Introduction
Primary liver cancer (PLC) is one of the most fatal
types of cancer in humans with a rising incidence worldwide. It
accounts for 70–90% of the total liver cancer burden and is the
third most common cause of cancer-associated mortality globally
(1). Long-term prognosis of PLC
remains poor, with the majority of patients succumbing to disease
due to recurrence. Therefore, understanding the pathogenesis of
primary PLC is crucial for improving the efficacy of current
treatment strategies.
The liver tumor microenvironment is an essential
contributor to PLC initiation and progression. It has been
demonstrated that various stromal cell types are recruited to
neoplasms, where they are activated and substantially promote the
proliferation, invasion and metastatic potential of cancer cells
(2,3).
Hepatic stellate cells (HSCs) belong to one of the most important
stromal cell types in the liver tumor environment. Numerous prior
studies have revealed that culturing hepatocytes and LX2 cells (a
spontaneous immortalized human HSC cell line) results in
bidirectional cross-talk, with LX2 cells promoting PLC
proliferation and migration, thus inducing an inflammatory reaction
(4,5).
Simultaneous in vivo subcutaneous implantation of human HSCs
and PLC cells in nude mice promotes tumor growth, invasiveness and
inhibits necrosis (6).
Neuropilin-1 (NRP-1) is a transmembrane receptor for
class 3 semaphorins (7) and vascular
endothelial growth factor isoforms (8). It is expressed in a wide range of
tissues and mediates diverse cellular functions, including
migration, adhesion, proliferation and apoptosis (9,10).
Recently, NRP-1 has been implicated in HSC activation and cirrhosis
progression (11). However, the
effect of HSCs on PLC cells following NRP-1 expression silencing
remains unclear. The present study demonstrated that silencing
NRP-1 expression of HSCs may inhibit the activation of HSCs, as
well as attenuate the malignant progression of PLC cells in
vitro and in vivo.
Materials and methods
Cell lines and culture
The LX2 human HSCs were provided by the American
Type Culture Collection (Manassas, VA, USA). HepG2 human
hepatoblastoma cells were provided by the Translation Medicine
Center of Xi'an Jiaotong University (Xi'an, China). LX2 and HepG2
cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
HyClone; GE Healthcare, Chicago, IL, USA) supplemented with 10%
fetal bovine serum (FBS; MP Biomedicals, Solon, OH, USA) and 1%
streptomycin/penicillin (100 IU/ml; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) with 5% CO2 at 37°C
for 24 h in imunofluorescence staining, ELISA, migration and
invasion assays, 48 h in western blot analysis, 72 h in lentivirus
transfection, MTT assay and in vivo experiments.
Expression constructs and
transfection
Lentivirus pGCSIL-RFPshNRP1 was constructed in
preliminary experiments (12). LX2
cells were transfected with non-targeting (NT) short hairpin
(sh)RNA lentiviruses (NT shRNA) or NRP-1 shRNA lentiviruses to
yield stable NRP-1 knockdown LX2 cells (LX2-NRP-1 shRNA) and stable
control LX2 cells (LX2-NT shRNA). Transfection of LX2 with viral
particles was performed by incubating cells with viral supernatant
(25%) supplemented with polybrene (5 µg/ml; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) overnight at 37°C. Following
48 h, the cells were harvested for further experiments. Lentiviral
transduction efficiency was determined by western blot analysis. In
order to prepare the conditioned medium (CM), the cells in each
group were washed twice with serum-free DMEM one day following
seeding into T25 flasks (2×106 cells), and subsequently
incubated for 24 h with serum-free DMEM at 37°C.
MTT assay
For the MTT assay, stable NRP-1 knockdown LX2 and
HepG2 cells were used. Briefly, cells were seeded into 96-well
plates at 1×104 cells/well and stained with 100 µl MTT
(0.5 mg/ml; BioTime, Inc., Alameda, CA, USA) for 4 h at 37°C.
Subsequently, the culture medium was removed and 150 µl dimethyl
sulfoxide (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was added
to each well. The absorbance was evaluated at 490 nm. Experiments
were performed in triplicate and repeated three times with
consistent results.
Migration and invasion assays
In order to assess the paracrine effects of HSCs on
tumor invasion and migration, LX2 cells with or without NRP-1
knockdown were serum starved and CM were collected. The Transwell
chambers (pore size, 8.0 µm; EMD Millipore, Billerica, MA, USA)
without (for the migration assay) or with Matrigel (for the
invasion assay; BD Biosciences, Franklin Lakes, NJ, USA) coatings
were inserted into a 24-well culture plate.
For the migration assay, the HepG2 cells (100 µl,
5×104) suspended in DMEM supplemented with 1% FBS were
placed in the upper chamber and 0.5 ml CM collected from LX2-NRP-1
shRNA, LX2-NT shRNA and LX2-control was added into each lower
chamber as a chemoattractant. The Transwell chambers were then
incubated for 24 h.
For the invasion assay, 8-µm pore chamber inserts
were coated with Matrigel. HepG2 cells in the log phase of growth
were cultured in 6-well plates (100 µl; 5×105/ml) in
medium supplemented with 1% FBS for 24 h. The remaining steps were
the same as for the migration assay. The Transwell chambers were
incubated for 48 h.
The migrated and invaded cells on the underside of
the filter were fixed in 37% methanol and stained with crystal
violet (Boster Biological Technology, Pleasanton, CA, USA). Cell
migration and invasion was determined by counting the stained cells
in 10 randomly selected fields using a light microscope
(magnification, ×100).
ELISA
To detect the expression levels of soluble
transforming growth factor (TGF)-β1 secreted by LX2 cells,
2×105 LX2 cells with or without NRP-1 knockdown were
seeded into 6-well plates, grown for 48 h and the supernatant was
harvested for ELISA analysis. The human TGF-β1
Quantikine® ELISA kit from RapidBio Systems, Inc., (cat.
no. DRE10098; Bedford, MA, USA) was used to perform ELISA TGF-β1
evaluation, according to the manufacturer's instructions. There
were 6 replicates for each evaluation and the assessment was
repeated three times.
In vivo experiments
Nude mice (n=24; age, 4 weeks; weight, 20±5 g;
male:female, 1:1) were purchased from the Shanghai Experimental
Animal Center (Shanghai, China). All nude mice were kept at the SPF
level Laboratory Animal Center of Xi'an Jiaotong University (Xi'an,
China). The housing conditions were as follows: Between 18 and 28°C
temperature, 50% relative humidity, 12/12 h lighr/dark cycle, 10
times/h of fresh air exchange, air flow speed <0.18 m/sec and
noise <60 dB. The required water was acidified after high
pressure disinfection (13). The food
was treated with ultraviolet radiation. HepG2 cells
(1×106) with or without LX2 (2×105) cell
suspensions were injected subcutaneously into the armpits of nude
mice to establish subcutaneous xenograft models without anesthesia
to avoid affecting HepG2 cell proliferation. Tumor sizes were
evaluated using calipers every 7 days. The size of PLC xenografts
were evaluated using the following formula: Volume =
AxB2 ×0.52 (A, length; B, width; all measurements were
in millimeters). All nude mice were sacrificed after 4 weeks by
decapitation following 5% isoflurane for induction of anesthesia
and 1.5% isoflurane for the maintenance of anesthesia. The maximum
tumor size was ~236 mm3. The present study was approved
by the Ethics Committee of Xi'an Jiaotong University.
Immunohistochemical (IHC)
staining
IHC staining assays were performed according to the
protocol described previously (14).
Formalin-fixed and paraffin-embedded tissue samples were cut into
4-µm thick sections, deparaffinized with xylene and rehydrated in
100, 95, 90, 80 and 75% ethanol. Following washing in PBS, the
tissue samples were boiled in antigen-retrieval buffer containing
0.01 M sodium citrate-hydrochloric acid (pH 6.0) for 15 min at
90–100°C. The slides were rinsed with PBS and blocked overnight at
4°C. Following three washes in PBS, the slides were incubated with
a mouse monoclonal antibody directed against α-smooth muscle actin
(α-SMA; 1:1,000; cat. no. EPR5368; Abcam, Cambridge, USA) at 4°C
overnight. Subsequently, the slides were incubated with a goat
anti-rabbit immunoglobulin G (IgG; cat. no. E031320-01; EarthOx,
LLC, San Francisco, CA, USA) antibody. The bound antibody was
visualized using horseradish-peroxidase-streptavidin conjugates.
The tissue sections were counterstained with hematoxylin,
dehydrated in 75, 80, 90, 95, 80 and 100% ethanol and 99% xylene
and then mounted and examined using a light microscope (CX31RTSF;
magnification, ×100; Olympus, Tokyo, Japan).
Immunofluorescence staining
For fluorescent immunocytochemistry, the LX2 cells
were fixed for 20 min in 4% paraformaldehyde in PBS, and the
endogenous peroxidase activity was blocked using 3% hydrogen
peroxide. The tissue samples were permeabilized with 0.3% Triton
X-100 supplemented with 1% normal goat serum (ZSGB-BIO, Beijing,
China) in PBS for 20 min on ice, pre-blocked for 30 min with bovine
serum albumin (MP Biomedicals) at 37°C and incubated with
anti-Neuropilin-1 antibody (1:100; cat. no. EPR3113; Abcam) or
anti-PDGFR-β (cat. no. sc-1627; Santa Cruz Biotechnology, Inc.)
overnight at 4°C. Staining was detected using
fluorescein-conjugated secondary antibodies (Red Donkey anti-rabbit
IgG, cat. no. A24421, and Green Donkey anti-goat IgG, cat. no.
A24231 both from Abbkine, Inc., Redlands, CA, USA). Cell nuclei
were counterstained with DAPI (1:1,000; Sigma-Aldrich; Merck KGaA).
Slides were mounted and examined using a Nikon Corporation confocal
microscope (magnification, ×200) (Nikon Corporation, Tokyo,
Japan).
Western blot analysis
The BCA assay kit (Shaanxi Pioneer Biotech Co.,
Ltd., Xi'an, China) was applied to detect protein concentrations.
The LX2-NRP-1 shRNA, LX2-NT shRNA and LX2-control cells were lysed
separately using cell lysis buffer (Shaanxi Pioneer Biotech Co.,
Ltd.) with protease inhibitors (Roche Diagnostics, Indianapolis, IN
USA). Protein samples (10 µl) were electrophoretically resolved via
denaturing SDS-PAGE and electro-transferred onto nitrocellulose
membranes. The membranes were initially blocked with 5% non-fat dry
milk in Tris-buffered saline for 2 h and subsequently probed with
antibodies against NRP-1, extracellular signal-related kinase
(ERK), phosphorylated-ERK and α-SMA. Following co-incubation with
the primary antibodies at 4°C overnight, the membranes were
hybridized with the appropriate goat anti-mouse or anti-rabbit
secondary antibody (Abcam) for 1 h at room temperature. The
proteins were normalized to β-actin. The probed proteins were
detected using enhanced chemiluminescence (EMD Millipore) and
quantified by Image-pro plus 6.0 (Media Cybernetics, Rockville, MD,
USA).
Statistical analysis
All data were analyzed using SPSS v.13.0 software
(SPSS, Inc., Chicago, IL, USA). Data are presented as the mean ±
standard deviation. Differences among the groups were compared with
one-way ANOVA and the LSD-t test. Categorical data were compared
with χ2 tests. All statistical tests were two-tailed and
P<0.05 was considered to indicate a statistically significant
difference.
Results
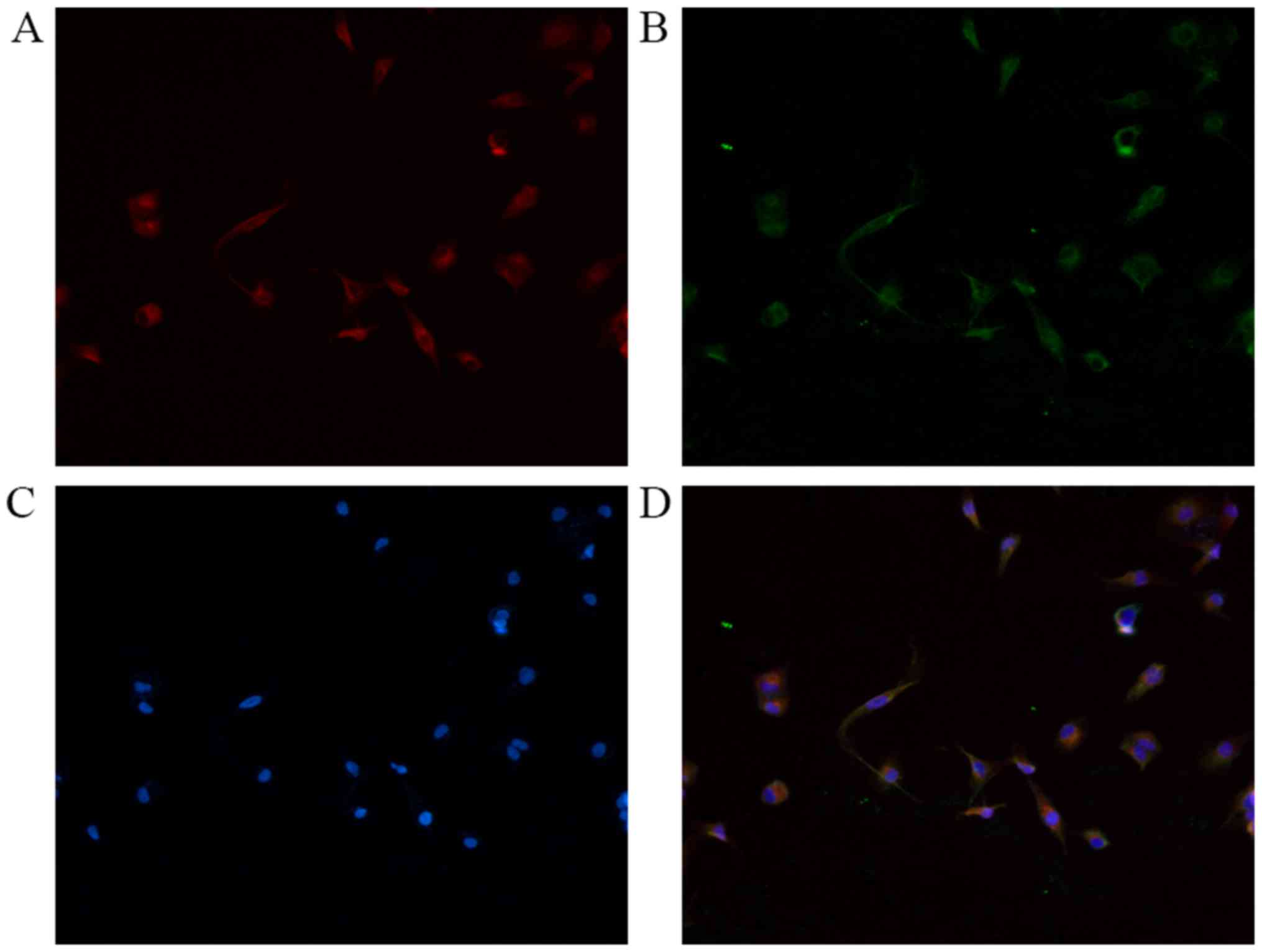
NRP-1 expression level and its
co-localization with platelet-derived growth factor receptor-β
(PDGFR-β) in LX2 cells
Immunofluorescence double staining was performed for
NRP-1 with PDGFR-β. As presented in Fig.
1, NRP-1 and PDGFR-β were detected on the surface of LX2 cells.
The results confirmed that LX2 cells expressed NRP-1 and
co-expressed with PDGFR-β in the cell membrane.
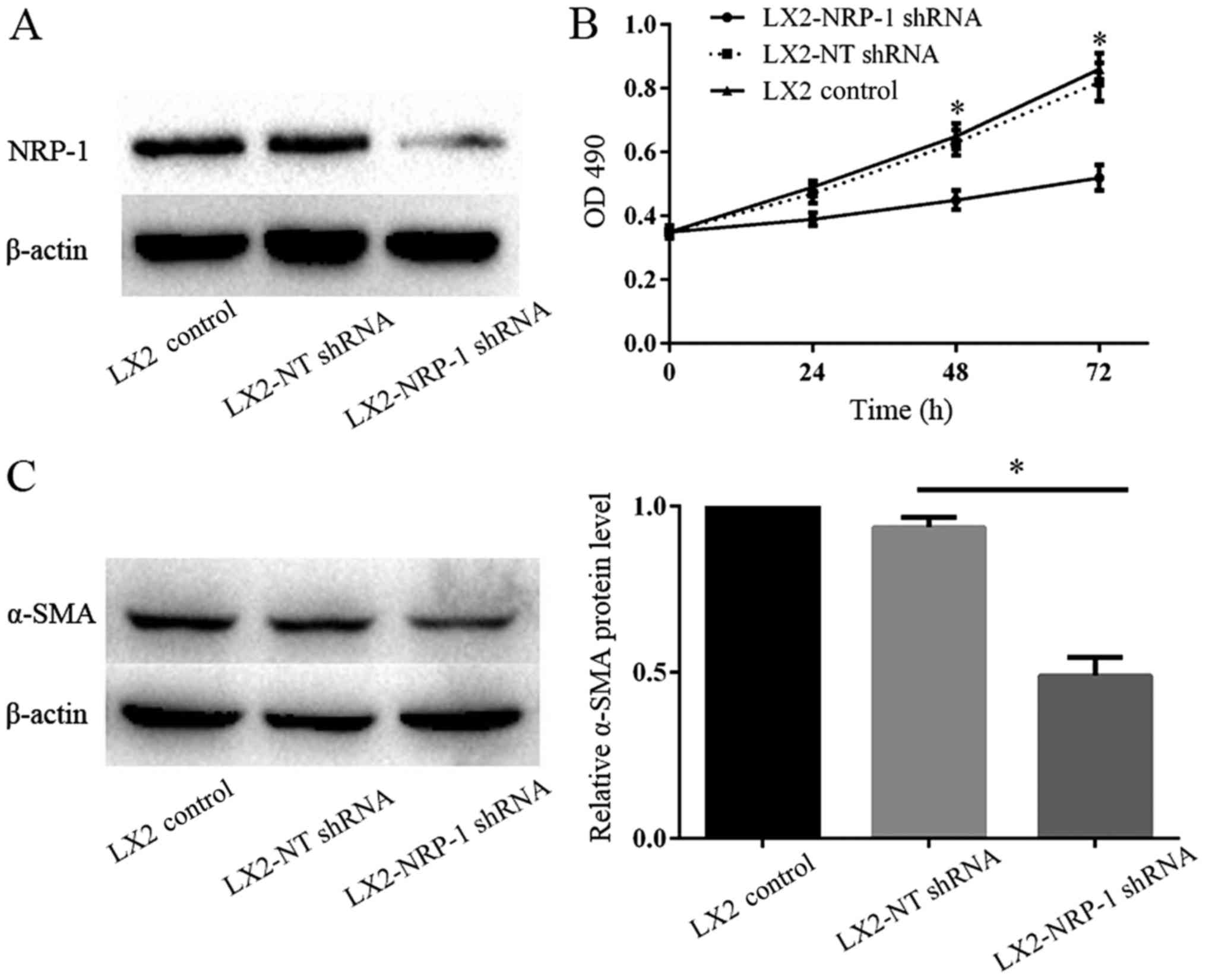
Viability of LX2 cells decreases
following silencing of NRP-1 expression
The lentiviral-based shRNA transduction system is
known to provide a high transduction rate and stable, long-term
gene knockdown in HSCs (15). Western
blot analysis verified that NRP-1 expression in LX2-NRP-1 shRNA
cells was significantly low when compared with that in LX2-NT shRNA
cells (Fig. 2A). MTT assays were
performed to detect LX2 cell proliferation following the silencing
of NRP-1. The results revealed that there were significant
differences in the optical density at various time points (24, 48
and 72 h) between the LX2-NRP-1 shRNA and the LX2-NT shRNA groups
(Fig. 2B). The expression of α-SMA is
an important indicator of HSC activation; therefore, the present
study compared the α-SMA protein expression levels in the control
and NRP-1-knockdown LX2 cells using western blot analysis. As
presented in Fig. 2C, the expression
level of α-SMA in LX2-NRP-1 shRNA cells was significantly low when
compare with in the LX2-NT shRNA cells. Thus, silencing NRP-1
expression in LX2 cells inhibited the activation of LX2 cells.
NRP-1 knockdown impairs the paracrine
effects of LX2 cells on tumor cell proliferation, migration and
invasion in vitro
CM from LX2-NRP-1 shRNA cells or LX2-NT shRNA cells
was collected and used as a stimulant for tumor cells in the MTT
and Transwell chamber assays. CM from the control and NRP-1
knockdown LX2 cells promoted HepG2 proliferation, migration and
invasion in vitro, compared with the basal medium (Fig. 3A-C). However, CM of NRP-1 knockdown
LX2 cells was less effective compared with that of the control LX2
cells (Fig. 3A-C). Therefore, the
knockdown of NRP-1 impaired the effects of LX2 cells on the
promotion of tumor cell proliferation, migration and invasion in
vitro. To elucidate the potential molecular mechanisms
underlying this process, an ELISA was performed and the results
demonstrated there were markedly higher expression levels of
soluble TGF-β1 in LX2-NT shRNA CM, as compared with in the CM from
LX2-NRP-1 shRNA cells (Fig. 3D).
Western blotting revealed that, compared with the LX2 and NRP-1
shRNA groups, CM from LX2-NT shRNA cells induced a high
phosphorylation level of p42/p44 mitogen-activated protein kinases
in HepG2 cells (Fig. 3E).
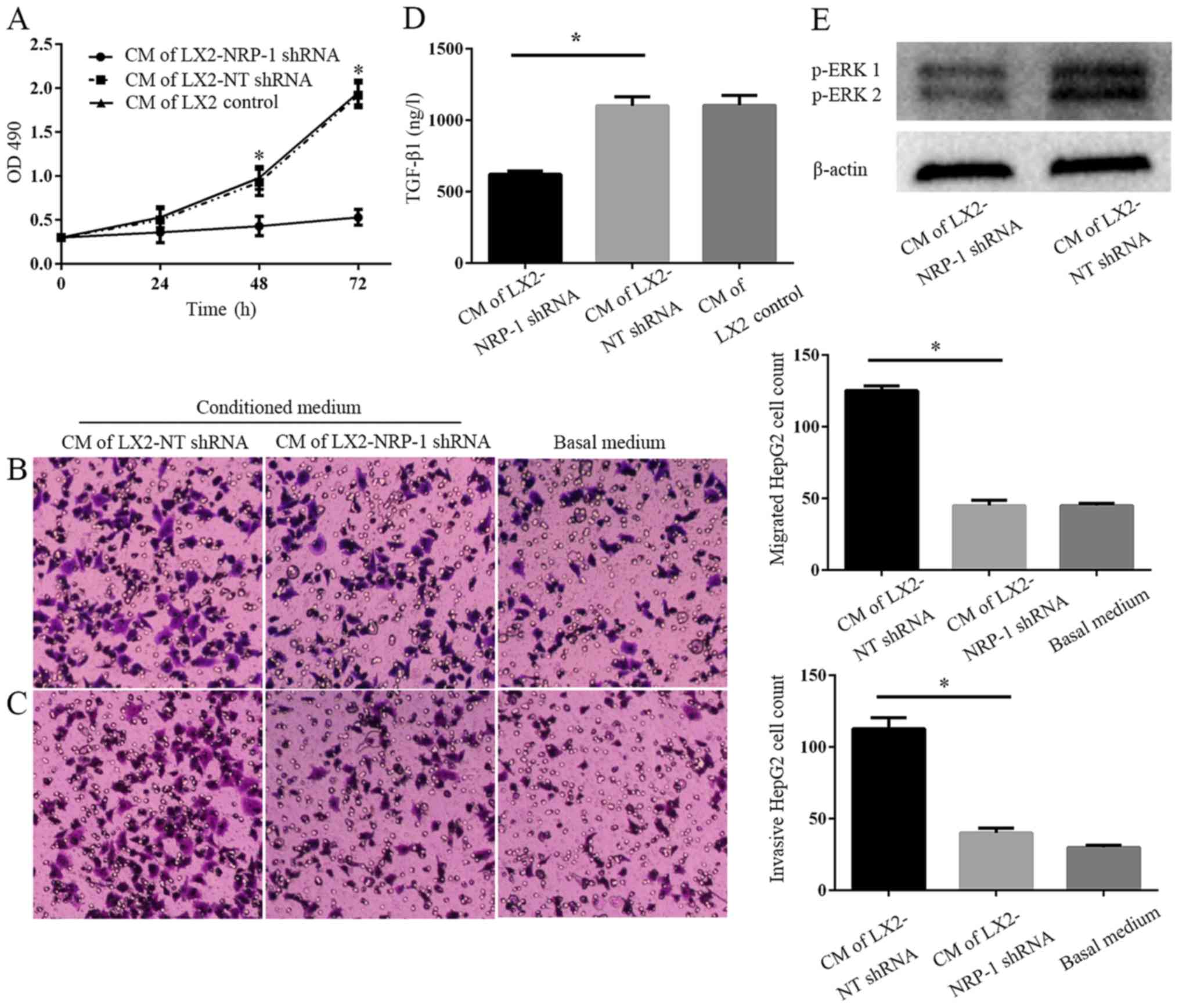 | Figure 3.NRP-1 knockdown impairs the effects of
LX2 cells on tumor proliferation, migration and invasion in
vitro. (A) CM were collected from control LX2 cells (transduced
with NT shRNA lentiviruses) and NRP-1 knockdown LX2 cells
(transduced with NRP-1 shRNA lentiviruses). HepG2 cells cultured in
LX2 cells CM were subjected to MTT assay analysis. CM of NRP-1
knockdown LX2 cells was less effective in promoting HepG2
proliferation compared with that of control LX2 cells. *P<0.05
by analysis of variance; n=3 repeats with similar results. CM
described in (A) was used as the chemoattractant in the Transwell
chamber assays. CM of NRP-1 knockdown LX2 cells was less effective
at promoting HepG2 (B) migration and (C) invasion in comparison
with that of control LX2 cells. *P<0.05 by analysis of variance;
n=3 repeats (magnification, ×100). (D) There was a markedly higher
expression level of soluble TGF-β1 in CM from LX2-NT shRNA compared
with LX2-NRP-1 shRNA CM (*P<0.05) as assessed by ELISA; n=3. (E)
Western blot analysis using an antibody against phosphorylated ERK,
p44 and p42, ERK1 and ERK2; n=3. CM, conditioned media; NT,
non-targeting; shRNA, short hairpin RNA; NRP-1, neuropilin-1; ERK,
extracellular signal-related kinase; OD, optical density; TGF-β1,
transforming growth factor β1. |
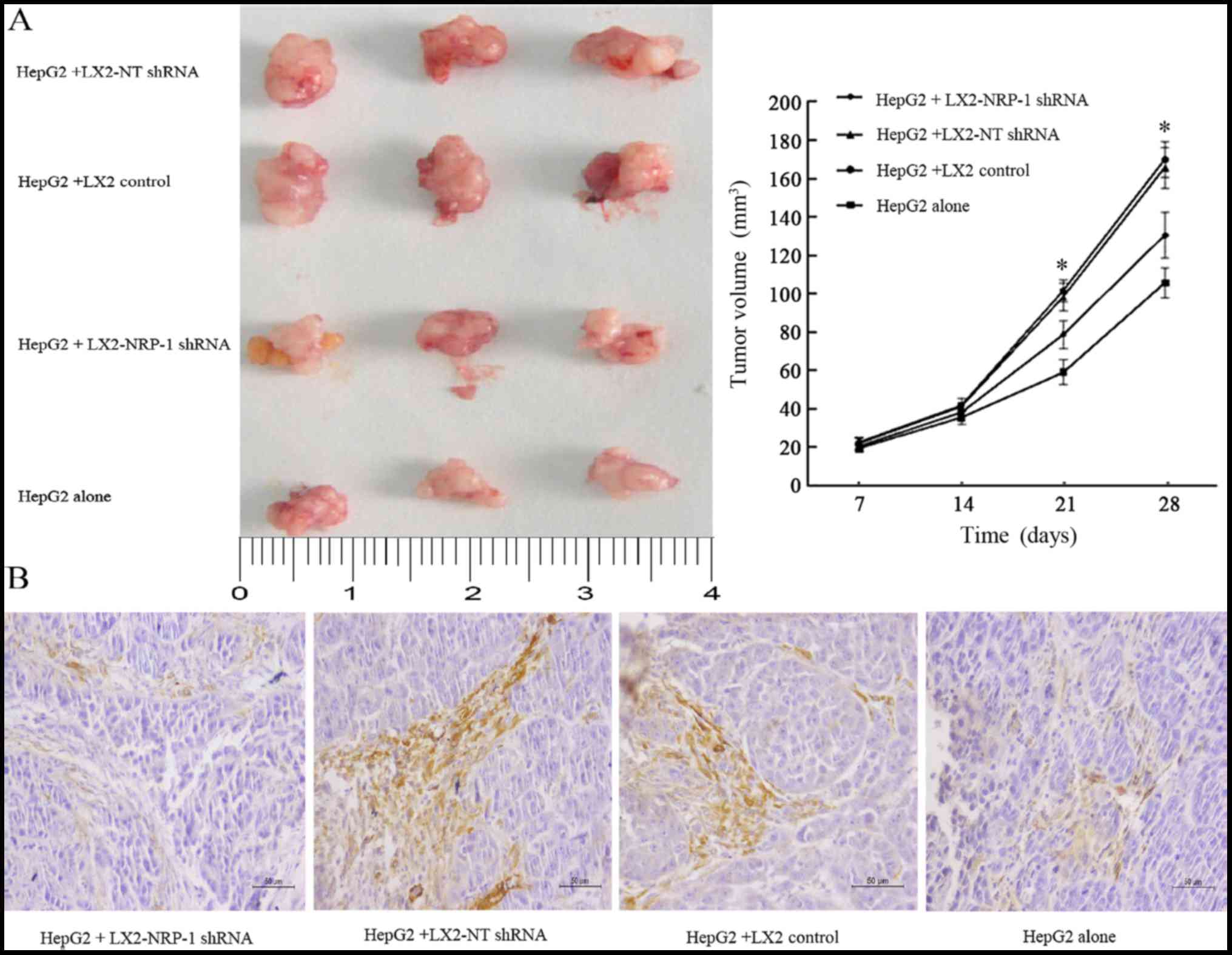
NRP-1 knockdown reduces the
stimulatory effect of LX2 cells on tumor growth in mice
The present study co-implanted HepG2 and LX2 cells
into mice in order to investigate whether NRP-1 in LX2 cells
influenced tumor growth in mice. A mixture of HepG2 cells
(1×106) and LX2 cells (2×105) cells
expressing either NT shRNA or NRP-1 shRNA were implanted into nude
mice via subcutaneous injection. Tumor growth curves revealed that
control and NRP-1-knockdown LX2 cells promoted HepG2 tumor growth
in mice (Fig. 4A). The average tumor
size of NRP-1 knockdown LX2 cells was less than that of the control
LX2 cells (Fig. 4A). Knockdown of
NRP-1 attenuated the effect of LX2 cells with regard to promoting
tumor growth in mice.
Expression of α-SMA weakens in
xenograft tumors
IHC analysis of α-SMA, an established marker of
activated HSC, revealed that the expression level of α-SMA in the
stroma of tumors formed by the LX2-NRP-1 shRNA group were lower
compared with in the LX2-NT shRNA and the LX2 control group,
whereas the expression was low or absent in the HepG2 alone group
(Fig. 4B).
Discussion
HCC is the most common type of primary tumor in the
liver (16). Although advances have
been made regarding the cellular and molecular mechanisms
underlying liver carcinogenesis, PLC has one of the highest
mortality rates worldwide; therefore, the development of innovative
therapeutic options is required. An increasing number of studies
have highlighted the crucial role of the tumor microenvironment in
the pathogenesis of cancer. Thus, targeting the tumor
microenvironment is now viewed as a promising therapeutic strategy
for treating cancer in a variety of organs, including the liver
(17,18). As an important stromal cell type in
the liver tumor environment, HSCs may represent attractive targets
in the design of innovative therapeutic strategies against liver
carcinogenesis.
It has previously been reported that PDGF, the
strongest cytokine promoting HSC activation, may bind with its
receptor, PDGFR-β, and promote HSC activation, whereas NRP-1
contributes to the promotion of PDGF-mediated activation of HSCs
(11). The present study revealed
that NRP-1 was expressed on the membrane of LX2 cells and
co-localized with PDGFR-β. Furthermore, it was demonstrated that
silencing of NRP-1 expression of LX2 cells inhibited the
proliferation and α-SMA expression levels of LX2 cells, thus the
activation of LX2 cells was inhibited.
HSC promotion of the proliferation, migration and
invasion of PLC cells relies on the mechanisms underlying activated
HSC-mediated transformation into myofibroblasts, which secrete
large amounts of cytokines, extracellular matrix proteins and
integrin-metalloproteinase-9 (ADAM9) (19). These factors include TGF-β, PDGF,
hepatocyte growth factor, stromal cell-derived factor-1 and ADAM9,
which are able to promote the proliferation of and trigger
epithelial-mesenchymal transition in PLC cells, resulting in the
enhancement of cell migration and invasion (20,21). In
the present study, following NRP-1 expression silencing, the effect
of LX2 cells on the promotion of HepG2 cell proliferation was
reduced. Similarly, the effect of CM on promoting the migration and
invasion of HepG2 cells was also significantly reduced. Therefore,
the present study suggested that the attenuated effects of HepG2
cell proliferation, migration and invasion may be attributed to the
reduction in cytokines and matrix components in the CM.
Subsequently, the present study performed an ELISA and revealed
that the expression of TGF-β1 protein in LX2-NT shRNA CM was
increased compared with that in LX2-NRP-1 shRNA CM. The present
study suggested that combinations of these factors, and others
factors not discussed herein, that are secreted by activated HSCs
may induce the tumorigenic effects on HepG2 cells observed in the
present and prior study (22).
In addition to the soluble factors released by
activated HSC, further mechanisms may be involved in the observed
carcinogenic effects. For example, Coulouarn and Clément (23) revealed that HSCs affect the
progression and metastasis of hepatic tumors through extracellular
matrix remodeling. A previous study investigating other types of
tumors demonstrated that tumor cells further induced the expression
of tumorigenic factors via activated HSCs, indicating a mutual
interaction of cancer cells and stromal myofibroblasts in the
promotion of carcinogenesis (24).
As a potential molecular mechanism underlying the
effect of LX2 cells on HepG2 cells, the present study identified
that the activity of ERK in HepG2 cells induced by CM collected
from LX2-NRP-1 shRNA cells was decreased as compared with the
LX2-NT shRNA. It has previously been demonstrated that aberrant
activation of the mitogen activated protein kinase/ERK signaling
pathway was involved in the progression of human PLC (25). Increased ERK activation is known to
induce HCC cell proliferation and to protect HCC cells from
apoptosis (26). Furthermore,
increased ERK activity has been revealed to affect the migratory
activity and invasiveness of HCC cells, suggesting that this
molecular pathway may be critical in the intrahepatic metastasis of
HCC (27).
In vivo experiments confirmed that HSCs
promotes the growth of PLC and that this effect was attenuated
following NRP-1 expression silencing. Subsequent analysis of the
interaction between HSCs and HepG2 cells in vivo
demonstrated α-SMA expression levels in the LX2-NRP-1 shRNA group
were lower compared with in the LX2-NT shRNA and LX2 control
groups, whereas the expression levels were low or absent in the
HepG2 alone group. This finding indicated that the activation of
LX2 cells was altered and α-SMA expression was downregulated
following NRP-1 expression silencing, which was consistent with the
results of the in vitro experiments.
The HepG2 cell line was used to investigate primary
liver cancer in this study, it was originally thought to be a
hepatocellular carcinoma cell line but has been revealed to derive
from hepatoblastoma (28). However,
it is a suitable in vitro and in vivo model for the
study of hepatocarcinogenesis, and such misidentification is
unlikely to affect the outcome of the study.
To conclude, the present study observed NRP-1
expression in and confirmed the co-localization of NRP-1 and
PDGFR-β in LX2 cells. Following silencing of NRP-1 expression in
LX2 cells, the activation of LX2 cells was inhibited, and the
effect on the promotion of proliferation, migration and invasion of
HepG2 cells was reduced. Finally, in vivo experiments
demonstrated that silencing NRP-1 expression in HSCs may attenuate
the growth of PLC, highlighting the contribution of NRP-1 as a
potential target for PLC therapy.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81572420).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics. CA Cancer
J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Albini A and Sporn MB: The tumour
microenvironment as a target for chemoprevention. Nat Rev Cancer.
7:139–147. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yan XL, Fu CJ, Chen L, Qin JH, Zeng Q,
Yuan HF, Nan X, Chen HX, Zhou JN, Lin YL, et al: Mesenchymal stem
cells from primary breast cancer tissue promote cancer
proliferation and enhance mammosphere formation partially via
EGF/EGFR/Akt pathway. Breast Cancer Res Treat. 132:153–164. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coulouarn C, Corlu A, Glaise D, Guénon I,
Thorgeirsson SS and Clément B: Hepatocyte-stellate cell cross-talk
in the liver engenders a permissive inflammatory microenvironment
that drives progression in hepatocellular carcinoma. Cancer Res.
72:2533–2542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Zijl F, Mair M, Csiszar A, Schneller
D, Zulehner G, Huber H, Eferl R, Beug H, Dolznig H and Mikulits W:
Hepatic tumor-stroma crosstalk guides epithelial to mesenchymal
transition at the tumor edge. Oncogene. 28:4022–4033. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Amann T, Bataille F, Spruss T, Mühlbauer
M, Gäbele E, Schölmerich J, Kiefer P, Bosserhoff AK and Hellerbrand
C: Activated hepatic stellate cells promote tumorigenicity of
hepatocellular carcinoma. Cancer Sci. 100:646–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kolodkin AL, Levengood DV, Rowe EG, Tai
YT, Giger RJ and Ginty DD: Neuropilin is a semaphorin III receptor.
Cell. 90:753–762. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soker S, Takashima S, Miao HQ, Neufeld G
and Klagsbrun M: Neuropilin-1 is expressed by endothelial and tumor
cells as an isoform-specific receptor for vascular endothelial
growth factor. Cell. 92:735–745. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pellet-Many C, Frankel P, Jia H and
Zachary I: Neuropilins: Structure, function and role in disease.
Biochem J. 411:211–226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zachary IC, Frankel P, Evans IM and
Pellet-Many C: The role of neuropilins in cell signalling. Biochem
Soc Trans. 37:1171–1178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao S, Yaqoob U, Das A, Shergill U,
Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E,
et al: Neuropilin-1 promotes cirrhosis of the rodent and human
liver by enhancing PDGF/TGF-beta signaling in hepatic stellate
cells. J Clin Invest. 120:2379–2394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng JB, Geng ZM, Chen Q and Wang L:
Construction of recombinant lentiviral vector containing shRNA for
human neuropilin-1 gene. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi.
28:1200–1203. 2012.(In Chinese). PubMed/NCBI
|
|
13
|
Tusnio A, Taciak M, Barszcz M,
Paradziej-Łukowicz J, Olędzka I, Wiczkowski W, Szumska M and
Skomiał J: Thermal sterilization affects the content of selected
compounds in diets for laboratory animals. J Anim Feed Sci.
23:351–360. 2014. View Article : Google Scholar
|
|
14
|
Chen C, Shen H, Tao J, Song H, Ma L, Wang
L and Geng Z: Effect of cancer-associated fibroblasts on
proliferation and invasion of gallbladder carcinoma cells. Nan Fang
Yi Ke Da Xue Xue Bao. 35:1149–1154. 2015.(In Chinese). PubMed/NCBI
|
|
15
|
Liu C, Billadeau DD, Abdelhakim H, Leof E,
Kaibuchi K, Bernabeu C, Bloom GS, Yang L, Boardman L, Shah VH and
Kang N: IQGAP1 suppresses TβRII-mediated myofibroblastic activation
and metastatic growth in liver. J Clin Invest. 123:1138–1156. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Omata M, Lesmana LA, Tateishi R, Chen PJ,
Lin SM, Yoshida H, Kudo M, Lee JM, Choi BI, Poon RT, et al: Asian
pacific association for the study of the liver consensus
recommendations on hepatocellular carcinoma. Hepatol Int.
4:439–474. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Junttila MR and de Sauvage FJ: Influence
of tumour micro-environment heterogeneity on therapeutic response.
Nature. 501:346–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hernandez-Gea V, Toffanin S, Friedman SL
and Llovet JM: Role of the microenvironment in the pathogenesis and
treatment of hepatocellular carcinoma. Gastroenterology.
144:512–527. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Carloni V, Luong TV and Rombouts K:
Hepatic stellate cells and extracellular matrix in hepatocellular
carcinoma: More complicated than ever. Liver Int. 34:834–843. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mikula M, Proell V, Fischer AN and
Mikulits W: Activated hepatic stellate cells induce tumor
progression of neoplastic hepatocytes in a TGF-beta dependent
fashion. J Cell Physiol. 209:560–567. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fischer AN, Fuchs E, Mikula M, Huber H,
Beug H and Mikulits W: PDGF essentially links TGF-beta signaling to
nuclear beta-catenin accumulation in hepatocellular carcinoma
progression. Oncogene. 26:3395–3405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gupta DK, Singh N and Sahu DK: TGF-β
mediated crosstalk between malignant hepatocyte and tumor
microenvironment in hepatocellular carcinoma. Cancer Growth
Metastasis. 7:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coulouarn C and Clément B: Stellate cells
and the development of liver cancer: Therapeutic potential of
targeting the stroma. J Hepatol. 60:1306–1309. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lu Y, Lin N, Chen Z and Xu R:
Hypoxia-induced secretion of platelet-derived growth factor-BB by
hepatocellular carcinoma cells increases activated hepatic stellate
cell proliferation, migration and expression of vascular
endothelial growth factor-A. Mol Med Rep. 11:691–697. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Min L, He B and Hui L: Mitogen-activated
protein kinases in hepatocellular carcinoma development. Semin
Cancer Biol. 21:10–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takeishi K, Taketomi A, Shirabe K, Toshima
T, Motomura T, Ikegami T, Yoshizumi T, Sakane F and Maehara Y:
Diacylglycerol kinase alpha enhances hepatocellular carcinoma
progression by activation of Ras-Raf-MEK-ERK pathway. J Hepatol.
57:77–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chan LK, Chiu YT, Sze KM and Ng IO:
Tensin4 is up-regulated by EGF-induced ERK1/2 activity and promotes
cell proliferation and migration in hepatocellular carcinoma.
Oncotarget. 6:20964–20976. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: Hep G2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|