Introduction
Lung cancer is the malignancy with the highest
morbidity and mortality among men and women worldwide (1,2). Non-small
cell lung cancer (NSCLC) accounts for approximately 80–85% of
cases, and can be classified into distinct histological subtypes
(3), including adenocarcinoma and
squamous cell carcinoma (3).
Epidermal growth factor receptor-tyrosine kinase inhibitors
(EGFR-TKI) such as gefitinib are the first-line agent for treating
advanced NSCLC (4). The majority of
patients relapse within 6–12 months of treatment due to acquired
resistance to EGFR-TKIs, with a 5-year survival rate of just 11%
(5,6).
Secondary resistance to EGFR-TKIs is a major factor limiting the
success of lung cancer treatment. Therefore, developing novel
strategies to resensitize lung tumors to these drugs is essential
for improving the survival rate of patients.
Up to 90% of the genome is transcribed into
non-coding RNA (ncRNA) (7,8), including long ncRNAs (lncRNA), which are
over 200 nt long (9,10) and participate in a variety of
biological processes (11,12). Although lncRNAs account for more than
68% of all ncRNAs, our knowledge of their functions is limited
(13). Clarifying the roles of
cancer-related lncRNAs can improve the survival rate of patients,
especially those with tumor recurrence.
The lncRNA Homeobox (Hox) transcript antisense RNA
(HOTAIR) is encoded by the antisense strand of the HoxC gene
(14). HOTAIR recruits polycomb
repressive complex 2 and the lysine-specific histone
demethylase/repressor element-1 silencing transcription
factor (REST)/CoREST complex for trimethylation and dimethylation
of histone H3 on lysines 27 and 4, respectively, leading to target
gene silencing. Previous studies have demonstrated that HOTAIR is
overexpressed in multiple types of cancer, including lung cancer,
which is correlated with metastasis and poor prognosis (15–18).
However, the mechanism by which HOTAIR mediates gefitinib
resistance in human lung adenocarcinoma is not known.
To address this issue, we investigated the role of
HOTAIR in a gefitinib-resistant lung adenocarcinoma PC-9 cell line
(RPC-9) in vitro and in vivo. We found that HOTAIR
silencing restored gefitinib sensitivity by activating B cell
lymphoma 2-associated X protein (Bax)/Caspase-3 and suppressing
transforming growth factor (TGF)-α/epidermal growth factor receptor
(EGFR) signaling pathways, suggesting that it is a novel
therapeutic target for lung cancer treatment.
Materials and methods
Cell lines and reagents
PC-9 human lung adenocarcinoma cells were purchased
from the American Type Culture Collection (Manassas, VA, USA) and
cultured in Roswell Part Memorial Institute (RPMI)-1640 medium
(HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; BI, Beit-Haemek, Israel), 2.05 mM l-glutamine, 100 U/ml
penicillin, and 100 µg/ml streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) at 37°C and 5%
CO2/95% humidified air. Gefitinib was from Selleck
Chemicals (Houston, Texas, USA). The gefitinib-resistant cell line
RPC-9 was established by exposing PC-9 cells to increasing
concentrations of gefitinib (0, 1.25, 2.5, 5, and 10 µM) for 1
month; cell viability was evaluated with the
5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
salt (MTS) assay.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from cells using TRIzol
reagent (Invitrogen) according to the manufacturer's instructions,
and 1 µg was reverse transcribed into cDNA with the PrimeScript RT
Reagent kit with gDNA Eraser (RR047A; Takara Biotechnology Co.,
Ltd., Dalian, China). PCR was performed using SYBR Premix EX Taq II
(RR820A; Takara Biotechnology Co., Ltd.) on an Mx3000P QPCR system
(Agilent Technologies, Inc., Santa Clara, CA, USA) using the
following forward and reverse primers synthesized by Sangon Biotech
(Shanghai, China): HOTAIR, 5′-GGTAGAAAAAGCAACCACGAAGC-3′ and
5′-ACATAAACCTCTGTCTGTGAGTGCC-3′; and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), 5′-TGCCTCCTGCACCACCAACT-3′ and
5′-CCCGTTCAGCTCAGGGATGA-3′. The reaction conditions were as
follows: 95°C for 30 sec, and 40 cycles of 95°C for 5 sec and 60°C
for 34 sec. Melting curve analysis was performed and relative gene
expression was calculated using the 2−ΔΔCt method, with
GAPDH used as the reference gene. The experiment was performed
using triplicate samples.
Lentivirus (LV) packaging and
transduction
The LV vector GV113 (HU6-MCS-CMV-RFP) constructed by
Shanghai Genechem Co., Ltd., (Shanghai, China) was used for stable
knockdown of HOTAIR expression in RPC-9 cells. Short hairpin RNAs
(shRNAs) used to target HOTAIR were: HOTAIR-sh1,
5′-AGAAATGCCACGGCCGCGTCC-3′; HOTAIR-sh2,
5′-ATGAGGAAAAGGGAAAATCTA-3′; and HOTAIR-sh3,
5′-CCAGTACCGACCTGGTAGAAA-3′. A negative control (NC) shRNA
(5′-TTCTCCGAACGTGTCACGT-3′) was used as a control. Cells were
infected with LV in enhanced infection solution supplemented with
polybrene according to the manufacturer's instructions and selected
with puromycin (Sigma-Aldrich, St. Louis, MO, US) for 3 weeks to
obtain stable cell lines.
Cell viability assay
The CellTiter 96 Aqueous Cell Proliferation Assay
(Promega, Madison, WI, USA) was used according to the
manufacturer's protocol to evaluate the sensitivity of PC-9 and
RPC-9 cells to gefitinib. Cells were seeded in a 96-well cell
culture plate at a density of 1×104 cells per well in
200 µl of medium for 24 h and allowed to adhere overnight. On the
following day, cells were treated with different concentrations of
gefitinib or with RPMI-1640 medium as a negative control for 24,
48, 72, or 96 h. A 20-µl volume of MTS reagent was added to each
well, followed by incubation for an additional 4 h at 37°C and 5%
CO2. The absorbance at 490 nm was measured on a Tecan
Infinite M200 microplate reader (Tecan Group, Ltd., Mannedorf,
Switzerland). The percentage of viable cells was calculated
relative to untreated control cells.
Annexin V-allophycocyanin
(APC)/7-aminoactinomycin D (7-AAD) apoptosis assay
Apoptosis was evaluated using the Annexin
V-APC/7-AAD Apoptosis Detection kit (Nanjing KeyGen Biotech Co.,
Ltd., Nanjing, China) according to the manufacturer's protocol.
Briefly, cells (1×105/well) were seeded in a 96-well
cell culture plate in RPMI-1640 medium with 10% FBS and incubated
overnight at 37°C. They were then treated on the following day with
10 µM gefitinib or left untreated at 37°C in 5% CO2 and
95% humidified air for 48 h. Both adherent and suspended cells were
harvested and washed twice with cold 1× phosphate-buffered saline
(PBS), then resuspended in 500 µl binding buffer. Annexin V-APC (5
µl) and 7-AAD (5 µl) were added to 500 µl of the cell suspension,
followed by incubation for 15 min in the dark. Samples were
analyzed within 1 h on a Novocyte flow cytometer (ACEA Biosciences,
San Diego, CA, USA). The experiment was performed using triplicate
samples.
Cell cycle analysis
After treatment with 10 µM gefitinib or incubation
without treatment for 48 h, cells (2×106) were collected
with trypsin-EDTA, washed with 1× PBS, fixed with 4 ml chilled 70%
ethanol, and store overnight at −20°C. Fixed cells were washed with
PBS, treated with 100 µl RNase A at 37°C for 30 min, and stained
with 400 µl propidium iodide (PI) at 4°C for 30 min in the dark.
Cell cycling was analyzed by flow cytometry.
Terminal deoxynucleotidyl transferase
(TdT) dUTP nick end labeling (TUNEL) assay
The TUNEL assay was carried out using an in
situ colorimetric TUNEL Apoptosis Assay kit (Beyotime Institute
of Biotechnology, Shanghai, China) according to the manufacturer's
instructions. Briefly, RPC-9 cells (5×103) were seeded
on coverslips and grown to 70–80% confluence, then treated with 10
µM gefitinib or left untreated at 37°C for 48 h. The cells were
fixed in 4% paraformaldehyde at 37°C for 60 min and rinsed with PBS
for 5 min. After incubation with 0.1% Triton X-100 in PBS for 2 min
on ice followed by 0.3% H2O2 in methanol for
20 min and three rinses with PBS, the cells were incubated with TdT
enzyme and biotin-dUTP for 60 min at 37°C. The stop buffer was
added for 10 min, and cells were treated with horseradish
peroxidase (HRP)-streptavidin for 30 min, then stained with
diaminobenzidine (DAB) and imaged under a light microscope. The
percentage of TUNEL-positive cells was calculated in five random
fields for each group.
Tumor xenograft model
Female and male BALB/c athymic nude mice (4–6 weeks
old, weighing 15–20 g) were purchased from Shanghai SLAC Laboratory
Animal Co., Ltd., (Shanghai, China). Animal protocols were approved
by the Ethics Committee of Zhejiang Provincial People's Hospital.
Mice were subcutaneously injected in the left or right dorsal
region with RPC-9 cells (1×107) infected with
LV-HOTAIR-shRNA or LV-NC-shRNA resuspended in 100 µl PBS. After 1
week, when tumors were about 5 mm in diameter, gefitinib (20
mg/kg/day, n=4) or vehicle (0.05% Tween 80 as a control, n=5) was
administered by oral gavage once daily. Tumor diameter was measured
with digital calipers each week, and the tumor volume was
calculated with the formula V=π/6 (length × width2).
After 28 days, mice were sacrificed and tumors were excised for the
TUNEL assay and immunohistochemistry.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tumor tissue
samples were sectioned at a thickness of 5 mm. The sections were
mounted on Superfrost glass slides (Thermo Fisher Scientific, Inc.,
Pittsburgh, PA, USA), de-paraffinized, and rehydrated in a graded
series of ethanol. Antigen retrieval was performed in 0.1 M
trisodium citrate buffer at pH 6.0. To block endogenous peroxidase
activity, sections were treated with 3% H2O2
for 5 min, then blocked with 10% normal goat serum (Abcam,
Cambridge, MA, USA) before overnight incubation at 4°C with primary
antibody. After rinsing with PBS, sections were incubated with a
biotin-labeled secondary antibody for 20 min followed by
HRP-streptavidin for 20 min at room temperature. Following
treatment with DAB substrate, sections were counterstained with
hematoxylin, dehydrated, and mounted in Permount (Thermo Fisher
Scientific, Inc.). As a negative control, immunohistochemistry was
performed without primary antibodies.
Western blotting
Total protein was extracted from tissues with
radioimmunoprecipitation buffer supplemented with 1%
phenylmethylsulfonyl fluoride solution (Beyotime Institute of
Biotechnology). Protein concentration was determined with a
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology), and 20 µg of protein were separated by 8–12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis and transferred
to 0.2-µm polyvinylidene difluoride membrane that was blocked with
5% non-fat milk in Tris-buffered saline with Tween-20 (TBST) for 2
h at room temperature. The membrane was then incubated overnight at
4°C with primary antibodies against the following proteins: Bax
(1:1,000 dilution; Cell Signaling Technology, Inc., Danvers, MA,
USA), Caspase-3 (1:1,000 dilution; Cell Signaling Technology,
Inc.), EGFR (1:1,000 dilution; Cell Signaling Technology, Inc.),
TGF-α (1:1,000 dilution; Abcam, San Francisco, CA, USA), and Bcl-2
(1:1,000 dilution; Cell Signaling Technology, Inc.). After five
washes with 1× TBST, the membrane was incubated with HRP-conjugated
goat anti-rabbit IgG (Cell Signaling Technology, Inc.) for 1 h at
room temperature. Protein bands were detected by enhanced
chemiluminescence (GE Healthcare Life Sciences, Little Chalfont,
UK). GAPDH served as a loading control.
Statistical analysis. Data are presented as mean ±
SD from at least three independent triplicate experiments.
Differences between groups were evaluated by one-way analysis of
variance and the independent samples t-test using SPSS v.13.0
software (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
PC-9 and RPC-9 cells exhibit
differential sensitivity to gefitinib
PC-9 and RPC-9 cells were treated with different
concentrations of gefitinib for 24, 48, 72, or 96 h and
cytotoxicity was evaluated with the MTS assay. At treated with
gefitinib concentrations <10 µM for 24, 48, 72, or 96 h, the
viability of PC-9 cells was decreased in a dose-dependent manner,
whereas RPC-9 cells were unaffected (Fig.
1A).
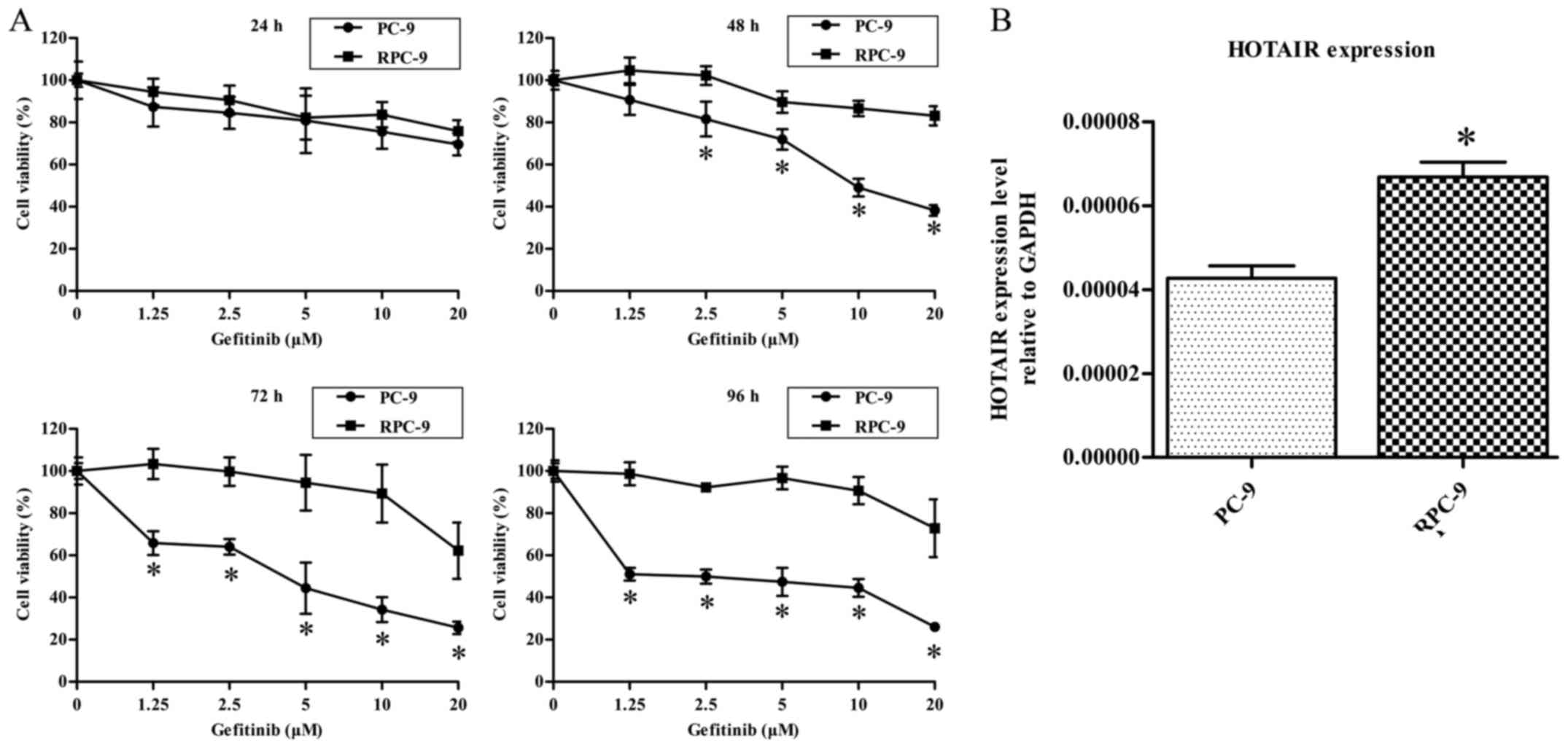 | Figure 1.(A) PC-9 and RPC-9 cells exhibit
differential sensitivity to gefitinib. Cells were treated with 0,
1.25, 2.5, 5, 10, or 20 µM gefitinib. After 24, 48, 72, or 96 h,
cell viability was evaluated with the MTS assay. Data represent
mean ± standard deviation (n=4). *P<0.05 vs. RPC-9 group. (B)
HOTAIR expression level was higher in RPC-9 than in PC-9 cells.
*P<0.05 vs. PC-9 group. HOTAIR, HOX transcript antisense
RNA. |
HOTAIR is more abundant in RPC-9 cells
than in PC-9 cells
We measured and compared HOTAIR expression levels in
RPC-9 and PC-9 cells by RT-qPCR. HOTAIR was more highly expressed
in RPC-9 than in PC-9 cells, indicating that HOTAIR may be involved
in mediating gefitinib resistance (P<0.05, Fig. 1B).
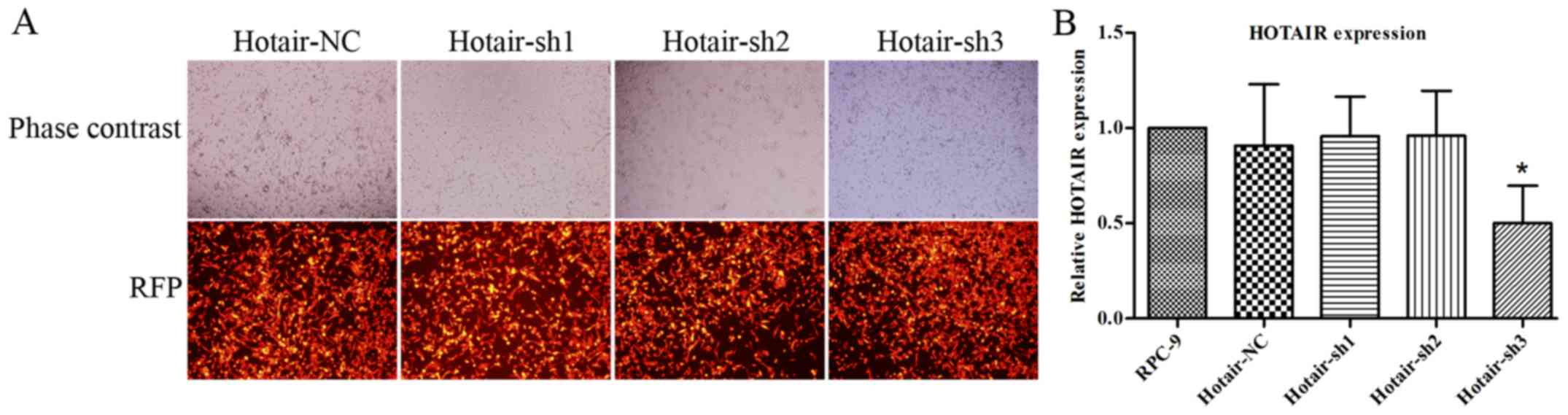
HOTAIR knockdown restores gefitinib
sensitivity to RPC-9 cells
To investigate the role of HOTAIR in acquired
gefitinib resistance, we silenced HOTAIR expression in RPC-9 cells
with red fluorescent protein (RFP)-carrying LV-HOTAIR-shRNAs and
visualized RFP expression by fluorescence microscopy 72 h post
infection (Fig. 2A). The red
fluorescent protein in Fig. 2A
represents the infection efficiency of LV-NC-shRNA or
LV-HOTAIR-shRNAs in RPC-9 cells. We found very high expression
levels of RFP in the LV-NC-shRNA and LV-HOTAIR-shRNA groups.
Therefore, PRC-9 cells were infected with LV-NC-shRNA or
LV-HOTAIR-shRNAs with high infection efficiency. HOTAIR knockdown
efficiency was confirmed by RT-qPCR (Fig.
2B). As shown in Fig. 2B, shRNA3
was one of the effective shRNAs in silencing HOTAIR. Therefore, we
chose LV-HOTAIR-sh3 for further in vitro and in vivo
experiments. In our preliminary experiments, LV-HOTAIR-sh1 and
LV-HOTAIR-sh2 could not silence HOTAIR lncRNA in RPC-9 cells,
restore gefitinib sensitivity to RPC-9 cells, or induce RPC-9 cell
apoptosis and cell cycle arrest in vitro. Hence, LV-NC-shRNA
resembled LV-HOTAIR-sh1 and LV-HOTAIR-sh2, and we chose only
LV-NC-shRNA as the negative control. MTS is a classical method for
testing the changes in cell viability after any cell treatment.
Thus, we used this method to test the response effects of PC-9 or
RPC-9 upon treatment with gefitinib or LV-HOTAIR-shRNAs. After
incubation with different concentrations of gefitinib or RPMI-1640
medium as a negative control for 24, 48, 72, or 96 h, we found that
gefitinib inhibited the proliferation of RPC-9 cells infected with
LV-HOTAIR-sh3 as compared to LV-NC-shRNA at 48 h (gefitinib
concentrations >2.5 µM), 72, and 96 h (Fig. 3), suggesting that HOTAIR is required
for the proliferation of gefitinib-resistant RPC-9 cells.
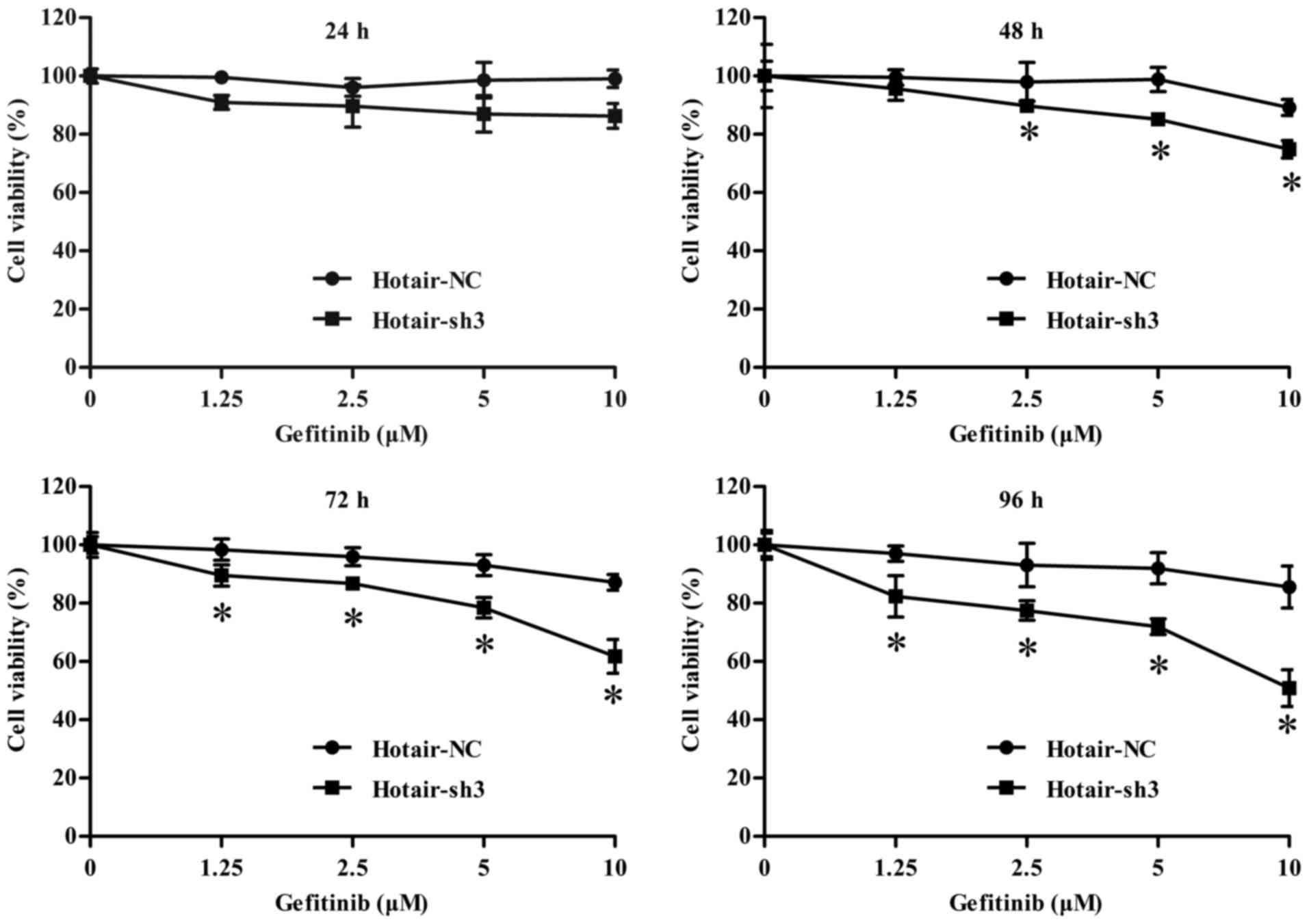 | Figure 3.Viability of LV-infected RPC-9 cells
treated with 0, 1.25, 2.5, 5, and 10 µM gefitinib for 24, 48, 72
and 96 h. Data represent mean ± standard deviation (n=3).
*P<0.05 vs. LV-NC-shRNA group. LV, lentivirus; HOTAIR, HOX
transcript antisense RNA; sh, short hairpin; NC, negative
control. |
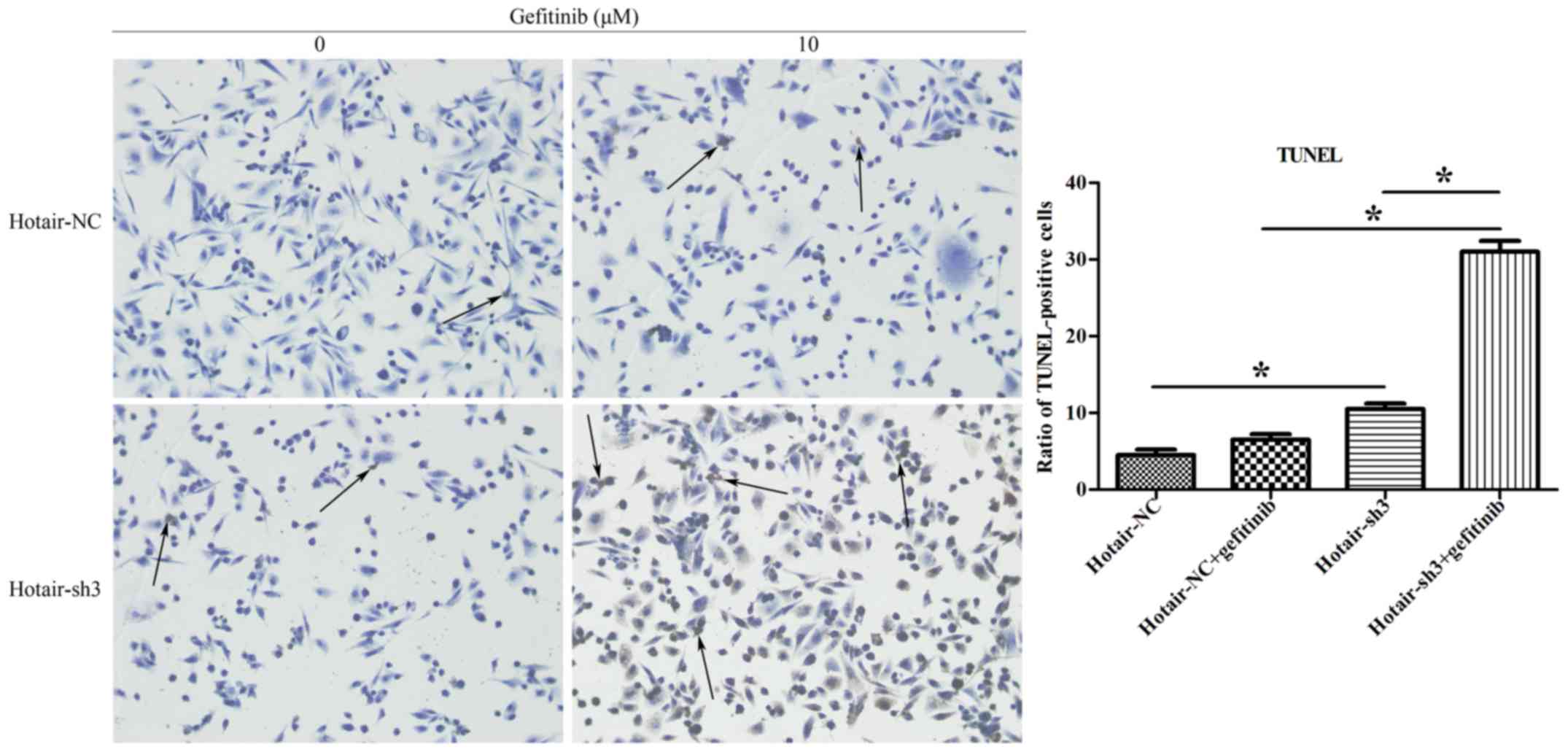
HOTAIR knockdown induces RPC-9 cell
apoptosis and cell cycle arrest
To confirm whether HOTAIR is required for RPC-9 cell
survival, cells were treated with 10 µM gefitinib for 48 h after
infection with LV-HOTAIR-sh3 or LV-NC-shRNA, and apoptosis was
evaluated with the TUNEL assay. The bar plot in Fig. 4 shows the ratio of TUNEL-positive
cells in various treatment groups. There were more TUNEL-positive
cells in the LV-HOTAIR-sh3 + gefitinib group than in the
LV-NC-shRNA + gefitinib group (31±1.41% vs. 6.5±0.71%; P<0.05)
(Fig. 4), indicating that loss of
HOTAIR increased apoptosis of RPC-9 cells. This was confirmed by
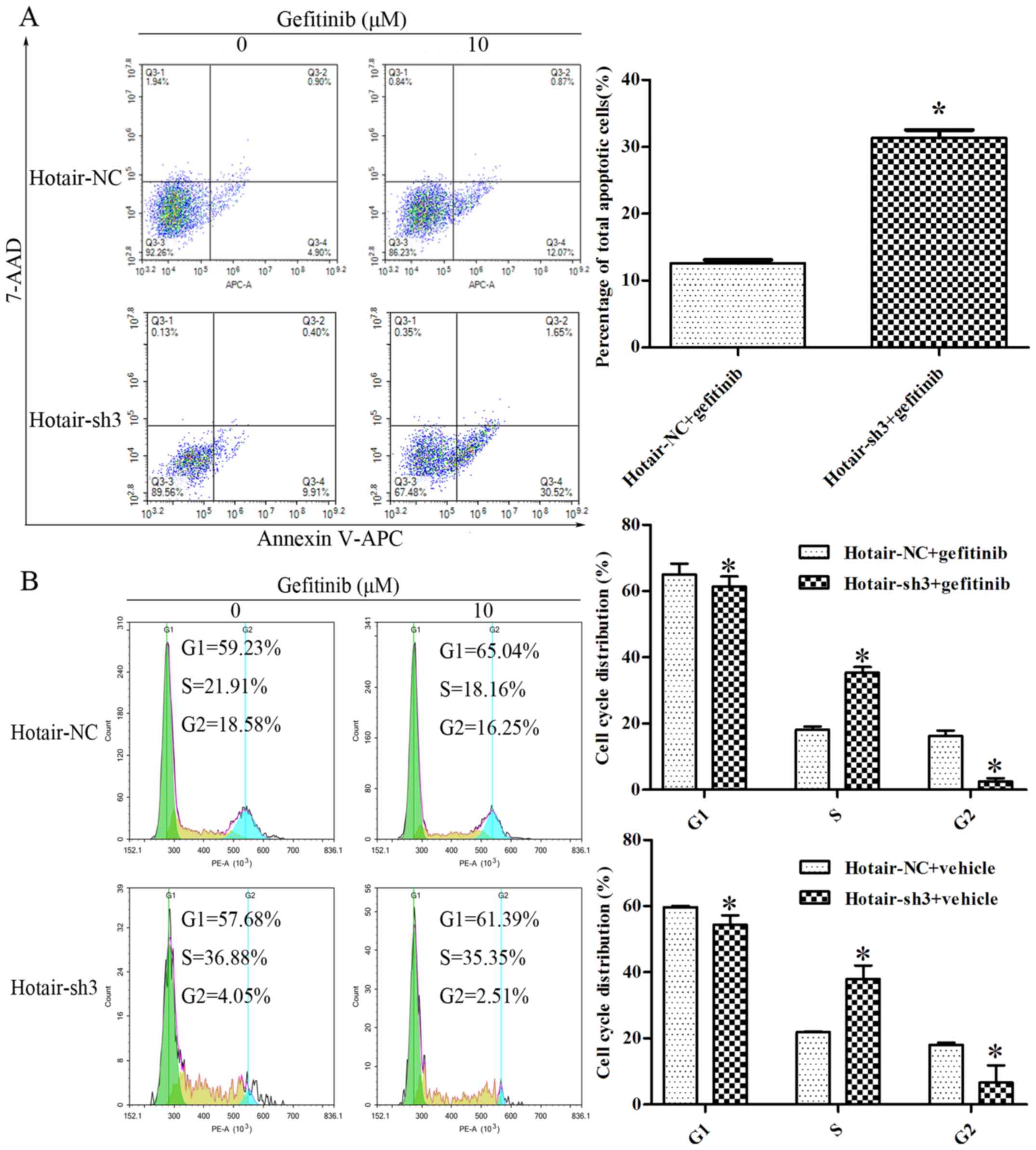
annexin-V-APC and 7-AAD double staining followed by flow cytometry
analysis; after treatment with 10 µM gefitinib for 48 h,
LV-HOTAIR-sh3 infection increased the fraction of apoptotic RPC-9
cells as compared to infection with LV-NC-shRNA (32.17±1.61% vs.
12.94±0.65%; P<0.05) (Fig. 5A).
Loss of HOTAIR inhibited RPC-9 cell proliferation by modulating
cell cycling, as evidenced by the increased S-phase fraction in
LV-HOTAIR-sh3 + gefitinib as compared to LV-NC-shRNA + gefitinib
group (35.36±0.07% vs. 18.08±0.16%; P<0.05) detected by flow
cytometry analysis (Fig. 5B).
Moreover, HOTAIR silencing decreased the G1-phase (61.39±0.06% vs.
65.09±0.77%; P<0.05) and G2-phase (2.46±0.07% vs. 16.25±0.62%;
P<0.05) fractions in the LV-HOTAIR-sh3 + gefitinib group as
compared to the LV-NC-shRNA + gefitinib group. Similar results were
found in the LV-HOTAIR-sh3 + vehicle and LV-NC-shRNA + vehicle
groups (Fig. 5B). Hence, it seems
that the cell cycle changes occurred mainly because of HOTAIR
silencing, and additional treatment of gefitinib seemed to have no
effect. The data of one representative experiment are shown in the
quadrants of Fig. 5. Thus, HOTAIR
knockdown inhibits RPC-9 cell proliferation by inducing cell cycle
arrest and apoptosis.
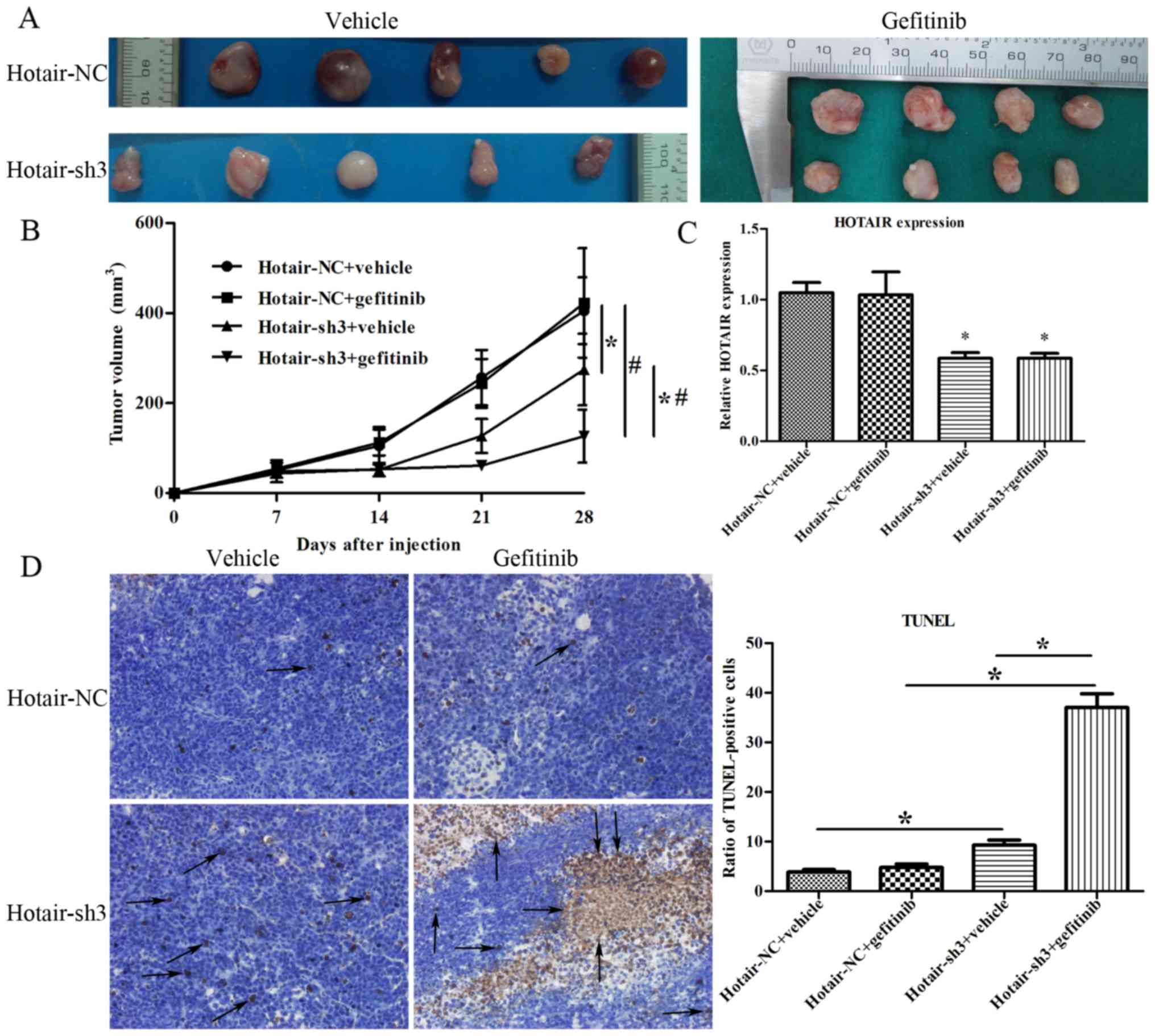
HOTAIR knockdown suppresses the
tumorigenicity of RPC-9 cells in vivo
To assess the antitumor effects of HOTAIR silencing
in vivo, RPC-9 cells infected with LV-HOTAIR-sh3 or
LV-NC-shRNA were subcutaneously injected into nude mice and growth
of the resultant tumors was compared. After 21 days of gefitinib
administration, the growth of tumors with HOTAIR knockdown was
slower as compared to those infected with the negative control
(P<0.05; Fig. 6A, B). Moreover,
the growth of tumors in the LV-HOTAIR-sh3 + gefitinib group was
slower than that in the LV-HOTAIR-sh3 + vehicle group, indicating
that HOTAIR silencing could effectively restore the sensitivity of
RPC-9 cells to gefitinib (P<0.05; Fig.
6A, B). There was no significant difference in the growth of
tumors between the LV-NC-shRNA + vehicle and LV-NC-shRNA +
gefitinib groups (P>0.05). HOTAIR is a lncRNA, which does not
encode a protein; thus, RT-qPCR was implemented to confirm HOTAIR
expression in LV-HOTAIR-sh3 tumors (19). RT-qPCR confirmed that HOTAIR
expression was similarly downregulated in LV-HOTAIR-sh3 + vehicle
and LV-HOTAIR-sh3 + gefitinib tumors as compared to LV-NC-shRNA +
vehicle tumors (Fig. 6C). The bar
plot in Fig. 6D shows the ratio of
TUNEL-positive cells in various treatment groups. The ratio of
TUNEL-positive cells in LV-HOTAIR-sh3 + vehicle tumors was
significantly higher than that in LV-NC-shRNA + vehicle tumors
(9.25±1.06% vs. 3.85±0.49%; P=0.023 <0.05). This is in
accordance with the tumor growth data in Fig. 6B, as well as the upregulation of
Caspase-3 in Fig. 7. Moreover, tumors
derived from RPC-9 cells infected with LV-HOTAIR-sh3 showed higher
ratio of TUNEL-positive cells than those originating from
LV-NC-shRNA-infected cells following gefitinib treatment
(P<0.05, Fig. 6D). The ratio of
TUNEL-positive cells in the LV-HOTAIR-sh3 + gefitinib group was
higher than that in the LV-HOTAIR-sh3 + vehicle group, indicating
that HOTAIR silencing could effectively restore the sensitivity of
RPC-9 cells to gefitinib (P<0.05; Fig.
6D). These results indicate that HOTAIR knockdown restores
gefitinib sensitivity in vivo.
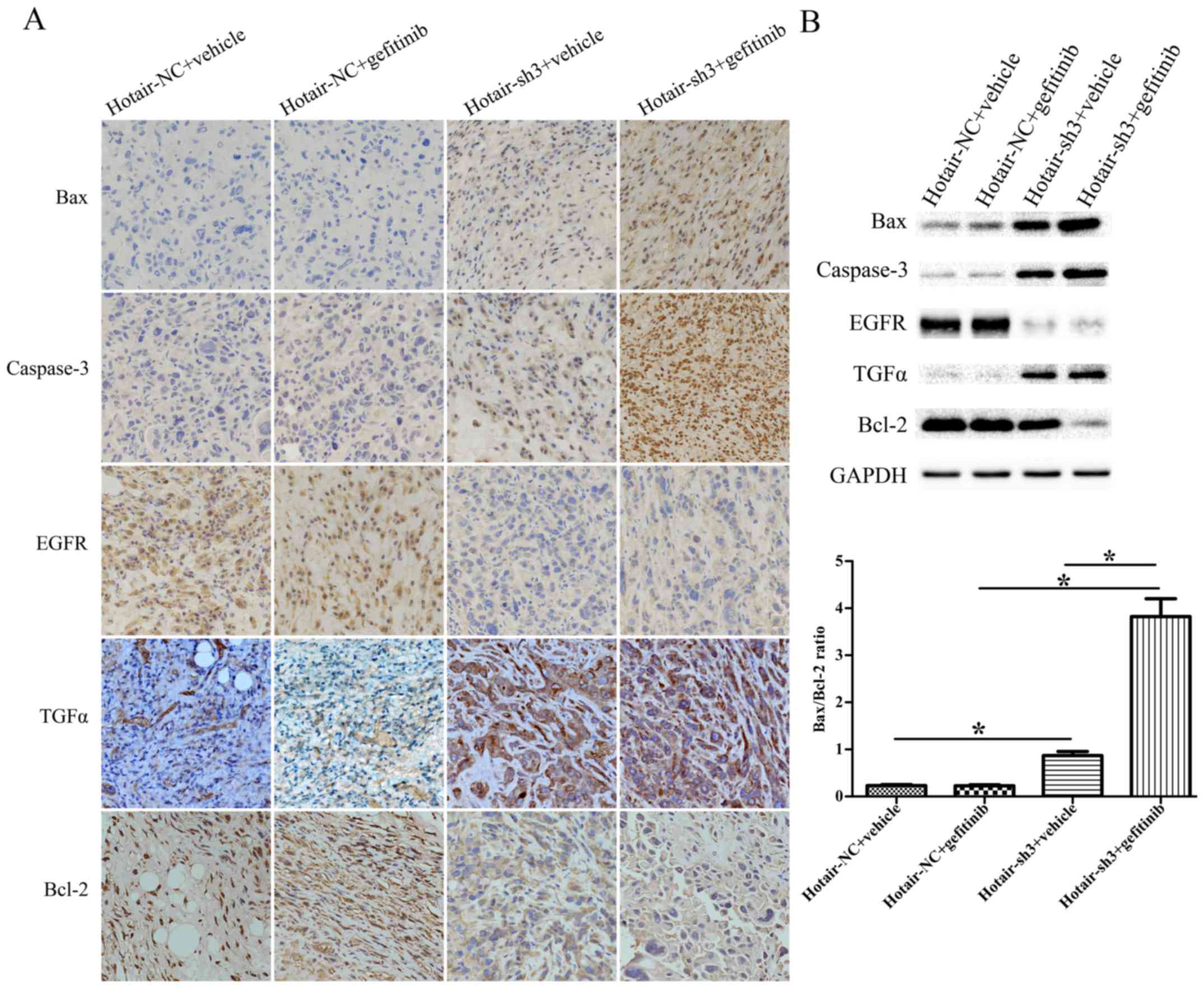 | Figure 7.(A) Immunohistochemical analysis of
Bax, Caspase-3, EGFR, TGF-α, and Bcl-2 expression in xenograft
tumors originating from RPC-9 cells infected with LV-HOTAIR-sh3 or
LV-NC-shRNA (400× magnification). (B) Western blot analysis of Bax,
Caspase-3, EGFR, TGF-α, and Bcl-2 expression in xenograft tumors
originating from RPC-9 cells infected with LV-HOTAIR-sh3 or
LV-NC-shRNA. *P<0.05. HOTAIR, HOX transcript antisense RNA; sh,
short hairpin; NC, negative control; APC, allophycocyanin; Bax,
B-cell lymphoma 2-associated X protein; TGF-α, Transforming growth
factor-α; EGFR, epithelial growth factor receptor. |
HOTAIR knockdown restores gefitinib
sensitivity by activating Bax/Caspase-3 and suppressing TGF-α/EGFR
signaling
To clarify the mechanism by which HOTAIR silencing
restores gefitinib sensitivity to RPC-9 cells, we examined the
expression of genes related to apoptosis (Bax, Caspase-3, Bcl-2)
and EGFR signaling (TGF-α, EGFR) in xenograft tumors by
immunohistochemistry and western blotting. The Bax/Bcl-2 ratio and
Caspase-3 level were upregulated in LV-HOTAIR-sh3 + gefitinib
tumors compared to LV-NC-shRNA + gefitinib tumors. Moreover, the
Bax/Bcl-2 ratio and Caspase-3 level were upregulated in
LV-HOTAIR-sh3 + gefitinib tumors compared to LV-HOTAIR-sh3 +
vehicle tumors. EGFR levels were downregulated in tumors
originating from RPC-9 cells infected with LV-HOTAIR-sh3 as
compared to LV-NC-shRNA; the opposite trend was observed for TGF-α
expression. There was no significant difference in the Bax/Bcl-2
ratio or levels of Caspase-3, EGFR, and TGF-α between the
LV-NC-shRNA + vehicle and LV-NC-shRNA + gefitinib groups
(P>0.05) (Fig. 7A, B). Thus,
gefitinib resistance in RPC-9 cells can be overcome by HOTAIR
knockdown, which induces apoptosis via activation of Bax/Caspase-3
and blocks cell proliferation via modulation of TGF-α/EGFR
signaling.
Discussion
Various mechanisms of EGFR-TKI resistance have been
reported in NSCLC, including EGFR T790M mutation (20,21), MET
amplification (22), human epidermal
growth factor receptor 2 amplification (23), hepatocyte growth factor overexpression
(24),
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha mutation (25), and histologic
transformation to small-cell lung cancer (26), among others. About 50–60% of cases of
acquired resistance are attributed to T790M mutation (27). Second-generation EGFR-TKIs such as
afatinib that target T790M-induced resistance lack clinical
efficacy due to on-target toxicity and side effects (28). Third-generation TKIs including AZD9291
(osimertinib), CO-1686 (rociletinib), and HM61713 (olmutinib) that
target T790M while having no effect on wild-type EGFR are currently
in clinical trials. However, it is possible that resistance to
these drugs will eventually emerge. Therefore, novel therapeutic
strategies that reverse acquired resistance are needed.
HOTAIR is associated with chemoresistance in lung,
breast, and ovarian cancers (29–31). For
example, inhibiting HOTAIR reverses the resistance of lung
adenocarcinoma to cisplatin via downregulation of p21 expression
(29). In the present study, we found
that HOTAIR knockdown blocked the proliferation of RPC-9 cells and
restored their sensitivity to gefitinib in vitro and in
vivo. These effects were accompanied by increases in cell
apoptosis and cell cycle arrest. In a xenograft model, loss of
HOTAIR resulted in tumor shrinkage and restored gefitinib
sensitivity. Bax is a pro-apoptotic signaling molecule (32), whereas Caspase-3 is an effector in the
terminal stages of apoptosis (33).
In serous ovarian cancer, HOTAIR knockdown was found to induce
Caspase-3 expression (34); this is
consistent with our observation that Bax and Caspase-3 expression
was upregulated by silencing HOTAIR. Zou et al demonstrated
that the knockdown of Bcl-2 using siRNAs increases the sensitivity
to gefitinib in a gefitinib-resistant H1975 lung cancer cell line
(35). In our present study, we found
that Bcl-2 was downregulated by silencing HOTAIR (Fig. 7), which is in accordance with the
study of Zou et al.
Aberrant EGFR expression and signaling contribute to
the malignant transformation of various human cancers, including
lung cancer (36). HOTAIR knockdown
was found to suppress EGFR expression by inhibiting of miR-545
levels in colorectal cancer (37).
Ishikawa and Masago et al reported that TGF-α is a serum
biomarker for gefitinib resistance in patients with advanced NSCLC
(38,39). TGF-α is a ligand of EGFR, and the
function of EGFR somehow depends on the ligand quantity around
cells (40). In the present study,
HOTAIR silencing suppressed the expression of EGFR and induced that
of its ligand TGF-α in the cytoplasm. This leads us to believe that
HOTAIR silencing inhibits the release of TGF-α into the
extracellular environment, which is in accordance with the study of
Ishikawa and Masago et al. HOTAIR lncRNA might be restoring
gefitinib sensitivity by suppressing TGF-α/EGFR signaling. The
underlying molecular mechanism needs to be explored further.
Thus, inhibiting HOTAIR can reverse acquired
resistance to gefitinib by activating Bax/Caspase-3-mediated
apoptosis and suppressing TGF-α/EGFR signaling.
Although Shien et al have demonstrated that
the knockdown of EGFR using siRNAs suppressed RPC-9 cell
proliferation (41), the role HOTAIR
in gefitinib resistance in human lung adenocarcinoma is unknown.
The original aim of our study was to investigate the role of HOTAIR
in lung adenocarcinoma, but during our experiment, we found that
HOTAIR silencing could effectively restore the sensitivity of RPC-9
against gefitinib. Therefore, this is a novel study about the
relationship of HOTAIR lncRNA and gefitinib sensitivity in lung
adenocarcinoma.
In conclusion, these results demonstrate for the
first time that HOTAIR knockdown can reverse acquired resistance to
gefitinib in human lung adenocarcinoma. Based on these findings, we
propose that HOTAIR is a novel therapeutic target for NSCLC cases
exhibiting gefitinib resistance.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (nos. 81470109 and
81470241) and The Foundation of Science and Technology Department
of Zhejiang Province (no. 2014C37022).
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistic, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Herbst RS, Heymach JV and Lippman SM: Lung
cancer. N Engl J Med. 359:1367–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park K, Tan EH, O'Byrne K, Zhang L, Boyer
M, Mok T, Hirsh V, Yang JC, Lee KH, Lu S, et al: Afatinib versus
gefitinib as first-line treatment of patients with EGFR
mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase
2B, open-label, randomised controlled trial. Lancet Oncol.
17:577–589. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nguyen KS, Kobayashi S and Costa DB:
Acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in non-small-cell lung cancers dependent on the
epidermal growth factor receptor pathway. Clin Lung Cancer.
10:281–289. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Allemani C, Weir HK, Carreira H, Harewood
R, Spika D, Wang XS, Bannon F, Ahn JV, Johnson CJ, Bonaventure A,
et al: Global surveillance of cancer survival 1995–2009: Analysis
of individual data for 25, 676, 887 patients from 279
population-based registries in 67 countries (CONCORD-2). Lancet.
385:977–1010. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Djebali S, Davis CA, Merkel A, Dobin A,
Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F,
et al: Landscape of transcription in human cells. Nature.
489:101–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Batista PJ and Chang HY: Long noncoding
RNAs: Cellular address codes in development and disease. Cell.
152:1298–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okazaki Y, Furuno M, Kasukawa T, Adachi J,
Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al:
Analysis of the mouse transcriptome based on functional annotation
of 60,770 full-length cDNAs. Nature. 420:563–573. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Caley DP, Pink RC, Trujillano D and Carter
DR: Long noncoding RNAs, chromatin, and development.
ScientificWorldJournal. 10:90–102. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Moazed D: Small RNAs in transcriptional
gene silencing and genome defence. Nature. 457:413–420. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brosnan CA and Voinnet O: The long and the
short of noncoding RNAs. Curr Opin Cell Biol. 21:416–425. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Iyer MK, Niknafs YS, Malik R, Singhal U,
Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et
al: The landscape of long noncoding RNAs in the human
transcriptome. Nat Genet. 47:199–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rinn JL, Kertesz M, Wang JK, Squazzo SL,
Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E and
Chang HY: Functional demarcation of active and silent chromatin
domains in human HOX loci by Non-coding RNAs. Cell. 129:1311–1323.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY,
Zhang F and Zheng SS: Overexpression of long non-coding RNA HOTAIR
predicts tumor recurrence in hepatocellular carcinoma patients
following liver transplantation. Ann Surg Oncol. 18:1243–1250.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kogo R, Shimamura T, Mimori K, Kawahara K,
Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, et al:
Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin
modification and is associated with poor prognosis in colorectal
cancers. Cancer Res. 71:6320–6326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhuang Y, Wang X, Nguyen HT, Zhuo Y, Cui
X, Fewell C, Flemington EK and Shan B: Induction of long intergenic
non-coding RNA HOTAIR in lung cancer cells by type I collagen. J
Hematol Oncol. 6:352013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cai B, Song XQ, Cai JP and Zhang S:
HOTAIR: A cancer-related long non-coding RNA. Neoplasma.
61:379–391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Eng J Med. 352:786–792. 2005. View Article : Google Scholar
|
|
21
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med. 2:e732005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takezawa K, Pirazzoli V, Arcila ME, Nebhan
CA, Song X, de Stanchina E, Ohashi K, Janjigian YY, Spitzler PJ,
Melnick MA, et al: HER2 amplification: A potential mechanism of
acquired resistance to EGFR inhibition in EGFR-mutant lung cancers
that lack the second-site EGFRT790M mutation. Cancer discov.
2:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yano S, Yamada T, Takeuchi S, Tachibana K,
Minami Y, Yatabe Y, Mitsudomi T, Tanaka H, Kimura T, Kudoh S, et
al: Hepatocyte growth factor expression in EGFR mutant lung cancer
with intrinsic and acquired resistance to tyrosine kinase
inhibitors in a Japanese cohort. J Thorac Oncol. 6:2011–2017. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Whyte DB and Holbeck SL: Correlation of
PIK3Ca mutations with gene expression and drug sensitivity in
NCI-60 cell lines. Biochem Biophys Res Commun. 340:469–475. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piotrowska Z, Niederst MJ, Karlovich CA,
Wakelee HA, Neal JW, Mino-Kenudson M, Fulton L, Hata AN, Lockerman
EL, Kalsy A, et al: Heterogeneity underlies the emergence of
EGFRT790 wild-type clones following treatment of T790M-positive
cancers with a third-generation EGFR inhibitor. Cancer discov.
5:713–722. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu HA, Arcila ME, Rekhtman N, Sima CS,
Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M and Riely GJ:
Analysis of tumor specimens at the time of acquired resistance to
EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers.
Clin Cancer Res. 19:2240–2247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Miller VA, Hirsh V, Cadranel J, Chen YM,
Park K, Kim SW, Zhou C, Su WC, Wang M, Sun Y, et al: Afatinib
versus placebo for patients with advanced, metastatic
non-small-cell lung cancer after failure of erlotinib, gefitinib,
or both and one or two lines of chemotherapy (LUX-Lung 1): A phase
2b/3 randomised trial. Lancet Oncol. 13:528–538. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Z, Sun M, Lu K, Liu J, Zhang M, Wu W,
De W, Wang Z and Wang R: The long noncoding RNA HOTAIR contributes
to cisplatin resistance of human lung adenocarcinoma cells via
downregualtion of p21 (WAF1/CIP1) expression. PloS One.
8:e772932013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xue X, Yang YA, Zhang A, Fong KW, Kim J,
Song B, Li S, Zhao JC and Yu J: LncRNA HOTAIR enhances ER signaling
and confers tamoxifen resistance in breast cancer. Oncogene.
35:2746–2755. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li J, Yang S, Su N, Wang Y, Yu J, Qiu H
and He X: Overexpression of long non-coding RNA HOTAIR leads to
chemoresistance by activating the Wnt/β-catenin pathway in human
ovarian cancer. Tumour Biol. 37:2057–2065. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qiu JJ, Wang Y, Ding JX, Jin HY, Yang G
and Hua KQ: The long non-coding RNA HOTAIR promotes the
proliferation of serous ovarian cancer cells through the regulation
of cell cycle arrest and apoptosis. Exp Cell Res. 333:238–248.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zou M, Xia S, Zhuang L, Han N, Chu Q, Chao
T, Peng P, Chen Y, Gui Q and Yu S: Knockdown of the Bcl-2 gene
increases sensitivity to EGFR tyrosine kinase inhibitors in the
H1975 lung cancer cell line harboring T790M mutation. Int J Oncol.
42:2094–2102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yarden Y and Pines G: The ERBB network: At
last, cancer therapy meets systems biology. Nat Rev Cancer.
12:553–563. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang X and Lu S: MicroR-545 mediates
colorectal cancer cells proliferation through up-regulating
epidermal growth factor receptor expression in HOTAIR long
non-coding RNA dependent. Mol Cell Biochem. 431:45–54. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ishikawa N, Daigo Y, Takano A, Taniwaki M,
Kato T, Hayama S, Murakami H, Takeshima Y, Inai K, Nishimura H, et
al: Increases of amphiregulin and transforming growth factor-alpha
in serum as predictors of poor response to gefitinib among patients
with advanced non-small cell lung cancers. Cancer Res.
65:9176–9184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Masago K, Fujita S, Hatachi Y, Fukuhara A,
Sakuma K, Ichikawa M, Kim YH, Mio T and Mishima M: Clinical
significance of pretreatment serum amphiregulin and transforming
growth factor-alpha, and an epidermal growth factor receptor
somatic mutation in patients with advanced non-squamous, non-small
cell lung cancer. Cancer sci. 99:2295–2301. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Singh B and Coffey RJ: From wavy hair to
naked proteins: The role of transforming growth factor alpha in
health and disease. Semin Cell Dev Biol. 28:12–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shien K, Ueno T, Tsukuda K, Soh J, Suda K,
Kubo T, Furukawa M, Muraoka T, Maki Y, Tanaka N, et al: Knockdown
of the epidermal growth factor receptor gene to investigate its
therapeutic potential for the treatment of non-small-cell lung
cancers. Clin Lung Cancer. 13:488–493. 2012. View Article : Google Scholar : PubMed/NCBI
|