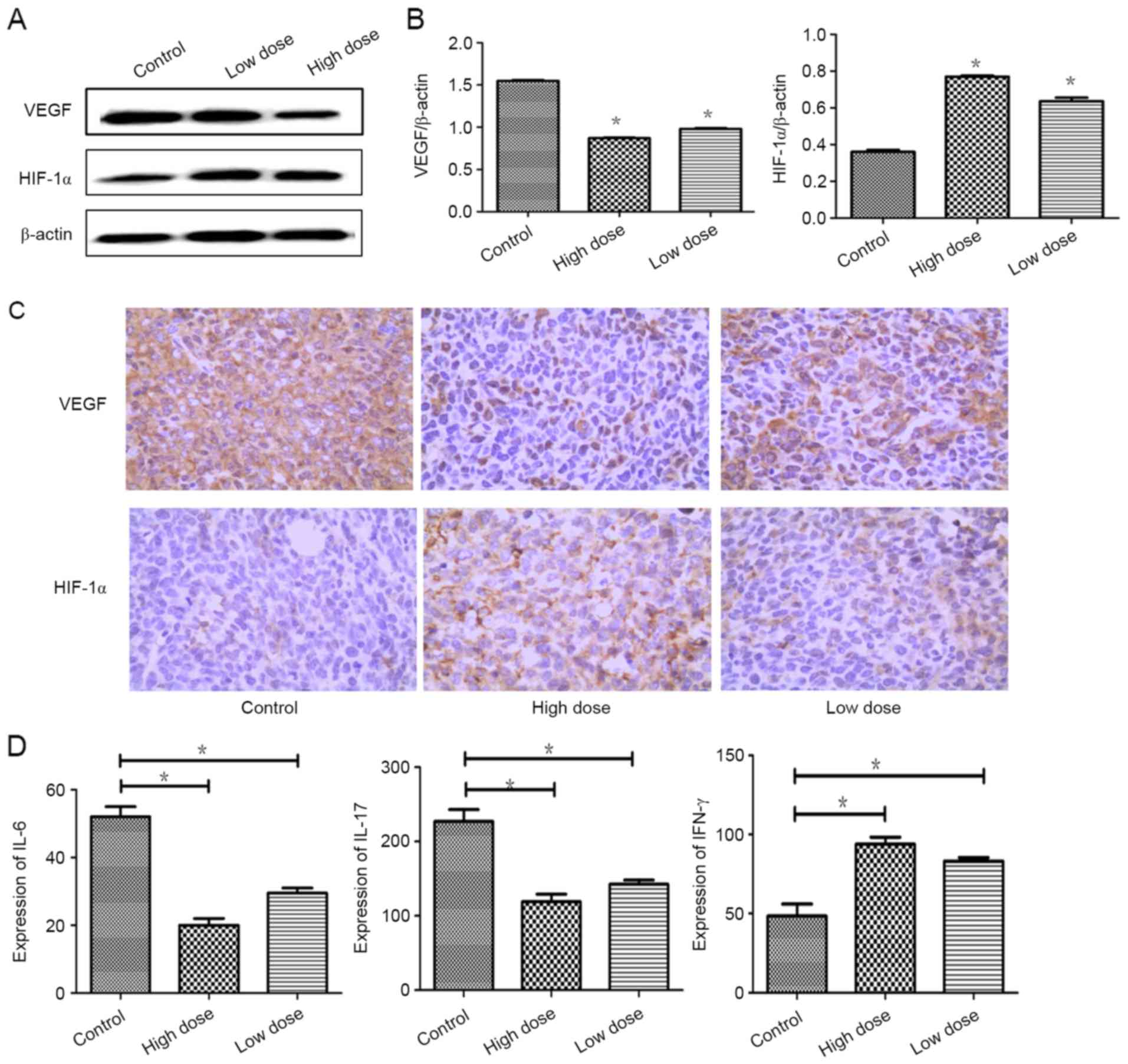
Factors of tumor angiogenesis were
strongly inhibited by endostatin therapy
To test the effect of endostatin therapy on tumor
angiogenesis and hypoxia, IHC were performed separately. Compared
with control group, the expression of VEGF was actively suppressed
by low dose of endostatin therapy (P=0.001), while the expression
of HIF-1α was significantly increased (P=0.000). Similarly, the
expression of VEGF was lower and the expression of HIF-1α was
increased markedly in high dose of endostatin therapy (P=0.001)
(Fig. 2A-C). Expressions of IL-6 and
IL-17 were reduced (P=0.012; P=0.029), and the expression of IFN-γ
was increased strongly (P=0.044) in low dose of endostatin therapy
by detection with ELISA. Similarly, the expressions of IL-6 and
IL-17 was decreased strongly (P=0.012; P=0.029) and the expression
of IFN-γ was increased strongly (P=0.036) in high dose of
endostatin therapy (Fig. 2D). These
data proved that endostatin exacerbated hypoxic conditions and
inhibited proangiogenic factors and increased expressions of
antiangiogenic factors.
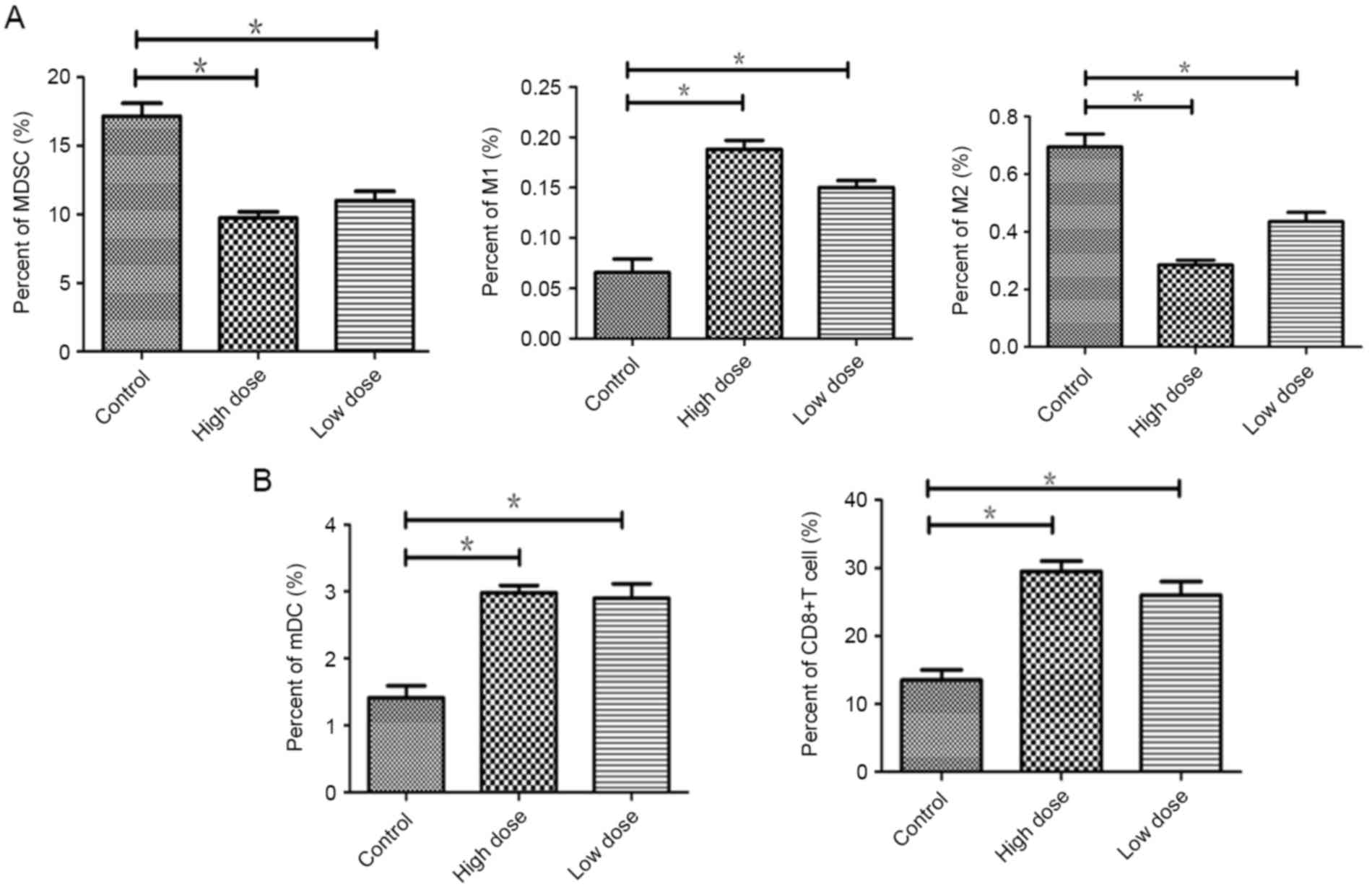
Endostatin effectively decreased the infiltration of
immunosuppression cell and promoted infiltration of DCs and CD8+T
cells in tumor. Compared to the controls, the proportion of MDSCs
was reduced in low dose (P=0.035) and less in high dose of
endostatin (P=0.019). Immune-enhanced M1 type of TAMs were
increased in low dose (P=0.031) and more in high dose (P=0.017).
Immunity suppressive M2 type of TAMs were declined in low dose
(P=0.042) and more in high dose group (P=0.01) (Fig. 3A).
Compare to the control, the infiltration of mDCs and
CD8+ T cells were increased in low dose (P=0.034;
P=0.038), which were significantly increased in the high dose group
(P=0.018; P=0.017) (Fig. 3B). There
was no significant difference between low and high dose group
(P>0.05). The study suggested that through downregulation of
immunosuppressive cells, endostatin enchanced M1 type of TAMs, the
cell infiltration of mature DCs and CD8+ T cells in
tumor, to reverse immunosuppression of tumor micro-environment.
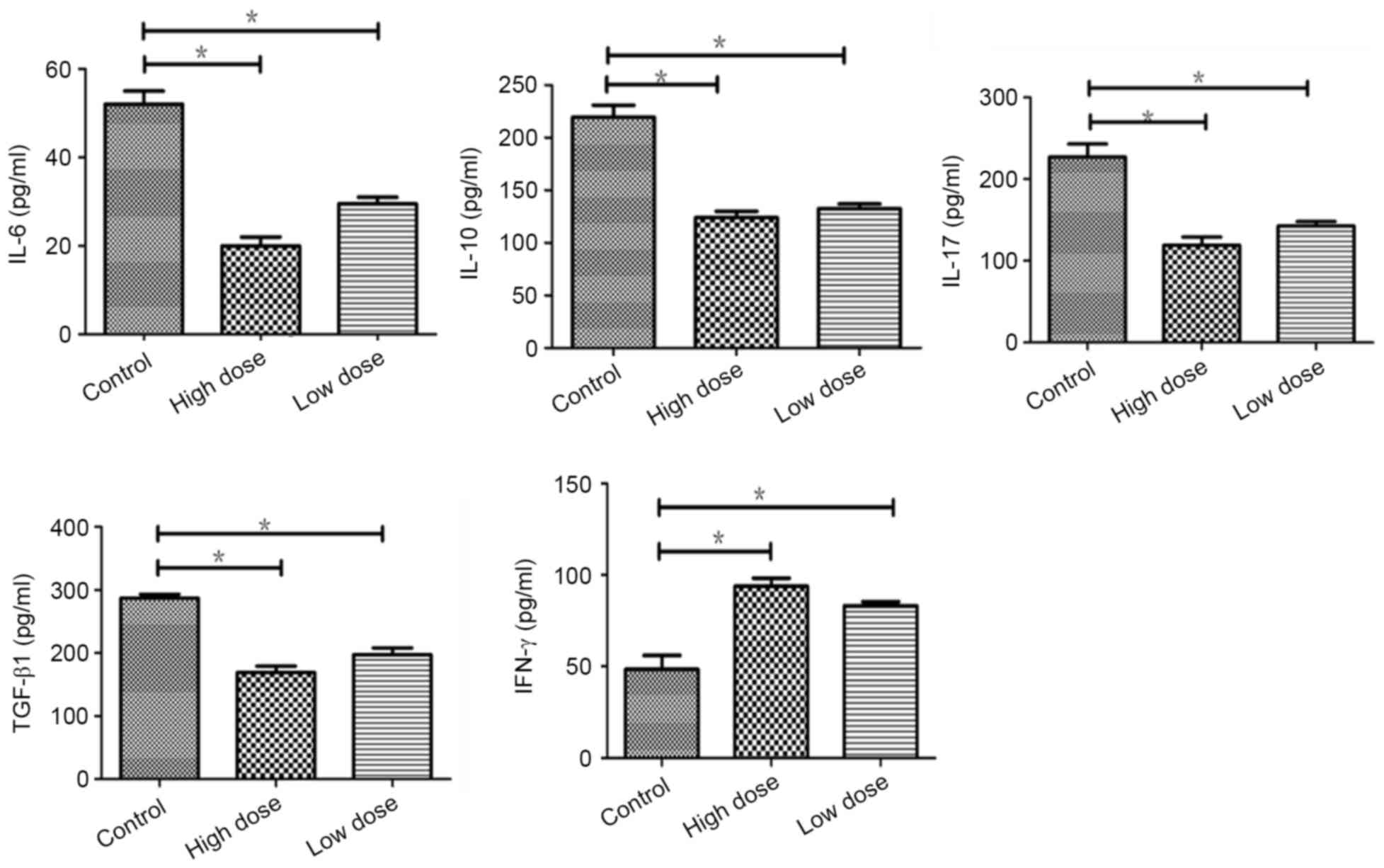
Endostatin increased the expression of
IFN-γ by inhibition of immunosuppressive cytokines in tumor
Compare to the control, immunosuppressive cytokines
including IL-6, IL-10, IL-17 and TGF-β were reduced significantly
(P=0.022, 0.020, 0.038, 0.018) in low dose of endostatin by
detected with ELISA, which was more significant in high dose group
(P=0.012,0.018,0.014,0.029). The expression of IFN-γ was increased
(P=0.044), which was more in high dose of endostatin (P=0.036).
There was no significant difference between low and high dose
(P>0.05) (Fig. 4). The study
suggested that endostatin repressed the expressions of
immunosuppressive factors to inhibit immunosuppression of tumor
micro-environment.
Our study proved that endostatin could inhibit the
expressions of immunosuppressive cells and cytokines, enhance the
M1 type of TAMs and IFN-γ, and accelerate the invasion of mature
DCs and CD8+ T cells in tumor, reversing
immunosuppression of tumor micro-environment, and play antitumor
effect collaboration with DC-T cell therapy.
Discussion
Studies have proved that inflammation play crucial
roles in the development of cancer. Inflammatory cells can prove
angiogenesis, accelerate cell proliferation, invasion and migration
(10). As one critical factor of
tumor, angiogenesis promotes tumor growth and migration to
surrounding tissue, and even spread to other organ (11). Folkman suggested that it is a good way
to inhibit tumor growth through arrest development of new vascular
formation (12). Endothelial cell can
be regarded as target of antitumor therapy. Angiogenesis is
produced by interaction of various type of cells in tumor
microenviroment (13). Inflammation
can promote tumor development by angiogenesis and organization
restructuring (2).
Inflammatory microenvironment is major structure of
all the tumors (14). About 90% of
tumor are related with mutation of somatic cell and environment
factor. In other words, cancer can be a never healed wound. TAMs
and T cells are most common in tumor microenvironment. TAMs could
enhance tumor growth, angiogenesis, invasion and migration.
Angiogenesis is dependent on recruitment of TAMs to angiopoietin 2
and VEGF (15). CXCL8/IL-8, CXCL1,
VEGF and HIF-1α could be regulated by TAMs, MDSCs, NF-κB, STAT3 and
AP-1 (4,16). The inactivation of NF-κb and STAT3,
neutralization of CCL2 or CXCL12, section of TAMs, inhibit
angiogenesis, arrest tumor growth, which indicate the critical role
of inflammation medium in tumor angiogenesis (17). There are all kinds of partial
differentiation of myeloid progenitor cells in tumor stroma, which
have the preference to promote tumor formation (18). Myeloid-derived suppressor cells (MDSC)
are a major host component contributing to the immune suppressive
environment. In addition to their inherent immune suppressive
function, MDSC amplify the immune suppressive activity of
macrophages and dendritic cells via cross-talk (2,19,20). Hypoxia, a reduction in the normal
level of tissue oxygen tension, occurs during acute and chronic
vascular disease, pulmonary disease and cancer (21). The chemokine CXCL12 and its receptor
CXCR4 are known to play an important role in cancer development and
progression (22). HIF-1α could
enhance the expression of CXCL12, which activates endothelial cells
apoptosis a induced by overexpression CXCR4 (23). HIF-1α also induced the expression of
VEGF in hypoxic condition and angiogenesis by collecting bone
marrow inhibitory cells from different sources (24,25).
Given that the gene stability of endothelial cells,
it has been a good way to inhibit the progress of tumor by
targeting angiogenesis (11). As one
of endogenous inhibitor, endostatin was found in epithelioid
hemangioendothelioma (26). By its
regulation to angiogenesis gene, endostatin can inhibit
proliferation, migration or invasion and apoptosis of endothelial
cells, to play antineoplastic activity (27). It has been approved to cure or
retreatment III/IV stage non-small cell lung cancer combined with
NP chemotherapy (NP regimen includes Navelbine (25
mg/m2, d1, d8) and DDP (25 mg/m2, d1-3),
repeated every 21 days) in September of 2005 by China's state food
and drug administration. Studies have suggested that VEGF inhibit
the maturation of DCs by regulation of VEGFR-1 induced κB dependent
signal (28,29). mDCs from peripheral blood of cancer
patients are associated with increased serum levels of VEGF.
Therefore, antiangiogenic therapy blocking VEGF pathway, promote
DCs maturation and the decline of Tregs numbers.
In our study, we found that endostatin reduced the
fraction of immunosuppressive MDSCs and M2 type of TAMs in Lewis
lung mouse model. The expressions of immunosuppressive factors
including IL-6, IL-10, IL-1 and TGF-β were downregulated by
endostatin. M1 type of TAMs and IFN-γ were upregulated by
endostatin, which promote invasion of mature DCs and
CD8+ T cell in tumor microenvironment. These results
proved endostatin effectively corrects immunosuppression of tumor
microenvironment. The mechanism about the effect of endostatin to
tumor microenvironment is rare. To explore the specific mechanism
of endostatin antitumor effect, and its intervention to tumor
microenvironment immune network, we can carry out therapy combined
endostatin and cellular immunotherapy to find theoretical basis. On
the other hand, our study have found that endostatin inhibited
IL-6, IL-17 and VEGF, upregulated the expressions of IFN-γ, which
supports that endostatin inhibits angiogenesis and enhances hypoxia
in tumor microenvironment. Our previous study have proved that
endostatin enhances the antitumor effect of DC-T cell immunity, and
corrects the immunity inhibition, which suggested endostatin has
close relationship with body immune system. Therefore, to study the
mechanism of endostatin antitumor, we will offer further therapy
data to find predictive markers with target therapy and cell
immunity.
Acknowledgements
This study was supported by the Natural Science
Youth Foundation of Shandong Province, P.R. China (no. ZR2013HQ017)
and the Shandong Provincial Natural Science Foundation, China (no.
ZR2010HL015; ZR2015HL024).
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Prueitt RL, Wallace TA, Glynn SA, Yi M,
Tang W, Luo J, Dorsey TH, Stagliano KE, Gillespie JW, Hudson RS, et
al: An immune-inflammation gene expression signature in prostate
tumors of smokers. Cancer Res. 76:1055–1065. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan Y, Jiang YC, Sun CK and Chen QM: Role
of the tumor microenvironment in tumor progression and the clinical
applications (Review). Oncol Rep. 35:2499–2515. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ham M and Moon A: Inflammatory and
microenvironmental factors involved in breast cancer progression.
Arch Pharm Res. 36:1419–1431. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Samples J, Willis M and Klauber-Demore N:
Targeting angiogenesis and the tumor microenvironment. Surg Oncol
Clin N Am. 22:629–639. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jayson GC, Kerbel R, Ellis LM and Harris
AL: Antiangiogenic therapy in oncology: Current status and future
directions. Lancet. 388:518–529. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tartour E, Pere H, Maillere B, Terme M,
Merillon N, Taieb J, Sandoval F, Quintin-Colonna F, Lacerda K,
Karadimou A, et al: Angiogenesis and immunity: A bidirectional link
potentially relevant for the monitoring of antiangiogenic therapy
and the development of novel therapeutic combination with
immunotherapy. Cancer Metastasis Rev. 30:83–95. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen HM, Ma G, Gildener-Leapman N,
Eisenstein S, Coakley BA, Ozao J, Mandeli J, Divino C, Schwartz M,
Sung M, et al: Myeloid-derived suppressor cells as an immune
parameter in patients with concurrent sunitinib and stereotactic
body radiotherapy. Clin Cancer Res. 21:4073–4085. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walia A, Yang JF, Huang YH, Rosenblatt MI,
Chang JH and Azar DT: Endostatin's emerging roles in angiogenesis,
lymphangiogenesis, disease, and clinical applications. Biochim
Biophys Acta. 1850:2422–2438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Potente M, Gerhardt H and Carmeliet P:
Basic and therapeutic aspects of angiogenesis. Cell. 146:873–887.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ackermann M and Konerding MA: Vascular
casting for the study of vascular morphogenesis. Methods Mol Biol.
1214:49–66. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Horsman MR and Vaupel P:
Pathophysiological basis for the formation of the tumor
microenvironment. Front Oncol. 6:662016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patel A and Sant S: Hypoxic tumor
microenvironment: Opportunities to develop targeted therapies.
Biotechnol Adv. 34:803–812. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sancakdar E, Guven AS, Uysal EB, Deveci K
and Gültürk E: Important of angiopoietic system in evaluation of
endothelial damage in children with crimean-congo hemorrhagic
fever. Pediatr Infect Dis J. 34:e200–e205. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rogers TL and Holen I: Tumour macrophages
as potential targets of bisphosphonates. J Transl Med. 9:1772011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sakurai T and Kudo M: Signaling pathways
governing tumor angiogenesis. Oncology. 81 Suppl 1:S24–S29. 2011.
View Article : Google Scholar
|
|
18
|
Morello S, Pinto A, Blandizzi C and
Antonioli L: Myeloid cells in the tumor microenvironment: Role of
adenosine. Oncoimmunology. 5:e11085152015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ostrand-Rosenberg S, Sinha P, Beury DW and
Clements VK: Cross-talk between myeloid-derived suppressor cells
(MDSC), macrophages, and dendritic cells enhances tumor-induced
immune suppression. Semin Cancer Biol. 22:275–281. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fukuda K, Kobayashi A and Watabe K: The
role of tumor-associated macrophage in tumor progression. Front
Biosci (Schol Ed). 4:787–798. 2012.PubMed/NCBI
|
|
21
|
Harris AL: Hypoxia-a key regulatory factor
in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ji RC: Hypoxia and lymphangiogenesis in
tumor microenvironment and metastasis. Cancer Lett. 346:6–16. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee HJ and Jo DY: The role of the
CXCR4/CXCL12 axis and its clinical implications in gastric cancer.
Histol Histopathol. 27:1155–1161. 2012.PubMed/NCBI
|
|
24
|
Henegan JC Jr and Gomez CR: Heritable
cancer syndromes related to the hypoxia pathway. Front Oncol.
6:682016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rapisarda A and Melillo G: Overcoming
disappointing results with antiangiogenic therapy by targeting
hypoxia. Nat Rev Clin Oncol. 9:378–390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Alahuhta I, Aikio M, Väyrynen O,
Nurmenniemi S, Suojanen J, Teppo S, Pihlajaniemi T, Heljasvaara R,
Salo T and Nyberg P: Endostatin induces proliferation of oral
carcinoma cells but its effect on invasion is modified by the tumor
microenvironment. Exp Cell Res. 336:130–140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen X, Zhang H, Zhu H, Yang X, Yang Y,
Yang Y, Min H, Chen G, Liu J, Lu J, et al: Endostatin combined with
radiotherapy suppresses vasculogenic mimicry formation through
inhibition of epithelial-mesenchymal transition in esophageal
cancer. Tumour Biol. 37:4679–4688. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Alevizakos M, Kaltsas S and Syrigos KN:
The VEGF pathway in lung cancer. Cancer Chemother Pharmacol.
72:1169–1181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hato T, Zhu AX and Duda DG: Rationally
combining anti-VEGF therapy with checkpoint inhibitors in
hepatocellular carcinoma. Immunotherapy. 8:299–313. 2016.
View Article : Google Scholar : PubMed/NCBI
|