Introduction
Even though the majority of cancers aren't linked to
germline mutations, for some kind of tumours an inherited
predisposition is known. In such situations, people carrying a
mutation in specific genes are at increased risk of developing
cancer. This is the case of the Hereditary Breast and Ovarian
Cancer (HBOC) syndrome, where heterozygous carriers of a mutation
in BRCA1 or BRCA2 genes face an increased risk of
developing cancer in certain organs, especially breast (cumulative
risk by age 70 years of 55 and 47% for BRCA1 and
BRCA2, respectively) and fallopian tubes/ovarian cancer
(cumulative risk by age 70 years of 39 and 19% for BRCA1 and
BRCA2, respectively) (1). Even
though heterozygous mutations in both BRCA1 and BRCA2
in the same individual have been seldom reported in HBOC syndrome,
it is instead really rarer to observe in those genes homozygous or
compound heterozygous carriers. To date the few described cases
show a Fanconi anemia (FA) phenotype. FA is an autosomal recessive
syndrome characterized by spontaneous chromosomal breaks and an
increased sensitivity to radiation due to impaired DNA repair. FA
patients usually present a wide spectrum of clinical features,
mainly congenital anomalies, progressive pancitopenia and
predisposition to malignancy. At present time, there are no less
than 20 different genes linked to FA (where FANCA is the
most frequently involved, and BRCA1 is one of the rarest)
and the list will probably grow in the future (2).
Because of the high genetic heterogenity of FA, in
the case of a patient with a phenotype suggestive of FA, physicians
usually prescribe laboratory test, like mitomycin C or
diepoxybutane (DEB) test in order to confirm their clinical
evaluation (3). If one of these tests
detects the presence of a higher chromosomal breaks, a sequential
study of each gene known to be causative of FA can be pursued until
one mutation is detected in one of the FA genes.
Genetic testing has become quite common only in the
last few years. Today we have a long way to go before learning
about all genetic variations present in human being. For this
reason, it is not so uncommon to detect a genetic variation never
reported previously in PubMed or in gene-specific databases. In the
5-classes IARC (International Agency for Research on Cancer)
classification, such variations belong to class 3, which means we
are waiting for research studies aimed to demonstrate if they are
pathogenic or likely pathogenic, a class 5 or class 4 variant, or
not.
Such molecular results, called variant of unknown
significance (VUS), represent a real challenge for clinicians.
Because of the lack of knowledge of a possible link between cancer
risk and the detected variation, clinical management usually should
not be based on the molecular result of the test but on the
patient's personal and family history of cancer (4).
Here we present the case of a female patient
referred to our cancer clinic because of her personal cancer
history. The genetic test revealed the presence of a homozygous
BRCA1 VUS. The clinical phenotype of the patient is not
linked to the typical FA features, and no complex rearrangements or
multiradial formation were detected by DEB test, so we tend to
consider that BRCA1 VUS as of little clinical significance.
Our data, together with further data that may be available in
future studies, may therefore contribute to correctly define the
clinical significance of this variant.
Medical history of our patient
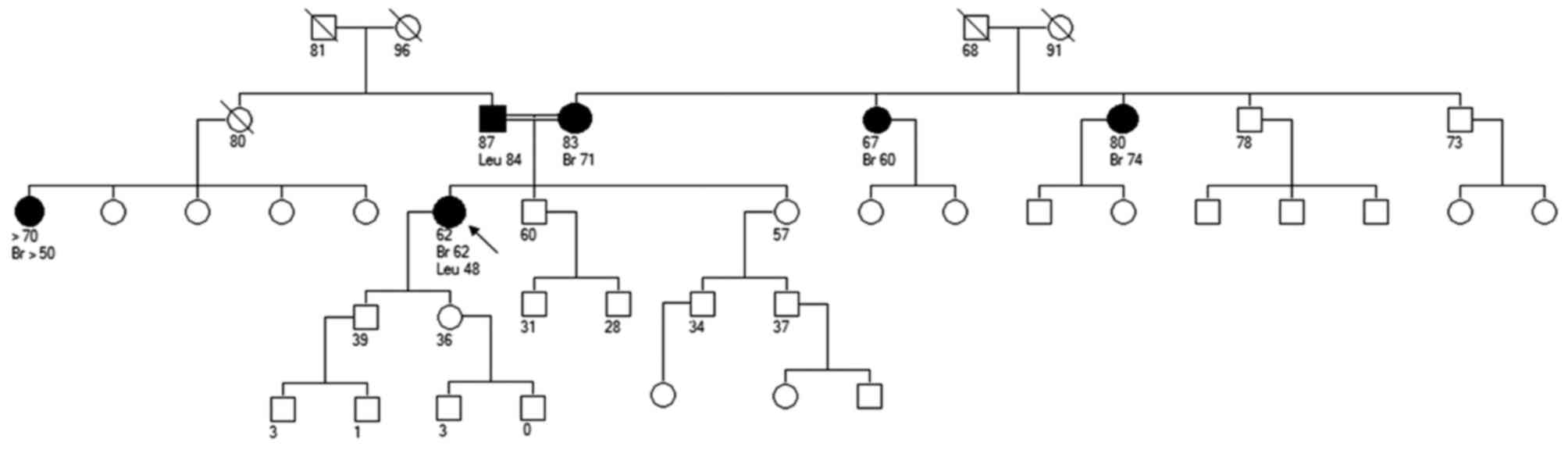
A 62-year-old woman was referred to our genetic
clinic because of her past tumour history. Chronic lymphatic
leukemia was diagnosed at age 48 and treated with chemotherapy and
anti-CD20 monocolonal antibodies. At age 50 she was treated with
hysterectomy and bilateral oophorectomy because of a huge uterine
polyp, while at age 62 she underwent left mastectomy and hormonal
therapy because of a diagnosis of multifocal lobular carcinoma
associated with lobular carcinoma in situ LIN2.
Her mother and both maternal aunts were diagnosed
with breast cancer (respectively, at 71, 74 and 60 years old)
(Fig. 1). Her father had leukemia at
age 84 and one paternal female cousin developed breast cancer aged
50+. Nothing else was relevant in the family, except referred
kindred between parents (even though they were not first
cousins).
BRCA mutation carrier probability was low (2.4%
according to BOADICEA, 0.22% according to CaGene), but because of
family history of breast cancers, we decided to offer her genetic
testing for BRCA1 and BRCA2. During counselling with
the geneticist, the patient was informed about BRCA testing and all
the possible personal and familial implications in case of the
detection of a BRCA mutation; the patient decided to sign informed
consent and underwent blood sample drawing.
Materials and methods
Sequencing and MLPA
Genomic DNA (gDNA) was extracted from blood sample
by MagCore Super (Diatech LabLine SRL, Jesi, Italy) using MagCore
Genomic DNA Whole Blood kit. Genomic DNA obtained from the patient
was used for BRCA1 and BRCA2 sequencing in order to
search for point mutations and small insertions/deletions. All
coding exons and the intron/exon boundaries of BRCA1 and
BRCA2 genes were amplified by PCR. All PCR fragments were
simultaneously amplified at the annealing temperature of 60°C, with
the AmpliTaq Gold kit (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Sequencing was performed on
purified PCR products by using BigDye® Terminator v.3.1
Cycle Sequencing kit (Thermo Fisher Scientific, Inc.) and run on
3730Xl DNA Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.), after purification with Agencourt
CleanSeq®-Beckman Coulter. Sequences were analyzed by
Mutation Surveyor® Software (v5.0.1; SoftGenetics, LLC.,
State College, PA, USA).
Because heterozygous large deletions or duplications
can go undetected by conventional PCR based sequencing of gDNA, we
searched for possible rearrangements by using the MLPA assay. MLPA
was performed by using SALSA P002(D1)-BRCA1 MLPA kit and
SALSA P045(B3)-BRCA2 MLPA kit (MRC-Holland, Amsterdam, The
Netherlands), following manufacturer's instructions. MLPA products
were run on the 3730Xl DNA Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) according to the Gene Mapper Module
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Results were
then analysed by the Gene Marker Software (v2.6.3; SoftGenetics,
LLC.).
In silico analysis of the missense
mutation
Pathogenicity of the variant was predicted with
bioinformatic tools based on the impact of aminoacid substitutions
on the structure and function of proteins and on the degree of
conservation of aminoacid residues along the same protein in
different species. The effect of the variant c.3082C>T
(p.Arg1028Cys) on BRCA1 protein was predicted using:
Mutation Taster (http://www.mutationtaster.org), PolyPhen (http://genetics.bwh.harvard.edu/pph), SIFT
(http://sift.jcvi.org/www/SIFT_enst_submit.html), Align
GVGD (http://agvgd.iarc.fr/agvgd_input.php) and HCI Breast
Cancer Genes Prior Probabilities (http://priors.hci.utah.edu/PRIORS). Possible effect on
mRNA splicing was predicted by using: MaxExScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html),
Human Splicing Finder (http://www.umd.be/HSF), Gene Splicer (http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml)
and NNSPLICE v.0.9 (http://www.fruitfly.org/seq_tools/splice.html).
DEB test
In order to highlight the possible increased rate of
chromosome breakage and radial forms, a DEB test was performed. The
analysis was carried out following the European and Italian
Guidelines (Specific Constitutional Cytogenetic Guidelines ECA July
2012-www.e-c-a.eu-, Linee guida diagnosi citogenetica
2013-www.sigu.net-). Peripheral blood lymphocyte
cultures in the absence or presence of a nontoxic concentration
(0.01 and 0.1 µg/ml) of DEB, chromosome preparation and QFQ
staining were made according to standard procedures (5). A total of 100 metaphases were analyzed
for each harvested cultures scoring: the rate of cells with
chromosome breakage, the mean of chromosome/chromatid breaks/gaps,
multiradial formations, acentric and dicentric fragments, rings,
endoreduplicated chromosomes and premature chromosome condensation
were evaluated for each metaphase. Case results were compared with
those derived from the normal control.
Results
Using Sanger sequencing and MLPA we were able to
find only the presence of the variant c.3082C>T (p.Arg1028Cys)
in BRCA1. Unexpectedly, this VUS was detected at homozygous
state. MLPA confirmed the presence of two alleles in the
corresponding exon and the variant was detectable also with a
different couple of primers in a further PCR reaction.
In our case, even though the presence of a
homozygous BRCA1 mutation was likely to be due to
consanguinity in her parents, the result was not compatible with
the typical clinical presentation of a FA, especially of
BRCA1 related-FA.
As we usually do, we invited the patient for a
second visit in order to discuss the result of her test. Because
clinical features of our patient suggested that the reported
BRCA1 variant was more likely a variant of little clinical
significance instead of a pathogenic one, we invited the woman to
undergo DEB test in order to definitively rule out FA.
The DEB testing was performed and a normal female
karyotype was detected in all analyzed metaphases. The chromosomal
instability has been evaluated in 100 metaphases without DEB, in
100 metaphases after incubation with 0.01 µg/ml DEB and in 100
metaphases after incubation with 0.1 µg/ml DEB. The frequency of
chromosomal breakage detected with and without DEB was below 1%,
and no complex rearrangement or multiradial formations were
detected. The standard of our laboratory of chromosomal breakage is
as high as 3%.
Discussion
The variant we detected is very rare and is now
reported in ExAC database (http://exac.broadinstitute.org) with a frequency of
0.0025%.
This VUS shows substantial disparity of
classification among open accessible database (6): in fact, it has been described as Likely
benign in ClinVar, as a VUS in BIC and UMD. In LOVD 3.0 the variant
has been reported as affecting function (+/?), according to
evolutionary conservation analysis (7).
Bioinformatics tools, used to predict the impact of
the VUS detected, gave conflicting results: Polyphen software
predicted that the variant is possibly damaging (with a score of
0.587). The Polyphen scoring system considers a variant as ‘benign’
below the score of 0.5, as ‘possibly damaging’ between 0.5 and
0.85, as ‘probably damaging’ over 0.85. Mutation Taster and SIFT
programs predicted that the variant is tolerated, based on sequence
homology and the physical properties of aminoacids. HCI Breast
Cancer Prior Probabilities predicted a weak or null probability of
pathogenicity from damage to the protein sequence. Align GVGD class
for this substitution is C15 (likely neutral). The tool combines
the biophysical characteristics of aminoacids and protein multiple
sequence alignments to predict where missense substitutions in
genes of interest fall in a spectrum from enriched deleterious to
enriched neutral. Possible effect on mRNA splicing was instead
excluded by all the bioinformatics tools.
For all unclassified variants, it is important to
collect useful data to correctly define the classification. The
presence of this variant at homozygous state may probably help this
difficult task.
Hypersensitivity to the clastogenic effect of DNA
cross-linking agents can be considered as a unique marker for the
diagnosis of FA. This cellular characteristic is still utilized as
a diagnostic test to identify the preanemic patient as well as the
patient with aplastic anemia or leukemia who may or may not have
the common physical stigmata associated with FA; in this situation,
we thought it could be useful also to provide additional data
regarding the possible effect of the detected mutation and,
therefore, its pathogenicity. A variety of chemical agents can be
used to test for DNA cross-link sensitivity but DEB analysis is the
preferred test for FA diagnosis, since it has the highest
sensitivity and specificity compared to other agents (8,9).
Data obtained by DEB-test performed in our patient
does not support the hypothesis that she is affected by FA. As ever
with a laboratory test, also DEB-test has its own limits. For
example, results interpretation of the chromosome breakage test may
be complicated by the presence of a cellular mosaicism, a condition
in which there are two cell populations, one with increased
sensitivity to DEB and the other with normal levels of chromosome
breakage.
Clinical features of our patient supported this
negative DEB result. Therefore, we didn't advise the patient to
undergo more intensive breast screening but to continue standard
five years follow-up. Direct gene testing was not offered to other
family members because the detection of that BRCA1 VUS would
not change their clinical management.
We decided to perform a literature review on FA, in
order to see what are the typical presentations of a
BRCA1-linked FA. Despite that the presence of a
homozygous/compound heterozygous pathogenic mutation in
BRCA1 is commonly considered embryonic lethal, we were able
to find just few case reports of carriers of such genetic
defect.
The first ever reported patient (10) with a homozygous biallelic BRCA1
mutation (c.2681_2682delAA; p.Lys894Thrfs*8) is a Scottish woman
who developed breast cancer at age 32 and subsequently
contralateral breast cancer. Nonetheless, Greenberg's equipe
recently re-sequenced her DNA and the mutation was found only at
heterozygous state. Therefore this Scottish woman actually does not
have a homozygous biallelic BRCA1 mutation.
The second report (11) is a woman who developed papillary
serous ovarian carcinoma at 28 years old; the cancer was found to
be extremely sensitive to the interstrand crosslinking agents like
carboplatin. Additional features were adult height of 150 cm,
developmental delay with limited speech at 4 years of age and
dysmorphic features.
This woman had multiple relatives who developed
cancer. The sister of the paternal grandfather and one paternal
aunt were affected by breast cancer, while the sister of the
maternal grandfather had bilateral breast cancer as well as ovarian
cancer.
From the molecular point of view, this woman had a
known deleterious mutation in BRCA1 (c.2457delC;
p.Asp821Ilefs*25), a VUS in trans on BRCA1
(c.5209T>C; p.Val1736Ala) as well as a VUS in BRCA2
(c.971G>C; p.Arg324Thr). Even though the co-occurrence in
BRCA1 of a VUS in trans with a pathogenic variant
suggests that the VUS should not be considered clinically relevant,
residue conservation across vertebrate species, loss of
heterozygosity, DNA double strand break data lead Greenberg's
equipe to consider the c.5209T>C variant as hypomorphic and
therefore supported their hypothesys of a biallelic heterozygous
mutations in BRCA1.
The last report (12)
is still from Greenberg, who described the case of a woman with
multiple dysmorphic features and anomalies, developmental delay,
intellectual disability and a personal history of breast cancer at
age 23.
The cancer familiy history was significant in the
maternal side, since both the mother and one maternal aunt had
ovarian cancer at the age of 50.
This woman was found to carry a frameshift mutation
(c.594_597del; p.Ser198Argfs*35) on one BRCA1 allele and a
missense mutation (c.5095C>T; p.Arg1699Trp), previously
described as pathogenic, on the other BRCA1 allele. DEB test
supported the diagnosis of FA and, therefore, the pathogenicity of
the frameshift mutation detected.
For our team, DEB test's result and literature
review supported the hypothesis that our patient was not affected
by FA and therefore we were pretty confident to exclude a possible
relationship between the BRCA1 mutation detected in the
woman and her personal history of cancer.
We offered to our patient a tailored surveillance
program according to her personal and family history of cancer
(rather than based to her molecular profile) and we did not suggest
testing to her relatives.
If our clinical case observation will be confirmed
by further evidence, this might contribute to better understanding
and more appropriate clinical management of similar families.
Acknowledgements
Authors wish to thank the patient, as she accepted
to trust in our lateral thinking plan (and undergo DEB testing) and
for allowing us to publish her data so as to help her family and
other people who may in future take advantage of the work that has
been done. The authors would like to to thank Edda Opack for
English revision of the study and Ms. Alessandra Rossi for
technical support.
References
|
1
|
Chen S and Parmigiani G: Meta-analysis of
BRCA1 and BRCA2 penetrance. J Clin Oncol. 25:1329–1333. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dufour C: How I manage patient with
Fanconi Anemia. Br J Haematol. 178:32–47. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Auerbach AD, Adler B and Chaganti RS:
Prenatal and postnatal diagnosis and carrier detection of Fanconi
anemia by a cytogenetic method. Pediatrics. 67:128–135.
1981.PubMed/NCBI
|
|
4
|
Miller-Samuel S, MacDonald DJ, Weitzel JN,
Santiago F, Martino MA, Namey T, Augustyn A, Mueller R, Forman A,
Bradbury AR and Morris GJ: Variants of uncertain significance in
breast cancer-related genes: Real-world implications for a clinical
conundrum. Part one: Clinical genetics recommendations. Semin
Oncol. 38:469–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Babu A and Verma RS: Human Chromosomes
Principles and Techniques. 2nd. McGraw Hill; Bronson, TX: 1995
|
|
6
|
Vail PJ, Morris B, van Kan A, Burdett BC,
Moyes K, Theisen A, Kerr ID, Wenstrup RJ and Eggington JM:
Comparison of locus-specific databases for BRCA1 and BRCA2 variants
reveals disparity in variant classification within and among
databases. J Community Genet. 6:351–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Burk-Herrick A, Scally M, Amrine-Madsen H,
Stanhope MJ and Springer MS: Natural selection and mammalian BRCA1
sequences: Elucidating functionally important sites relevant to
breast cancer susceptibility in humans. Mamm Genome. 17:257–270.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Auerbach AD: Fanconi anemia diagnosis and
the diepoxybutane (DEB) test. Exp Hematol. 21:731–733.
1993.PubMed/NCBI
|
|
9
|
Auerbach AD: Fanconi anemia and its
diagnosis. Mutat Res. 668:4–10. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boyd M, Harris F, McFarlane R, Davidson HR
and Black DM: A human BRCA1 gene knockout. Nature. 375:541–542.
1995. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Domchek SM, Tang J, Stopfer J, Lilli DR,
Hamel N, Tischkowitz M, Monteiro AN, Messick TE, Powers J, Yonker
A, et al: Biallelic deleterious BRCA1 mutations in a woman with
early-onset ovarian cancer. Cancer Discov. 3:399–405. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sawyer SL, Tian L, Kähkönen M,
Schwartzentruber J and Kircher M: University of Washington Centre
for Mendelian Genomics, FORGGE Canada Consortium. Majewski J,
Dyment DA, Innes AM, et al: Biallelic mutations in BRCA1 cause a
new Fanconi anemia subtype Cancer Discov. 5:135–142. 2015.
|