Introduction
In recent years, people have an increased risk of
hepatocellular carcinoma (HCC) due to living habits (heavy alcohol
drinking and tobacco smoking) and living in a worsening environment
(polluted air) (1). A previous study
demonstrated that liver cancer or primary hepatic cancer is the
fifth most common type of global malignancy and the third most
common cause of cancer-associated mortality globally (2). An effective chemotherapy for HCC cancer
has not yet been identified (3).
Sorafenib is the first-line treatment; however, this only has a
limited effect on increasing the survival time of patients with
HCC, with the median OS extended by approximately 3 months
(4). Therefore, it remains a
challenge to identify a novel effective therapeutic agent with low
toxicity for the treatment of HCC.
Betulin (BT), a member of pentacycliclupane-type
triterpenes primarily located in the white birch, has been
demonstrated to exhibit a number of biological functions including
anticancer, anti-human immunodeficiency virus and anti-inflammatory
effects (5,6). BT is a traditional medicine and has been
used for the treatment of actinic keratosis for a number of years
in Germany (7). A previous study
disproved the significance of BT in melanoma cells (8); however, subsequent studies have
demonstrated the anticancer activity of BT in a number of types of
human cancer including neuroblastoma (9), colon (10), breast (11), hepatocellular (12), lung (13), prostate (14), renal cell (15) and ovarian (16). In addition, it has been demonstrated
that the apoptotic properties of BT are due to modulation of the
B-cell lymphoma (Bcl-)2 family and cell cycle regulatory proteins
(12,13), and the activation of caspases and DNA
fragmentation (15,17).
To identify an agent which exhibit increased
inhibitory effects against distinct cancer cell lines and decreased
toxicity compared with BT, a variety of BT derivatives have been
synthesized (18–20). A previous study demonstrated that the
C-3 or C-28 positions serve a function in the pharmacological
activities of BT (21). On the basis
of this principle, in the present study, a library of semisynthetic
analogs of BT were synthesized, aiming at substituents with the C-3
or/and C-28 position. The results of the present study identified
that 3,28-di-(2-nitroxy-acetyl)-oxy-BT exhibited the most
significant effect on Huh7 cells. To the best of our knowledge, the
present study was the first to demonstrate that
3,28-di-(2-nitroxy-acetyl)-oxy-BT inhibited Huh7 cell growth, by
inducing mitochondrion-mediated cell apoptosis and G2/M
cell cycle arrest. Furthermore, the results of the present study
identified that 3,28-di-(2-nitroxy-acetyl)-oxy-BT inhibited the
phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) signaling
pathway. These results suggested that
3,28-di-(2-nitroxy-acetyl)-oxy-BT may be used for the clinical
treatment of HCC.
Materials and methods
Reagents
RPMI-1640 culture medium, fetal bovine serum (FBS),
trypsin, penicillin-streptomycin were purchased from Gibco; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). MTT, dimethyl sulfoxide
(DMSO), propidium iodide (PI) and RNase A were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). An Annexin
V-fluorescein isothiocyanate (FITC)/PI double staining kit was
purchased from Nanjing Institute of Biological Engineering
(Nanjing, China) and 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetramethyl
benzimidazolyl-carbocyanine iodide (JC-1) was obtained from
Invitrogen; Thermo Fisher Scientific, Inc. All antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA).
Synthesis of
3,28-di-(2-nitroxy-acetyl)-oxy-BT
BT (purity >95%) was obtained from XiaoGan
ShenYuan Chemical Co., Ltd. (XiaoGan, China). The reaction of BT
with bromoacetyl bromide (Thermo Fisher Scientific, Inc.) yielded
3,28-di-(2-bromo-acetyl)-oxy-BT. This compound reacted with silver
nitrate to form 3,28-di-(2-nitroxy-acetyl)-oxy-BT (Fig. 1A). The structure of
3,28-di-(2-nitroxy-acetyl)-oxy-BT was identified by infrared
spectroscopy (IR), 1D nuclear magnetic resonance (NMR) and
high-resolution mass spectrometry (HRMS). IR (KBr)/cm−1:
2918, 2850, 1742, 1655, 1467, 1384, 1292. 1H NMR (400
MHz, CDCl3) δ: 0.83, 0.85, 0.86, 0.87, 0.89, 1.04 (s,
18H, 6×CH3), 2.25 (m, 1H, H-19), 4.61 (d, 1H, J 7.5 Hz,
H-29b), 4.64 (d, 1H, J 7.5 Hz, H-29a), 3.83 (m, 2H), 4.89, 4.88 (s,
2×CH2ONO2). 13C NMR (100 MHz,
CDCl3) δ: 38.5 (C-1), 23.6 (C-2), 83.6 (C-3), 40.8
(C-4), 55.5 (C-5), 18.1 (C-6), 34.6 (C-7), 43.3 (C-8), 51.0 (C-9),
37.1 (C-10), 21.5 (C-11), 22.6 (C-12), 37.6 (C-13), 51.0 (C-14),
28.2 (C-15), 31.9 (C-16), 37.9 (C-17), 52.1 (C-18), 48.8 (C-19),
144.1 (C-20), 29.3(C-21), 34.9 (C-22), 27.9 (C-23), 16.7 (C-24),
16.5 (C-25), 15.5 (C-26), 14.1 (C-27), 67.0 (C-28), 109.6 (C-29),
21.0 (C-30), 165.6 (C-31, C-31′), 67.4 (C-32, C-32′). HRMS (m/z)
calculated for
C34H52N2O10, 648.3622;
identified 648.3621. 3,28-di-(2-nitroxy-acetyl)-oxy-BT was
dissolved in DMSO until use.
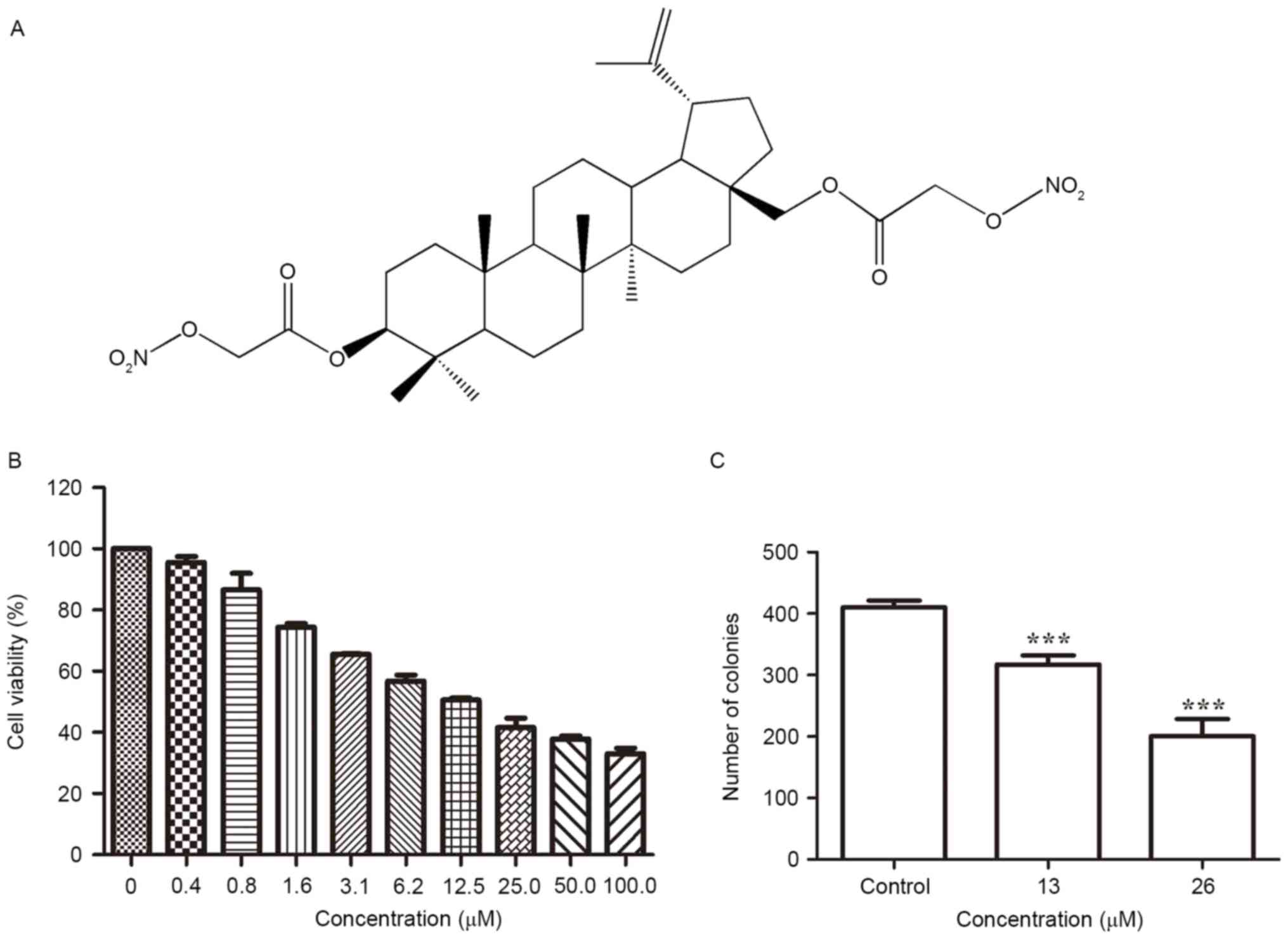 | Figure 1.Inhibitory effect of
3,28-di-(2-nitroxy-acetyl)-oxy-betulin on the viability of Huh7
cells. (A) Chemical structure of
3,28-di-(2-nitroxy-acetyl)-oxy-betulin. (B) Huh7 cells seeded in
96-well plates were exposed to
3,28-di-(2-nitroxy-acetyl)-oxy-betulin (0, 0.4, 0.8, 1.6, 3.1, 6.2,
12.5, 25.0, 50.0 and 100.0 µM) for 48 h. Cell viability was
determined using the MTT assay and demonstrated to decrease in a
concentration-dependent manner. (C) The clonogenicity of Huh7 cells
following treatment with 3,28-di-(2-nitroxy-acetyl)-oxy-betulin was
determine using a colony-formation assay. The number of colonies
decreased in a concentration-dependent manner. The data are
expressed as the mean ± standard deviation of three independent
experiments. ***P<0.001 vs. controlusing one-way analysis of
variance followed by Tukey's test. |
Cell culture
The human hepatocellular carcinoma cell line Huh7
was purchased from the Chinese Academy of Sciences (Shanghai,
China). Cells (passages <20) were cultured in 37°C in an
atmosphere containing 5% CO2 with RPMI-1640 medium,
supplemented with 10% FBS.
Cell viability assay
Huh7 cells (5×103 cells/well) in the
exponential growth phase (24 h following passage) were seeded in
96-well plates, cultured overnight in 37°C and added with various
concentrations of 3,28-di-(2-nitroxy-acetyl)-oxy-BT (0, 0.4, 0.8,
1.6, 3.1, 6.2, 12.5, 25.0, 50.0, 100.0 µM) at 37°C for 24 h.
Subsequently, MTT (5 mg/ml) was added to the cells and incubated in
the dark at 37°C for 4 h. The resulting formazan crystals were
dissolved using DMSO and the optical density was measured at 595 nm
to determine the half-maximal inhibitory concentration
(IC50) value, using the DTX 880 Multimode Detector
(Beckman Coulter, Inc., Brea, CA, USA).
Clonogenic assay
Cells (5×102 cells/well) in the
exponentialgrowth phase were seeded in 6-well plates and cultured
overnight at 37°C. The cells were treated with indicated
concentrations of 3,28-di-(2-nitroxy-acetyl)-oxy-BT (0, 13, 26 µM)
and cultured overnight at 37°C. Subsequently, the cells were
maintained for 14 days with fresh RPMI-1640 medium. Plates were
washed with PBS, and subsequently fixed with 75% methanol at 37°C
for 30 min and stained with 1% crystal violet at 37°C for 30 min.
The mixture was removed and the plates were washed with PBS and
allowed to dry at room temperature. The number of colonies >0.5
mm in diameter with 5 fields of view were counted under an inverted
phase-contrast IX51 microscope using ×10 magnification (Olympus
Corporation, Tokyo, Japan).
DNA content analysis
DNA content analysis was performed using
afluorescent probe, PI (Sigma-Aldrich, Merck KGaA), according to
the manufacturer's protocol. Huh7 cells (3×104
cells/well) in the exponential growth phase were seeded in 6-well
plates, cultured at 37°C for 24 h and added various concentrations
of 3,28-di-(2-nitroxy-acetyl)-oxy-BT (0, 13, 26 µM) at 37°C for 24
h. The cells were selected, washed three times with PBS and fixed
with 75% ethanol at 4°C overnight. Each sample was incubated with
0.05 mg/ml PI and 0.1 mg/ml RNase A for 30 min at 37°C. The DNA
content was determined using an Epics XL flow cytometer
(excitation, 488 nm; emission, 620 nm). The data were analyzed with
ModFit LT software (version 3.2; BD Biosciences, Franklin Lakes,
NJ, USA).
Apoptosis detection using Αnnexin
V-FITC/PI
The Αnnexin V-FITC/PI staining assay kit wasused to
evaluate the apoptosis rate (Nanjing Institute of Biological
Engineering, Nanjing, China). Huh7 cells (3×104
cells/well) were inoculated in 6-well plates and incubated with
various concentrations of 3,28-di-(2-nitroxy-acetyl)-oxy-BT (0, 13,
26 µM) at 37°C for 24 h. Subsequently, the cells were selected,
washed three times with PBS and incubated with Αnnexin V-FITC/PI at
37°C in the dark for 15 min. The apoptosis ratio was determined
using the Epics XL flow cytometer (Αnnexin V-FITC: Excitation, 488
nm; emission, 525 nm. PI: Excitation, 488 nm; emission, 620
nm).
Detection of mitochondrial membrane
potential
The JC-1 kit (Thermo Fisher Scientific, Inc.) was
used to detect mitochondrial membrane potential. Huh7 cells
(3×104 cells/well) in the exponential growth phase were
seeded in 6-well plates and incubated with various concentrations
of 3,28-di-(2-nitroxy-acetyl)-oxy-BT (0, 13, 26 µM) at 37°C for 24
h. The cells were selected, washed three times with PBS and
incubated with 10 µg/ml JC-1 in 37°C for 30 min in the dark.
Subsequently, the mitochondrial membrane potential alterations were
determined using the Epics XL flow cytometer (J-aggregates:
Excitation, 488 nm; emission, 575 nm. JC-1 monomers: Excitation,
488 nm; emission, 525 nm).
Western blot analysis
Huh7 cells (6×105 cells/100 mm dish) in
the exponential growth phase were incubated overnight with various
concentrations of 3,28-di-(2-nitroxy-acetyl)-oxy-BT. Following
treatment, the cells were selected, incubated with
radioimmunoprecipitation assay buffer (0.1 M phenylmethylsulfonyl
fluoride protease and phosphatase inhibitor cocktail;
Sigma-Aldrich; Merck KGaA) for 30 min on ice, centrifuged at 10,000
× g at 4°C for 15 min, and the supernatant was stored at −80°C. To
isolate the cytosolic fraction, the cells were selected, lysed in
cytosolic lysis buffer [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 300 mM
sucrose, 0.5% NP-40, 10 mMKCl] with protease and phosphatase
inhibitor cocktail (Sigma-Aldrich; Merck KGaA) for 30 min on ice
and centrifuged at 10,000 × g at 4°C for 15 min. Supernatant fluid
was selected as part of the cytoplasm. To isolate the mitochondrial
fraction, cell pellets were lysed in mitochondrial lysis buffer (50
mMTris-HCl, 150 mMNaCl and 1% NP-40) with protease and phosphatase
inhibitor (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) on ice
for 30 min, centrifuged at 10,000 × g at 4°C for 15 min and the
supernatant was selected. The cell lysates (50 µg) were separated
using 10% SDS-PAGE and transferred onto a polyvinylidene difluoride
membrane, blocked at 37°C for 1 h with 5% bovine serum albumin
(Beyotime Institute of Biotechnology, Haimen, China) and with the
primary antibody [cyclin B1, cat. no. 12231; CDK1, cat. no.
ab133327 (Abcam, Cambridge, MA, USA); CDC25C, cat. no. 4688; PARP,
cat. no. 9542; C-PARP, cat. no. 5625; caspase 3, cat. no. 9662;
caspase 9, cat. no. 9502; Bcl-2, cat. no. 7382; Bax, cat. no. 5023;
cytochrome c, cat. no. 11940; VDAC, cat. no. 4866; PI3K p110α, cat.
no. 4249; Akt, cat. no. 4691; p-AKT (Thr308), cat. no. 2965; p-AKT
(Ser473), cat. no. 4060; β-actin, cat. no. 4967; Cell Signaling
Technology, Inc.] at 4°C overnight (dilution, 1:1,000) and the
secondary antibody (anti-rabbit IgG, HRP-linked antibody, cat. no.
7074; dilution, 1:2,000; Cell Signaling Technology, Inc.).
Immunoreactive bands were visualized using an enhanced
chemiluminescence substrate (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) and a X-ray film processor (Kodak, Rochester, NY,
USA).
Statistical analysis
The data are presented as the mean ± standard
deviation. GraphPad Prism (version 5.0; GraphPad Software, Inc., La
Jolla, CA, USA) was used for statistical analysis. A one-way
analysis of variance followed by Tukey's test was performed.
P<0.05 was considered to indicate a statistically significant
difference.
Results
3,28-Di-(2-nitroxy-acetyl)-oxy-BT
inhibits the growth of Huh7 cells in vitro
Determined using an MTT assay,
3,28-di-(2-nitroxy-acetyl)-oxy-BT led to an anti-proliferative
effect on Huh7 cells and markedly decreased the viability of Huh7
cells in a dose-dependent manner (Fig.
1B). The IC50 value was identified to be 13.1±1.37
µM, following 24 h of treatment. To evaluate the long-term effect
on cell survival, a colony-formation assay was performed (Fig. 1C). The inhibitory effect of
3,28-di-(2-nitroxy-acetyl)-oxy-BT on colony formation was
demonstrated to be concentration-dependent, which validated the
cytotoxic effect of 3,28-di-(2-nitroxy-acetyl)-oxy-BT against Huh7
cells.
3,28-Di-(2-nitroxy-acetyl)-oxy-BT
induces cell cycle arrest and is associated with cell cycle
regulatory proteins
In order to determine whether the anti-proliferative
effect of 3,28-di-(2-nitroxy-acetyl)-oxy-BTon Huh7 cells was as a
result of cell cycle arrest, induced by
3,28-di-(2-nitroxy-acetyl)-oxy-BT, the cell cycle phase ratio was
determined using flow cytometry and PI staining. The results of the
present study demonstrated that, following exposure to 26 µM
3,28-di-(2-nitroxy-acetyl)-oxy-BT exposure (Fig. 2A), compared with the control, the
proportion of cell population accumulation in G2/M phase
increased (10.6 vs. 28.3%, respectively). Furthermore, compared
with the control, the proportion of sub-G1 cells,
following treatment with 26 µM 3,28-di-(2-nitroxy-acetyl)-oxy-BT,
increased (0.4 vs. 17.95%, respectively), indicating apoptosis. To
investigate the underlying molecular mechanism by which
3,28-di-(2-nitroxy-acetyl)-oxy-BT induced G2/M phase
arrest, the expression of proteins involved in cell cycle
regulation were identified using western blot analysis (Fig. 2B). The results revealed that
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment inhibited cyclin B1
expression and decreased the expression of cyclin-dependent kinase
(CDK)1, compared with the control. In addition, the expression
level of cell division cycle 25C (CDC25C), which acts an upstream
regulator of the CDK-cyclin complex, was inhibited by
3,28-di-(2-nitroxy-acetyl)-oxy-BT.
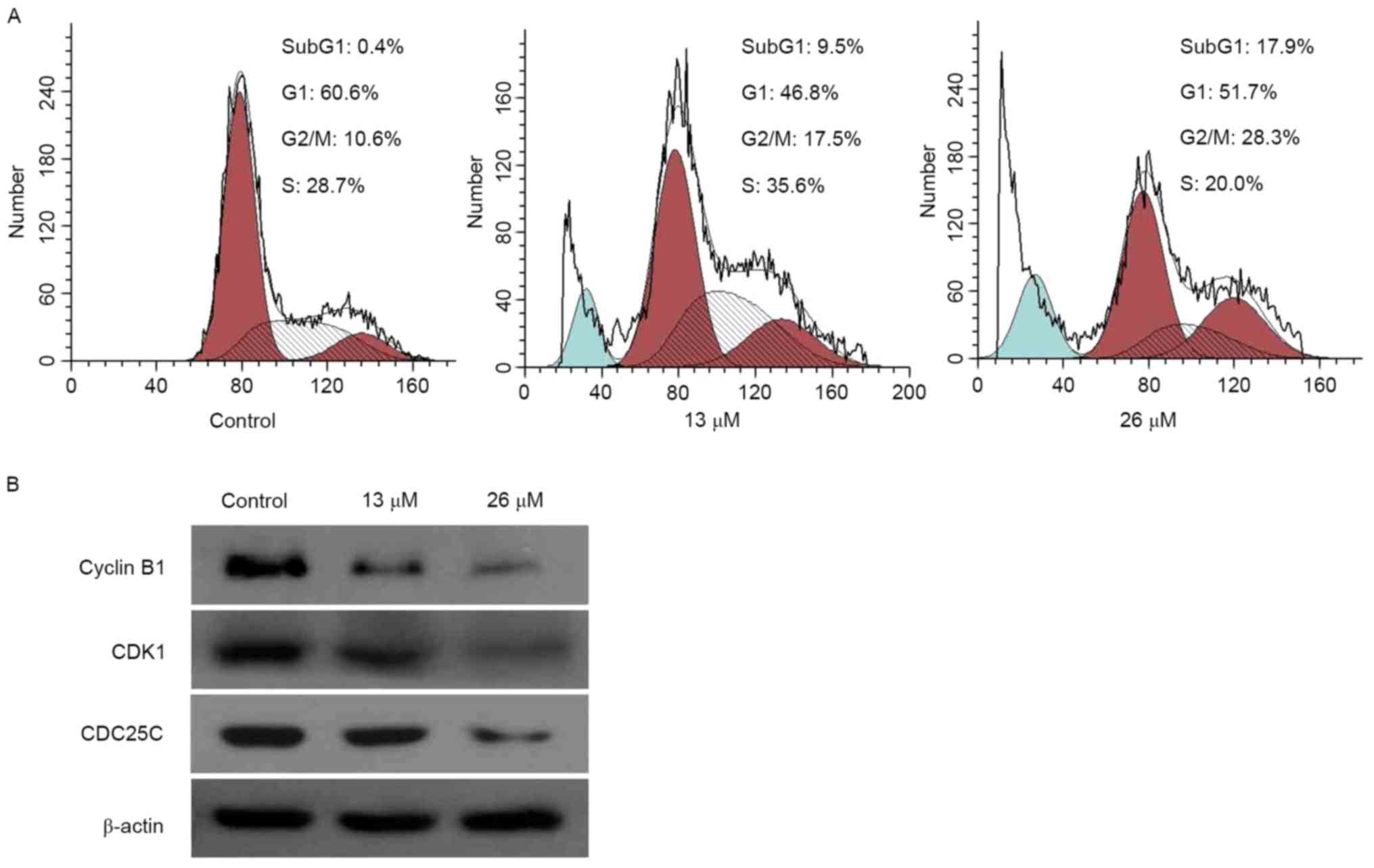 | Figure 2.G2/M cell cycle arrest is
induced by 3,28-di-(2-nitroxy-acetyl)-oxy-betulin in Huh7 cells.
(A) Cell cycle profiles, determined using flow cytometry, following
treatment of the cells with 0, 13 and 26 µM
3,28-di-(2-nitroxy-acetyl)-oxy-betulin. The proportion of the cell
population accumulated in G2/M and the proportion of
sub-G1 cells following treatmentincreased, indicating
apoptosis. (B) Huh7 cells were treated with 0, 13 and 26 µM
3,28-di-(2-nitroxy-acetyl)-oxy-betulin. Western blot analysis was
performed to determine cyclin B1, CDK1 and CDC25C. β-actin was used
as a loading control. Treatment inhibited cyclin B1 expression, and
decreased the expression levels of CDK1 and CDC25C. Represented
images are presented from two independent experiments. CDK1,
cyclin-dependent kinase; CDC25C, cell division cycle 25C. |
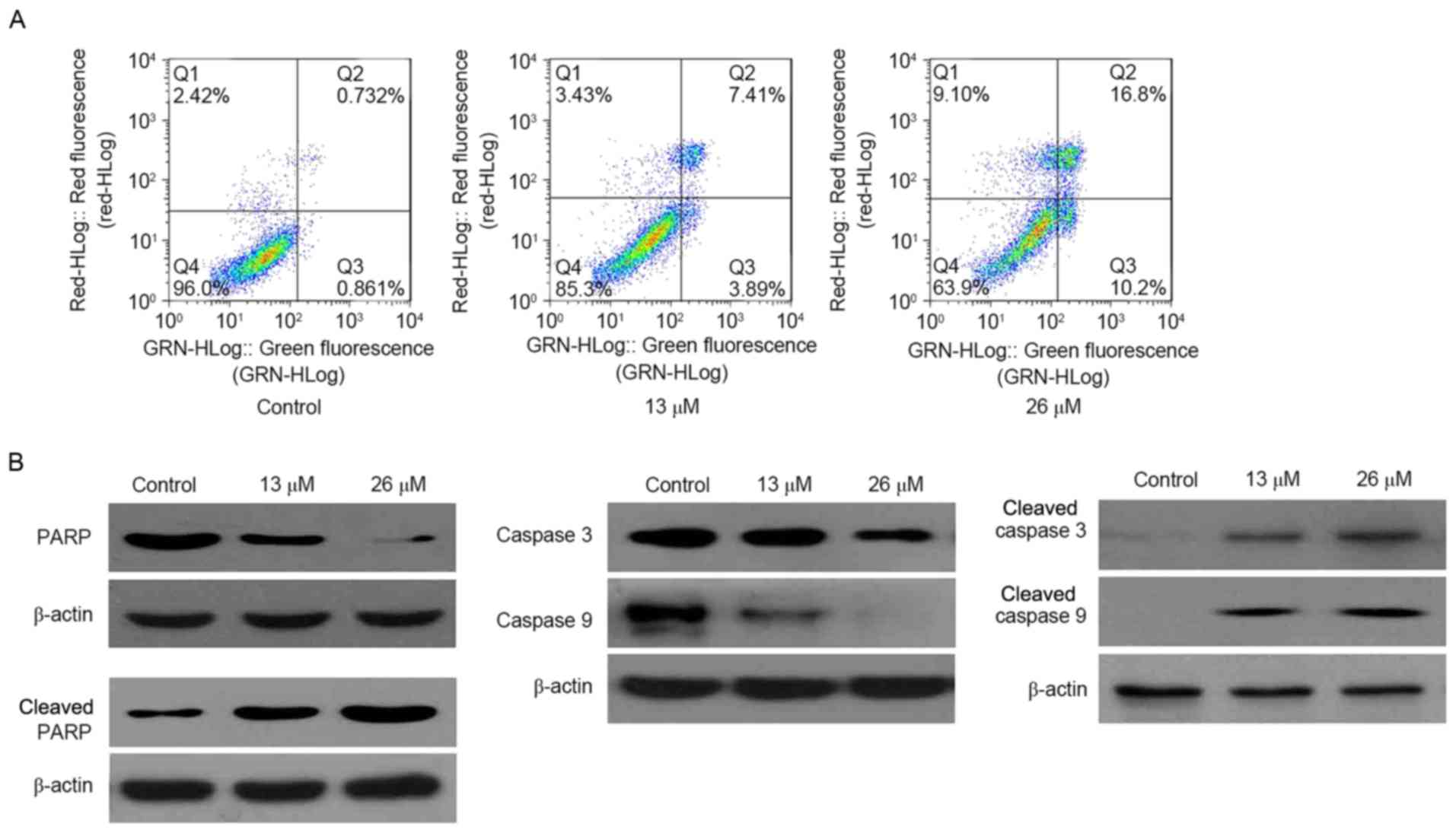
3,28-Di-(2-nitroxy-acetyl)-oxy-BT
induces caspase-dependent apoptosis
To validate the occurrence of apoptosis, an Annexin
V-FITC/PI double-staining assay was performed. As presented in
Fig. 3A, the proportion of apoptotic
cells (including early and late apoptotic cells) increased as the
concentration of 3,28-di-(2-nitroxy-acetyl)-oxy-BT increased (0 µM,
1.6%; 26 µM, 27.0%). Validated using western blotting, cleaved
poly(ADP-ribose) polymerase (PARP) was markedly increased in
3,28-di-(2-nitroxy-acetyl)-oxy-BT-treated Huh7 cells, compared with
the control. In addition, it was identified that cleaved caspase 3
and cleaved caspase 9 were markedly increased in
3,28-di-(2-nitroxy-acetyl)-oxy-BT-treated Huh7 cells (Fig. 3B).
3,28-Di-(2-nitroxy-acetyl)-oxy-BT
induces apoptosis through mitochondrial signaling pathways
To investigate the molecular mechanism underlying
3,28-di-(2-nitroxy-acetyl)-oxy-BT-induced apoptosis in Huh7 cells,
the loss of mitochondrial transmembrane potential was determined
using JC-1. As presented in Fig. 4A,
the green fluorescence of the JC-1 monomers increased, compared
with the control, following treatment with
3,28-di-(2-nitroxy-acetyl)-oxy-BT (control, 3.48%; 26 µM, 23.4%),
suggesting that 3,28-di-(2-nitroxy-acetyl)-oxy-BT induced a loss of
mitochondrial membrane potential in Huh7 cells in a
concentration-dependent manner. Subsequently, the expression level
of the Bcl-2 family of apoptosis regulator proteins were determined
using western blot analysis. As presented in Fig. 4B, Bcl-2 and Bcl-2-associated X protein
(Bax) were identified to be decreased and increased, respectively,
following treatment with 3,28-di-(2-nitroxy-acetyl)-oxy-BT,
compared with the control. In addition, the cytosolic cytochrome c
level was determined, which demonstrated that treatment with
3,28-di-(2-nitroxy-acetyl)-oxy-BT resulted in a marked increase,
compared with untreated cells (Fig.
4C). Furthermore, 3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment
increased the translocation of Bax from the cytosol to the
mitochondria.
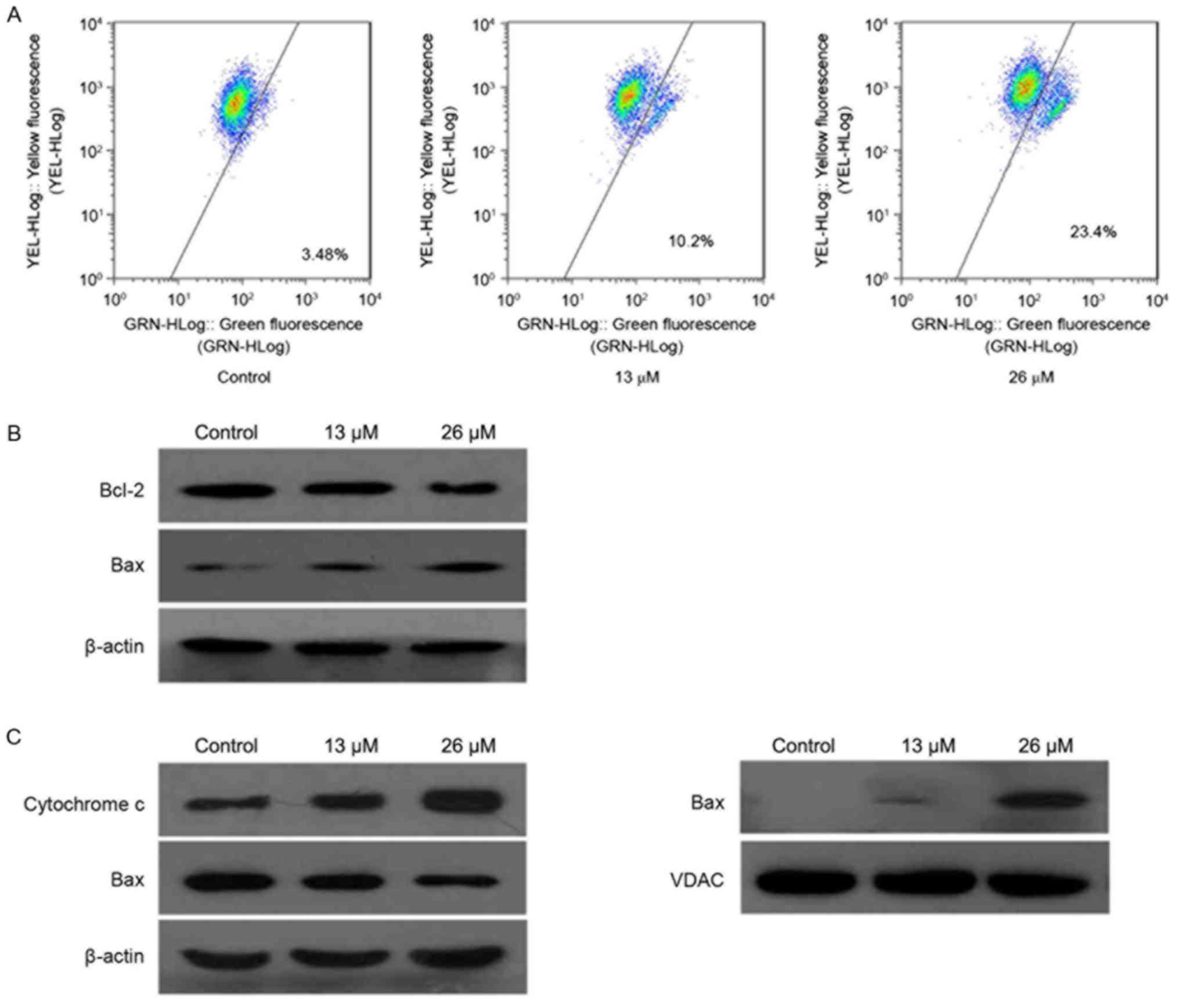 | Figure 4.Apoptosis is induced by
3,28-di-(2-nitroxy-acetyl)-oxy-betulin via the mitochondrial
pathways in Huh7 cells. (A) Decrease in mitochondrial potential
induced by 3,28-di-(2-nitroxy-acetyl)-oxy-betulin. Following
treatment with 3,28-di-(2-nitroxy-acetyl)-oxy-betulin, the cells
were stained with JC-1 and analyzed using flow cytometry. Changes
in mitochondrial potential were determined by the ratio of red
fluorescence with green fluorescence. (B) The total expression of
Bcl-2 and Bax in Huh7 cells treated with or without
3,28-di-(2-nitroxy-acetyl)-oxy-betulin were analyzed using western
blotting. Bcl-2 and Bax were identified to be decreased and
increased, respectively, following treatment. β-actin was used as a
loading control. (C) The cytosolic and mitochondrial levels of the
pro-apoptotic proteins, cytochrome c and Bax, in Huh7 cells with or
without 3,28-di-(2-nitroxy-acetyl)-oxy-betulin, determined using
western blot analysis. β-actin was used as the loading control.
Following treatment, the cytosolic cytochrome c level was markedly
increased and the translocation of Bax from the cytosol to the
mitochondria was increased. Represented images are presented from
two independent experiments. JC-1,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetramethyl
benzimidazolyl-carbocyanine iodide; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; VDAC, voltage-dependent anion
channel. |
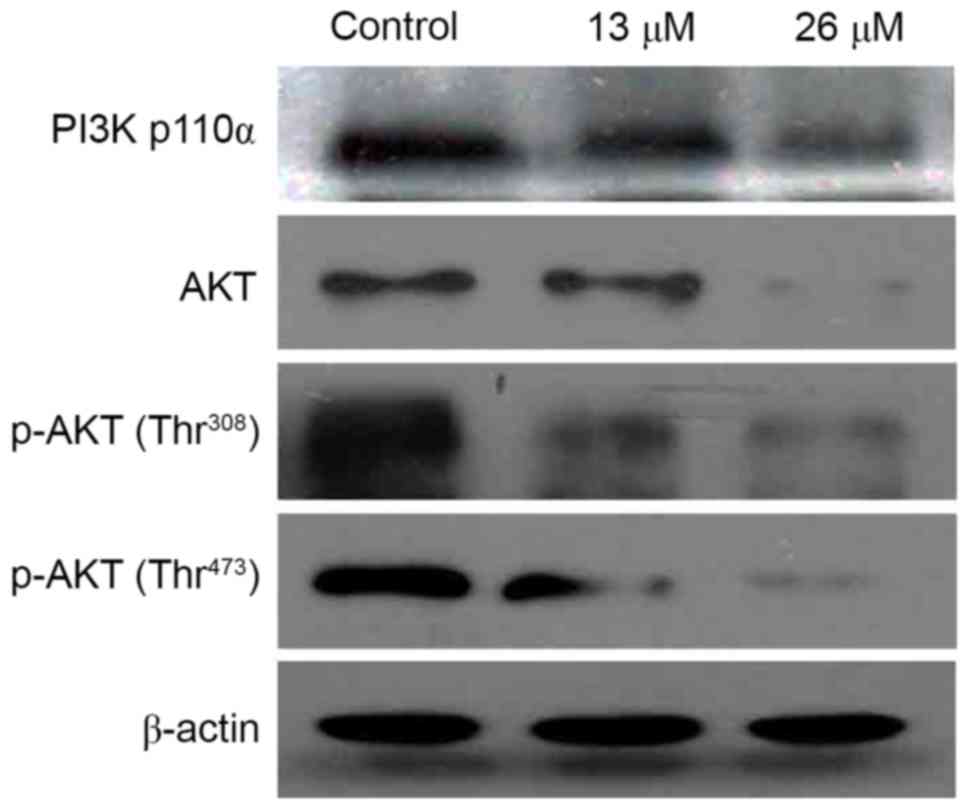
3,28-Di-(2-nitroxy-acetyl)-oxy-BT
inhibits the PI3K/Akt signaling pathway in Huh7 cells
As the PI3K/Akt signaling pathway is a critical
regulator of cellular survival and apoptosis, the effect of
3,28-di-(2-nitroxy-acetyl)-oxy-BT on this pathway, and whether
PI3K/Akt served a function in
3,28-di-(2-nitroxy-acetyl)-oxy-BT-induced apoptosis, was
investigated. As presented in Fig. 5,
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment decreased the level of
the catalytic (p110) subunit of PI3K, and the level of Akt and
phosphorylated (p-)Akt in a concentration-dependent manner,
compared with the controls.
Discussion
Despite advancements in the therapeutic strategies
for the majority of types of cancer, the systemic treatment for HCC
remains ineffective (22). Therefore,
identification of novel agents that may prevent the progression of
HCC is required. In the present study, a novel semi-synthetic
derivative of BT was developed to target tumor cells, to improve
patient outcome, was synthesized. To the best of our knowledge, the
present study was the first to investigate and demonstrate the
antitumor effect and cytotoxic mechanisms of
3,28-di-(2-nitroxy-acetyl)-oxy-BT against Huh7 cells.
Deregulation of cell cycle progression is a typical
hallmark of cancer (23). Therefore,
targeting the regulatory components of the cell cycle machinery has
been identified as an important strategy for the treatment of
cancer (24). Diverse natural
compounds inhibit cancer cell growth by arresting the cell cycle
(25,26). To determine whether
3,28-di-(2-nitroxy-acetyl)-oxy-BT induced cell cycle arrest of Huh7
cells, a DNA content assay was performed, which demonstrated that
Huh7 cells were arrested in G2/M phase following
treatment with 3,28-di-(2-nitroxy-acetyl)-oxy-BT. The cell cycle is
regulated by proteins which are divided into two classes of
molecule: CDKs, a family of serine/threonine kinases, and the
cyclin-binding partners (27). CDK1
and cyclin B1 serve regulatory functions in the G2/M
transition by forming the CDK1-cyclin B1 complex (28). In the present study, the
downregulation of CDK1 and cyclin B1 suggested that
3,28-di-(2-nitroxy-acetyl)-oxy-BT induced G2/M arrest
viathe modulation of CDK1 and cyclin B1. CDC25C is required for the
full activation of CDK1-cyclin B1 during the G2/M
transition (29). The decreased
expression of CDC25C, identified in the present study, indicated
that 3,28-di-(2-nitroxy-acetyl)-oxy-BT may decrease CDC25C and thus
suppress the activation of CDK1-cyclin B1, resulting in Huh7 cell
G2/M arrest.
Apoptosis is a process of programmed cell death,
which serves a function in maintaining cellular homeostasis between
cell division and cell death (30).
Previous studies have demonstrated that apoptosis is an important
mechanism by which chemotherapeutic agents kill susceptible cells
(31,32). As the results of the present study
demonstrated that 3,28-di-(2-nitroxy-acetyl)-oxy-BT caused a marked
sub-G1 apoptotic peak in Huh7 cells, the molecular
mechanisms underlying the anti-hepatic effect of
3,28-di-(2-nitroxy-acetyl)-oxy-BT on Huh7 cells were investigated.
Annexin V-FITC/PI double staining assay indicated that 27.0% of
Huh7 cells underwent apoptosis following treatment with 26 µM
3,28-di-(2-nitroxy-acetyl)-oxy-BT for 24 h. Additionally, activated
caspases 9 and 3, the initiator and executioner caspases in the
mitochondrial apoptotic signaling pathway (33), were determined, using western blot
analysis, in Huh7 cells following 3,28-di-(2-nitroxy-acetyl)-oxy-BT
treatment. A previous study demonstrated that once the specificity
substrate, including cleaved PARP, has been cleaved by caspases,
apoptosis will be induced (34). In
the present study, western blot analysis revealed that PARP was
cleaved from a 116 kDa fragment to an 85 kDa fragment during
3,28-di-(2-nitroxy-acetyl)-oxy-BT-induced apoptosis. Notably, a
decreased proportion of apoptotic cells (11.3%) was observed at the
IC50 concentration, which may be because cell death
processes, induced by 3,28-di-(2-nitroxy-acetyl)-oxy-BT, are
complex and include necrosis and autophagy. Additional study is
required to investigate the involvement of other types of cell
death in 3,28-di-(2-nitroxy-acetyl)-oxy-BT-treated cells, including
necrosis and autophagy.
Mitochondria are important organelles which regulate
cell apoptosis (35). In order to
clarify the underlying molecular mechanism by which
3,28-di-(2-nitroxy-acetyl)-oxy-BT induces apoptotic cell death, the
mitochondria-mediated apoptotic signaling pathway was evaluated.
The results of the present study indicated that the mitochondrial
transmembrane potential was decreased following
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment, compared with the
control. Previous studies have identified that the mitochondrial
membrane permeability is regulated by Bcl-2 family members, which
are the central regulators of caspase activation (36,37). Bax,
a pro-apoptotic member of the Bcl-2 family, serves as a gateway for
the release of apoptotic proteins, including cytochrome c (38). The results of the present study
demonstrated that Bax was translocated to the mitochondria in
marked amounts following 3,28-di-(2-nitroxy-acetyl)-oxy-BT
treatment. In addition, the Bax/Bcl-2 ratio was markedly increased
in treated cells which validated that the intrinsic mitochondrial
pathway triggered 3,28-di-(2-nitroxy-acetyl)-oxy-BT-induced Huh7
cell apoptosis. Furthermore, downregulated Bcl-2, an inhibitor of
mitochondrial cytochrome c release (39), observed following
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment, may participate in the
apoptosis of Huh7 cells. Therefore, it may be hypothesized that
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment resulted in the
decrease in the mitochondrial transmembrane potential and
subsequent apoptosis of Huh7 cells.
Activation of the PI3K/Akt signaling pathway is a
typical feature in a number of types of human cancer (40). Phosphorylation of the serine/threonine
kinase Akt is known to trigger the inactivation of proapoptotic
factors, which in turn confers a survival advantage on tumor cells
(41). The significance of the
PI3K/Akt signaling pathway and its potential as a therapeutic
target for cancer treatment have been investigated in preclinical
studies of several types of human cancer, including renal, lung,
breast, glioblastoma, neuroblastoma and HCC (42). The results of these studies suggested
that PI3K/Akt and its downstream signaling pathways are promising
targets for therapeutic intervention (43,44). The
PI3K/Akt pathway is known to serve a function in cell cycle
progression, apoptosis and tumorigenesis; therefore, it is
hypothesized that the PI3K/Akt signaling pathway may serve
functions in 3,28-di-(2-nitroxy-acetyl)-oxy-BT-induced apoptosis.
The results of the present study demonstrated that
3,28-di-(2-nitroxy-acetyl)-oxy-BT decreased the expression level of
the catalytic (p110α) subunit of PI3K, and the expression levels of
Akt and p-Akt, in a concentration-dependent manner. Furthermore,
treatment of Huh7 cells with 3,28-di-(2-nitroxy-acetyl)-oxy-BT
resulted in decreased expression of PI3K (p110α) and decreased
phosphorylation of Akt at Ser473 and
Thr308.
The results of the present study indicated that
3,28-di-(2-nitroxy-acetyl)-oxy-BT may trigger Huh7 cells to undergo
apoptosis, with the decreased expression level of the catalytic
(p110α) subunit of PI3K, Akt and p-Akt, in a
concentration-dependent manner. Additionally,
3,28-di-(2-nitroxy-acetyl)-oxy-BT treatment downregulated the
protein expression of Bcl-2 and resulted in a loss of mitochondrial
membrane potential, and consequent release of cytochrome c.
Therefore, the present study demonstrated the potential usefulness
of 3,28-di-(2-nitroxy-acetyl)-oxy-BT as an anti-liver cancer
therapeutic candidate.
Acknowledgements
The present study was supported by the Guangdong
Provincial Science and Technology Plan (grant no. 2012A030100015)
and Guangzhou University of Chinese Medicine (grant no.
XH20150103).
References
|
1
|
Bosetti C, Turati F and La Vecchia C:
Hepatocellular carcinoma epidemiology. Best Practice & Res Clin
Gastroenterol. 28:753–770. 2014. View Article : Google Scholar
|
|
2
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fares N and Peron JM: Epidemiology,
natural history and risk factors of hepatocellular carcinoma. Rev
Prat. 63:216–217. 2013.(In French). PubMed/NCBI
|
|
4
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: SHARP Investigators Study Group. Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alakurtti S, Makela T, Koskimies S and
Yli-Kauhaluoma J: Pharmacological properties of the ubiquitous
natural product betulin. European journal of pharmaceutical
sciences: official journal of the European Federation for
Pharmaceutical Sciences. 29:1–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tolstikov GA, Flekhter OB, Shultz EE,
Baltina LA and Tolstikov AG: Betulin and its derivatives. Chemistry
and biological activity. Chemistry for Sustainable Development.
13:1–29. 2005.
|
|
7
|
Huyke C, Reuter J, Rodig M, Kersten A,
Laszczyk M, Scheffler A, Nashan D and Schempp C: Treatment of
actinic keratoses with a novel betulin-based oleogel. A
prospective, randomized, comparative pilot study. J Dtsch Dermatol
Ges. 7:128–133. 2009.(In English, German). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Soica C, Dehelean C, Danciu C, Wang HM,
Wenz G, Ambrus R, Bojin F and Anghel M: Betulin complex in
gamma-cyclodextrin derivatives: properties and antineoplasic
activities in in vitro and in vivo tumor models. Int J Mol Sci.
13:14992–15011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rzeski W, Stepulak A, Szymanski M,
Juszczak M, Grabarska A, Sifringer M, Kaczor J and
Kandefer-Szerszen M: Betulin elicits anti-cancer effects in tumour
primary cultures and cell lines in vitro. Basic Clin Pharmacol
Toxicol. 105:425–432. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hwang BY, Chai HB, Kardono LB, Riswan S,
Farnsworth NR, Cordell GA, Pezzuto JM and Kinghorn AD: Cytotoxic
triterpenes from the twigs of Celtis philippinensis.
Phytochemistry. 62:197–201. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hsu RJ, Hsu YC, Chen SP, Fu CL, Yu JC,
Chang FW, Chen YH, Liu JM, Ho JY and Yu CP: The triterpenoids of
Hibiscus syriacus induce apoptosis and inhibit cell migration in
breast cancer cells. BMC Complement Altern Med. 15:652015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, He K, Huang Y, Zheng D, Gao C, Cui L
and Jin YH: Betulin induces mitochondrial cytochrome c release
associated apoptosis in human cancer cells. Mol carcinogene.
49:630–640. 2010.
|
|
13
|
Li XD, Zhang YJ and Han JC: Betulin
inhibits lung carcinoma proliferation through activation of AMPK
signaling. Tumour biol. 35:11153–11158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gauthier C, Legault J, Lavoie S, Rondeau
S, Tremblay S and Pichette A: Synthesis and cytotoxicity of
bidesmosidic betulin and betulinic acid saponins. J natural prod.
72:72–81. 2009. View Article : Google Scholar
|
|
15
|
Yim NH, Jung YP, Kim A, Kim T and Ma JY:
Induction of apoptotic cell death by betulin in multidrug-resistant
human renal carcinoma cells. Oncol Rep. 34:1058–1064. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dehelean CA, Soica C, Ledeti I, Aluas M,
Zupko IG, Luscan A, Cinta-Pinzaru S and Munteanu M: Study of the
betulin enriched birch bark extracts effects on human carcinoma
cells and ear inflammation. Chem Central J. 6:1372012. View Article : Google Scholar
|
|
17
|
Saudagar P and Dubey VK: Molecular
mechanisms of in vitro betulin-induced apoptosis of Leishmania
donovani. TAm J Trop Med Hyg. 90:354–360. 2014. View Article : Google Scholar
|
|
18
|
Kommera H, Kaluderovic GN, Dittrich S,
Kalbitz J, Drager B, Mueller T and Paschke R: Carbamate derivatives
of betulinic acid and betulin with selective cytotoxic activity.
Bio Organic Med Chem Letters. 20:3409–3412. 2010. View Article : Google Scholar
|
|
19
|
Drag-Zalesinska M, Kulbacka J, Saczko J,
Wysocka T, Zabel M, Surowiak P and Drag M: Esters of betulin and
betulinic acid with amino acids have improved water solubility and
are selectively cytotoxic toward cancer cells. Bio Organic Med Chem
Letters. 19:4814–4817. 2009. View Article : Google Scholar
|
|
20
|
Santos RC, Salvador JA, Marin S and
Cascante M: Novel semisynthetic derivatives of betulin and
betulinic acid with cytotoxic activity. Bio Organic Med Chem.
17:6241–6250. 2009. View Article : Google Scholar
|
|
21
|
Yang SJ, Liu MC, Xiang HM, Zhao Q, Xue W
and Yang S: Synthesis and in vitro antitumor evaluation of betulin
acid ester derivatives as novel apoptosis inducers. Eur J Med Chem.
102:249–255. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Llovet JM and Hernandez-Gea V:
Hepatocellular carcinoma: reasons for phase III failure and novel
perspectives on trial design. Clin Cancer Res. 20:2072–2079. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weber AM and Ryan AJ: ATM and ATR as
therapeutic targets in cancer. Pharmacol Ther. 149:124–138. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee YS, Choi KM, Kim W, Jeon YS, Lee YM,
Hong JT, Yun YP and Yoo HS: Hinokitiol inhibits cell growth through
induction of S-phase arrest and apoptosis in human colon cancer
cells and suppresses tumor growth in a mouse xenograft experiment.
J Nat Prod. 76:2195–2202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shin EM, Kim S, Merfort I and Kim YS:
Glycyrol induces apoptosis in human Jurkat T cell lymphocytes via
the Fas-FasL/caspase-8 pathway. Plantamedica. 77:242–247. 2011.
|
|
27
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tamura D, Arao T, Tanaka K, Kaneda H,
Matsumoto K, Kudo K, Aomatsu K, Fujita Y, Watanabe T, Saijo N, et
al: Bortezomib potentially inhibits cellular growth of vascular
endothelial cells through suppression of G2/M transition. Cancer
Sci. 101:1403–1408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Boutros R, Lobjois V and Ducommun B: CDC25
phosphatases in cancer cells: key players? Good targets? Nature
reviews. Cancer. 7:495–507. 2007.PubMed/NCBI
|
|
30
|
Thornberry NA and Lazebnik Y: Caspases:
enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brown JM and Attardi LD: The role of
apoptosis in cancer development and treatment response. Nature
reviews. Cancer. 5:231–237. 2005.PubMed/NCBI
|
|
32
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ghavami S, Hashemi M, Ande SR, Yeganeh B,
Xiao W, Eshraghi M, Bus CJ, Kadkhoda K, Wiechec E, Halayko AJ, et
al: Apoptosis and cancer: mutations within caspase genes. J Med
Gene. 46:497–510. 2009. View Article : Google Scholar
|
|
35
|
Sola S, Morgado AL and Rodrigues CM: Death
receptors and mitochondria: Τwo prime triggers of neural apoptosis
and differentiation. Biochim Biophys Acta. 1830:2160–2166. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sasi N, Hwang M, Jaboin J, Csiki I and Lu
B: Regulated cell death pathways: new twists in modulation of BCL2
family function. Mol Cancer Ther. 8:1421–1429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Salvesen GS and Dixit VM: Caspase
activation: the induced-proximity model. Proc Natl Acad Sci USA.
96:10964–10967. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: Α requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Molecular cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yuan TL and Cantley LC: PI3K pathway
alterations in cancer: variations on a theme. Oncogene.
27:5497–5510. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Manning BD and Cantley LC: AKT/PKB
signaling: Νavigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mayer IA and Arteaga CL: The PI3K/AKT
pathway as a target for cancer treatment. Ann Rev Med. 67:11–28.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Neri LM, Cani A, Martelli AM, Simioni C,
Junghanss C, Tabellini G, Ricci F, Tazzari PL, Pagliaro P, McCubrey
JA, et al: Targeting the PI3K/Akt/mTOR signaling pathway in
B-precursor acute lymphoblastic leukemia and its therapeutic
potential. Leukemia. 28:739–748. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Slomovitz BM and Coleman RL: The
PI3K/AKT/mTOR pathway as a therapeutic target in endometrial
cancer. Clin Cancer Res. 18:5856–5864. 2012. View Article : Google Scholar : PubMed/NCBI
|