Introduction
Pancreatic cancer remains a highly lethal neoplasm.
Even with multimodality therapy for localized disease, patient
survival is measured in months. Although the FOLFIRINOX regimen has
produced substantial benefits in the treatment of metastatic
pancreatic cancer, it is associated with severe adverse effects
(1). The further development of
better therapeutic regimens for pancreatic cancer requires new and
potent anticancer agents.
Ras transformation renders cells sensitive to
reactive oxygen species (ROS)-induced cell death (2,3), and
pancreatic cancers exhibit an extremely high mutation rate of K-ras
(>90%) (4). Therefore, we
investigated the anti-proliferative effects of ROS generators on
pancreatic cancer cells. Phenethyl isothiocyanate (PEITC) is a
potent ROS generator (5–8). PEITC belongs to the family of natural
isothiocyanates, which are found in a variety of cruciferous
vegetables and released when the vegetables are cut or masticated
(5). PEITC has an inhibitory effect
on the growth of several types of cancer cells, and is now being
studied in phase 2 clinical trials for the prevention of lung and
oral cancer (3,5–8). ROS
results in the oxidation of cell constituents such as DNA, lipids
and proteins, and this oxidation may damage cancer cells and
ultimately lead to cell death.
ROS attack nucleotides present in the
deoxynucleoside triphosphate (dNTP) pool as well as within DNA, and
convert them into their oxidized forms. The dNTP used in DNA
synthesis are up to 13,000-fold more susceptible to oxidative
damage than bases in duplex DNA (9).
MuT homolog-1 (MTH1) is a pyrophosphatase that acts on oxidized
nucleotides and hydrolyzes 8-oxo-2′-deoxyguanosine triphosphate in
the dNTP pool to prevent its incorporation into nuclear and
mitochondrial DNA, which reduces cytotoxicity in cells (10,11).
MTH1 is overexpressed in cancer cells and prevents
oxidized nucleotides from being misincorporated into DNA. Although
MTH1 is non-essential in normal cells, cancer cells require MTH1
activity to avoid the incorporation of oxidized dNTPs, which would
result in DNA damage and cell death (12–14). By
aiming at a redox-adaptation mechanism, MTH1 inhibition represents
a new approach for a therapeutic strategy against cancer (15,16).
However, recent reports have indicated that growth inhibition by
MTH1 inhibitors may be due to off-target cytotoxic effects
(17,18). TH588 is a potent inhibitor of human
MTH1 with an IC50 value of 5 nM and inhibits the growth
of cancer cell lines (IC50s=2.48–6.37 µM) without
significant cytotoxicity toward immortalized cells
(IC50s≥20 µM) (17). It
exhibits >1000-fold selectivity for MTH1 over the related Nudix
hydrolase protein family members MTH2, NUDT5, NUDT12, NUDT14, and
NUDT16, as well as other proteins with known nucleoside
triphosphate pyrophosphatase activity (dCTPase, dUTPase, and ITPA)
(17). These results suggest that low
concentrations of MTH1 inhibitors may suppress the enzyme activity
without anti-proliferative activity. TH588 has good metabolic
stability and is available in vivo (17). In the present study, we examined the
combined effects of TH588 and PEITC on the growth of human
pancreatic cancer cells.
Materials and methods
Materials
TH588 was obtained from Selleckchem (Houston, TX,
USA). PEITC and
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma-Aldrich; Merck KGaA, Darmstadt, Germany.
General caspase inhibitor
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK) was
purchased from R&D Systems, Inc., Minneapolis, MN, USA.
Cells and cell culture
Human pancreatic carcinoma (MIAPaCa-2 and Panc-1),
lymphoma (BALM3), and leukemia (NB4) cells were cultured in RPMI
1640 medium supplemented with 10% fetal bovine serum and 80 µg/ml
gentamicin at 37°C in a humidified atmosphere of 5% CO2
in air.
Assay of cell growth and
viability
Cell numbers were counted in a Model Z1 Coulter
Counter (Beckman Coulter, Tokyo, Japan). The cells were seeded at
1×105 cells/ml in a 24-well multidish. After culture
with or without the test compounds for the indicated times, viable
cells were examined by a modified MTT assay (19) or a trypan blue dye exclusion test
using an automated cell counter (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using TRI reagent
(Sigma-Aldrich; Merck KGaA). Total RNA was converted to
first-strand cDNA primed with random hexamer in a reaction volume
of 20 µl using an RNA PCR kit (qPCR RT Master Mix; Toyobo Life
Science, Osaka, Japan), and 2 µl of this reaction was used as a
template in real time PCR. The primers were used as described
previously (20). The RT-qPCR
reaction was performed using an Takara TP860 Real-Time PCR system
(Takara Bio Inc., Tokyo, Japan) according to the manufacture's
instruction. The threshold cycle values were normalized to the
threshold value of glyceraldehydes-3-phosphate dehydrogenase.
Real-time PCR results were calculated according to the following
protocol: Relative expression level=2−∆Ct, where ∆Cq=Cq
(gene of interest)-Cq (housekeeping gene) (20).
Immunofluorescence microscopy
Cells were fixed with 4% paraformaldehyde in PBS for
10 min at room temperature, and then permeabilized with 0.3%
Tween-20 for 15 min. After fixation, cells were washed three times
with PBS and then blocked with blocking buffer (1% bovine serum
albumin in PBS for 60 min. Cells were incubated with anti-pH2AX
(Cell Signaling Technology Inc., Tokyo, Japan) for 60 min, washed
with the blocking buffer and then incubated for 60 min with Alexa
Fluor 594-conjugated anti-rabbit secondary antibodies (Cell
Signaling Technology Inc.) and FITC-labeled avidin (Vector
Laboratories, Inc., Burlingame, CA, USA). Avidin binds with high
specificity to 8-oxo-dG (21,22). Confocal images were obtained using an
inverted microscope (Olympus Corporation, Tokyo, Japan). All
immunofluorescence experiments were repeated three times.
Statistical analysis
The results are expressed as means ± standard
deviation (SD). Pairs of data were compared using Student's t-test.
Significant differences were considered to exist for probabilities
below 5% (P<0.05) and are indicated by asterisks (*). For
comparisons among multiple groups, an F-test using one-way analysis
of variance and a post hoc Tukey-Kramer test were performed to
demonstrate statistical significance. Again, significant
differences were considered to exist for probabilities below
5%.
Results
Combined effects of TH588 and
ROS-inducers on the growth of MIAPaCa-2 cells
The potent MTH1 inhibitor TH588
concentration-dependently inhibited the growth of pancreatic cancer
Panc-1 cells, but the antioxidant N-acetyl cysteine (NAC) did not
affect the growth inhibition (Fig.
1A), suggesting that the growth inhibition induced by TH588 is
independent of oxidized stress. These results are consistent with
reports that growth inhibition by TH588 may be due to off-target
cytotoxic effects (17,18). However, it is possible that TH588 may
act as an MTH1 inhibitor under oxidized stress conditions for
cytotoxic effects. Thus, we examined the effect of TH588 on the
growth of Panc-1 cells in the presence of hydrogen peroxide
(H2O2). A low concentration of TH588 alone
did not affect cell growth, but significantly enhanced
H2O2-induced growth inhibition (Fig. 1B). Next, we examined the combined
effects of TH588 and ROS inducers. A low concentration of TH588
effectively enhanced the growth inhibition of MIAPaCa-2 cells
induced by several ROS inducers, such as doxorubicin (23), MK615 (Japanese apricot extract)
(24), isopentenyl adenosine
(25) and isothiocyanates (3,5–8). Combined treatment with doxorubicin and
TH588 effectively inhibited the growth of MIAPaCa-2 cells (Fig. 1C). Among them, the combination of
PEITC and TH588 was the best for inhibiting the growth of MIAPaCa-2
cells (Fig. 1D). Similar results were
obtained when other pancreatic cancer cells were examined. TH588 at
1 µM alone hardly affected the growth of MIAPaCa-2 cells, but
significantly enhanced the growth inhibition induced by PEITC
(Fig. 1D). NAC effectively
counteracted the growth inhibition induced by PEITC alone (Fig. 2A), as well as that by PEITC plus TH588
(Fig. 2B). These results suggest that
ROS production is associated with the growth-inhibitory effects of
TH588 plus ROS inducers. When the cells were treated with 4 µM
PEITC and 2 µM TH588, the viability was markedly decreased and it
was significantly counteracted with the general caspase inhibitor
Z-VAD-FMK (Fig. 2C), suggesting that
the combined treatment induced caspase-dependent cell death. As
shown in Fig. 2D, MTH1 mRNA is
expressed in several malignant cell lines. The combined treatment
was also effective in leukemia and lymphoma cells (Fig. 2E and F). Although expression of MTH1
mRNA in NB4 leukemia cells was less than 35% in MIAPaCa-2 cells,
the combined treatment similarly effective to NB4 cells.
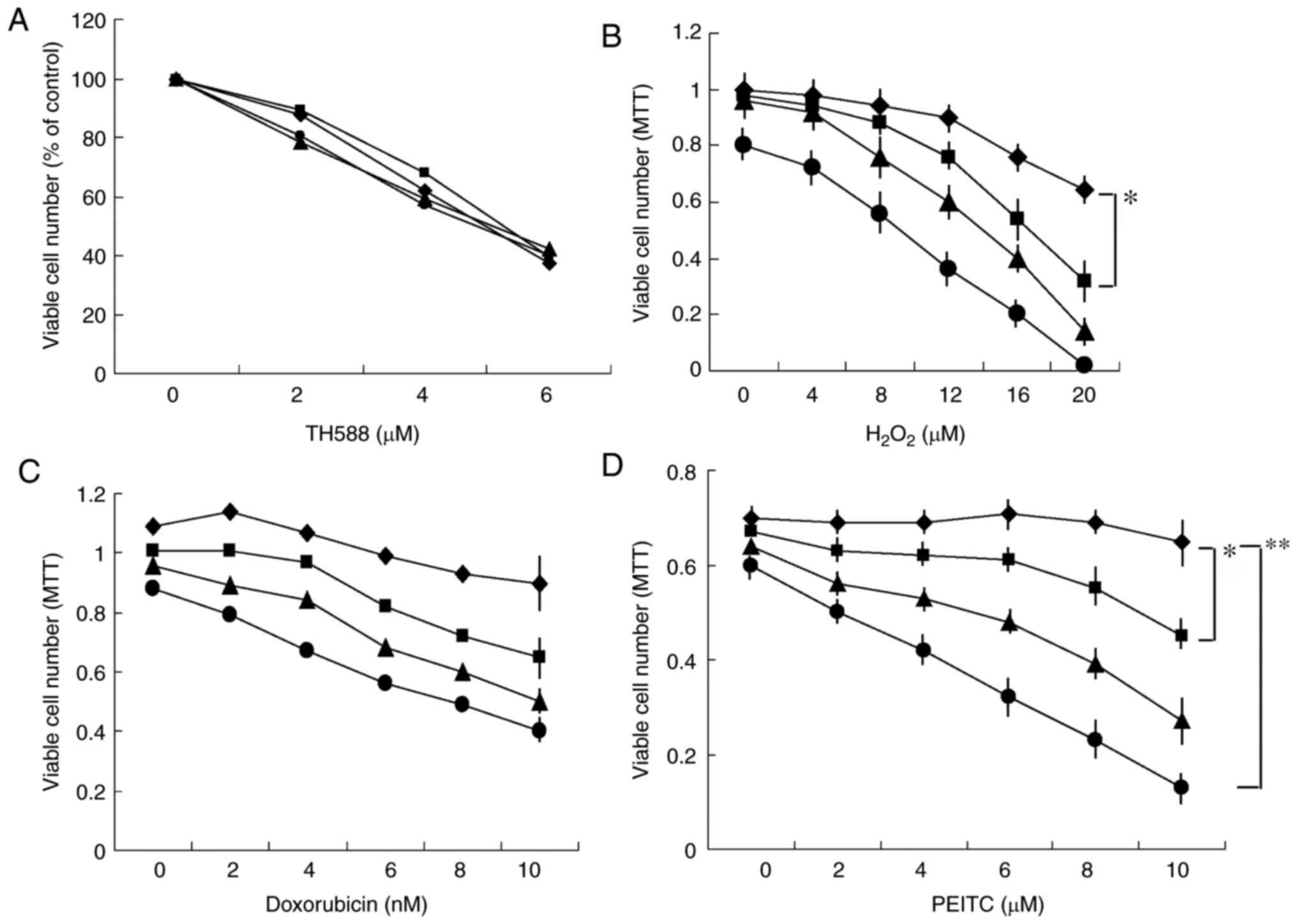 | Figure 1.(A) Effect of NAC on the
growth-inhibitory effect of TH588. Panc-1 cells were cultured with
TH588 in the presence of 0 (♦), 5 (■), 7.5 (▲), or 10 (●) mM NAC
for 5 days. The values are the means of 3 determinations. (B)
Combined effects of H2O2 and TH588 on growth
of Panc-1 cells. Cells were treated with H2O2
in the presence of 0 (♦), 1 (■), 2 (▲), or 3 (●) µM TH588 for 7
days. H2O2 was added on days 0, 3 and 5. The
values are the means ± SD of 3 determinations. (C and D) Combined
effects of TH588 and doxorubicin or PEITC on growth of MIAPaCa-2
cells. Cells were treated with doxorubicin (C) or PEITC (D) in the
presence of 0 (♦), 1 (■), 2 (▲), or 3 (●) µM TH588 for 5 days. The
values are the means ± SD of 3 determinations. *P<0.05,
**P<0.01. |
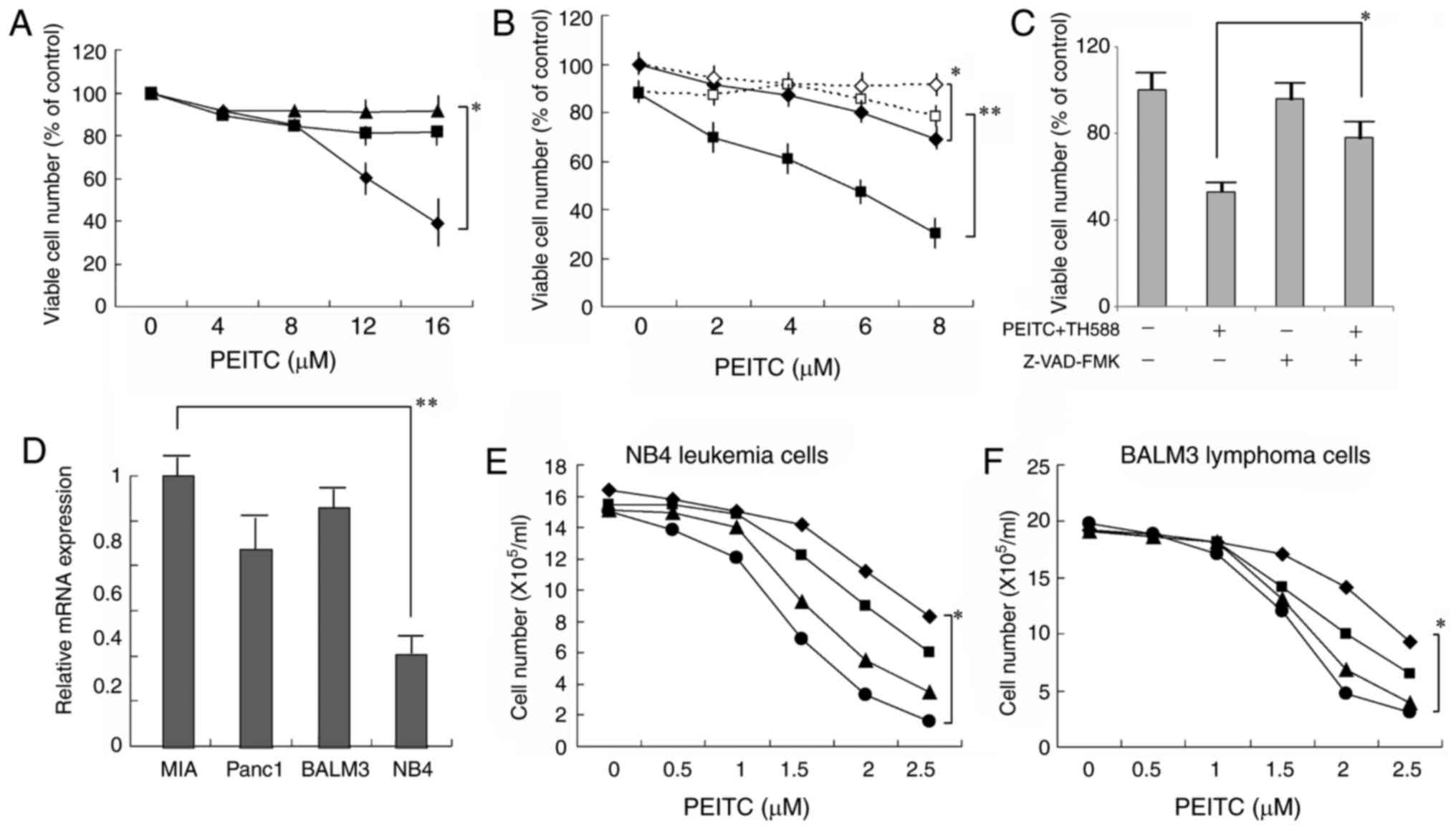 | Figure 2.(A) Effect of NAC on the
growth-inhibitory effect of PEITC. MIAPaCa-2 cells cultured with
PEITC in the presence of 0 (♦), 5 (■), or 7.5 (▲) mM NAC for 6
days. The values are the means ± SD of 3 determinations. (B) Effect
of NAC on the growth-inhibitory effect of PEITC plus TH588.
MIAPaCa-2 cells cultured with PEITC (♦, ◊) or PEITC plus 2 µM TH588
(■, □) in the presence (◊, □) or absence (♦, ■) of 7.5 mM NAC for 6
days. The values are the means ± SD of 3 determinations. (C)
Suppression by caspase inhibitor of the cell death induced by PEITC
and TH588. Cells were treated with 4 µM PEITC plus 2 µM TH588 in
the presence of 4 µM Z-VAD-FMK for 2 days. The values are means of
three separate experiments ± SD. (D) MTH1 mRNA expression in
several cancer cell lines. Gene expression was determined by RT-PCR
and normalized to MIAPaCa-2 cells. The values are means of three
separate experiments ± SD. (E, F) Combined effects of PEITC and
TH588 on the growth of leukemia or lymphoma cells. Cells were
treated with PEITC in the presence of 0 (♦), 0.2 (■), 0.4 (▲), or
0.6 (●) µM TH588 for 5 days. The values are the means of 3
determinations. *P<0.05, **P<0.01. |
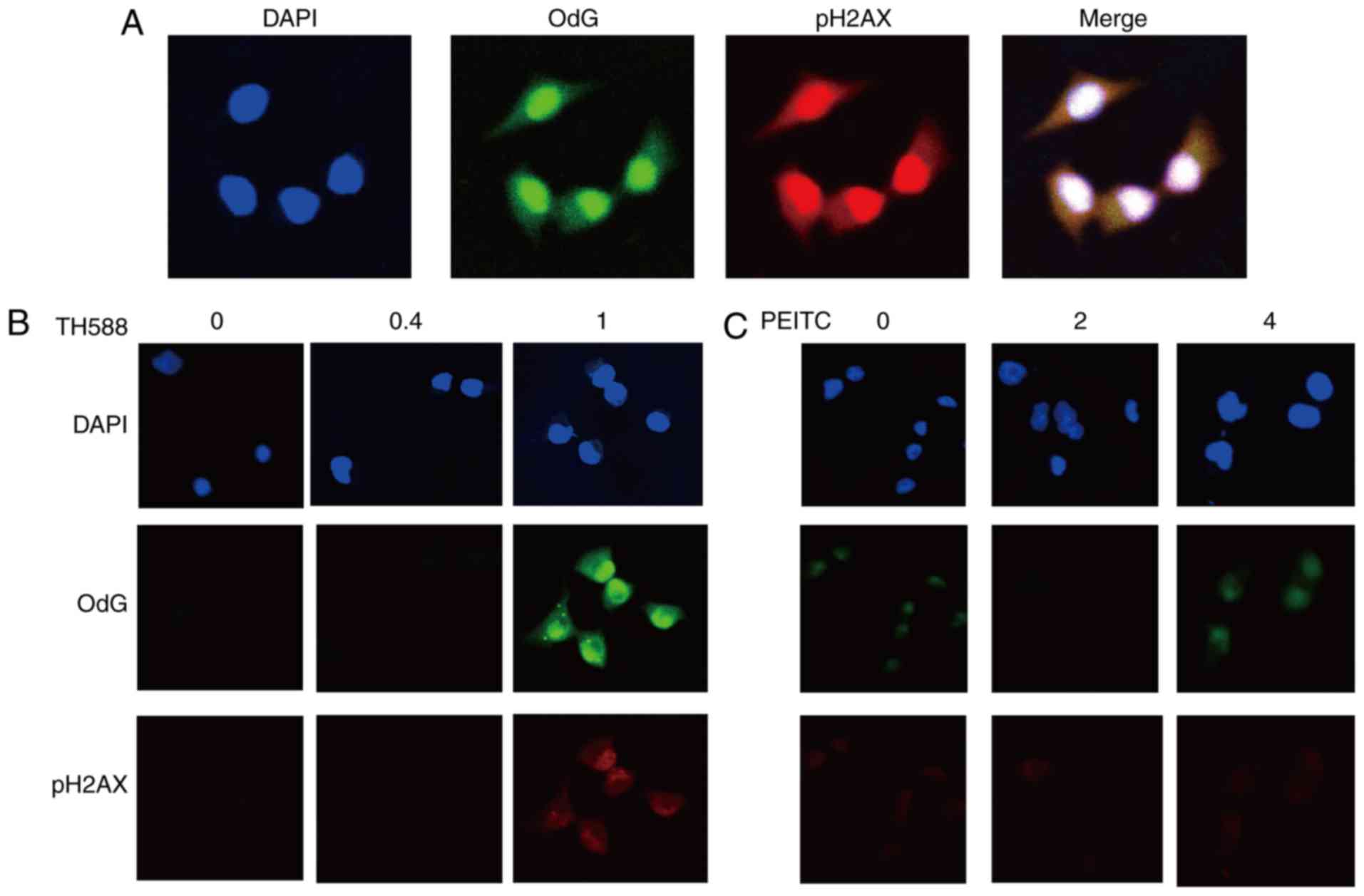
Enhancement of the formation of
8-oxo-dG and DNA damage by combined treatment with TH588 and
PEITC
MIAPaCa-2 cells treated with 2 µM TH588 exhibited
increases in both a marker of DNA damage, the phosphorylation of
histone 2A.X (pH2AX), and the formation of 8-oxo-dG (Fig. 3A). Both of these effects were mainly
localized in nuclei. Although TH588 at 1 µM hardly affected the
growth of MIAPaCa-2 cells (Fig. 1A),
formation of 8-oxo-dG and pH2AX was evident in treatment for 56 h
(Fig. 3B), suggesting that TH588
enhances 8-oxo-dG production. PEITC also concentration-dependently
induced the formation of 8-oxo-dG, and this effect was evident in
the treatment with 4 µM (Fig. 3C).
These results indicate that 8-oxo-dG formation and DNA damage were
induced by low concentrations of TH588 or PEITC, which hardly
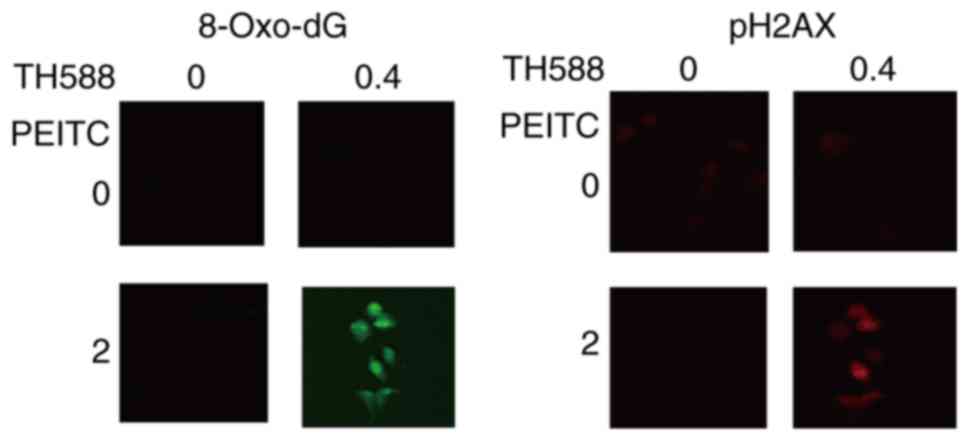
affected the growth of MIAPaCa-2 cells. Fig. 4 shows that low concentrations of TH588
and PEITC co-operatively induce the formation of 8-oxo-dG and DNA
damage in MIAPaCa-2 cells. Similar results were observed in Panc-1
cells treated with TH588 and PEITC.
Discussion
While the mechanism of the anticancer activity of
isothiocyanates has not yet been fully elucidated, numerous
pathways have been implicated. They include oxidative stress, cell
cycle arrest, inhibition of histone deacetylation, inhibition of
angiogenesis, and regulation of translation initiation (5). PEITC disables the glutathione
antioxidant system and causes ROS accumulation preferentially in
cancer cells (5). In the present
investigation, the effect of PEITC was completely counteracted by
treatment with NAC (Fig. 2A),
suggesting that the effects were mainly due to oxidative stress by
ROS generation. One of the mechanisms that protects cancer cells
from the cytotoxic effect of high levels of ROS is the expression
of MTH1, which sanitizes oxidized dNTP pools by converting them to
the corresponding monophosphates (10,11). TH588
is a potent and selective inhibitor of human MTH1, although the
growth-inhibiting effect of TH588 is not associated with the
inhibition of MTH1 (17,18). While MTH1 function may be critical to
survival in cells with elevated levels of ROS, MTH1 is dispensable
in cancer cells under normal culture conditions. Wang et al
(26) reported that the sensitivity
of melanoma cells to TH588 is correlated with the level of
endogenous ROS, although the cytotoxicity of TH588 is not
associated with its inhibitory effect on MTH1. In the present
study, TH588 enhanced the formation of 8-oxo-dG induced by PEITC
(Fig. 4). These results suggest that
TH588 enhances the formation of 8-oxo-dG in DNA and contributes to
growth inhibition in cancer cells with highly elevated levels of
ROS. However, MTH1 mRNA expression is not associated with the
combined effects of TH588 plus PEITC (Fig. 2). We cannot eliminate the possibility
that TH588 act the other site(s). These results suggest that the
combined treatment indicates the off-target effects, although low
concentrations of TH588 enhance the formation of 8-oxo-dG in
DNA.
MTH1 is overexpressed in glioblastoma, and MTH1
silencing inhibits colony formation and xenograft tumor growth
(27). A high expression of MTH1 was
significantly associated with deeper tumor invasion, advanced
cancer stage, and poor overall survival and disease-specific
survival compared with low MTH1 expression on esophageal squamous
cell carcinoma (28). These results
suggest that MTH1 may be a potential target for cancer therapy,
although MTH1 is dispensable for some malignancies. Further
investigation will be necessary to understand the role of MTH1 in
cancer therapy.
Since several reports have shown that PEITC has
little toxic effects on normal cells (5), oxidative stress caused by PEITC can be
used to treat drug-resistant cancer cells. However, there is no
evident dose-relationship between the cellular ROS level and its
cytotoxicity in cancer cells treated with several ROS inducers
(29), suggesting that many factors
may influence ROS-mediated cytotoxicity in cancer cells. Lactic
acidosis induced a much higher ROS level in cancer cells than
PEITC, but permitted the progressive growth of cancer cells
(29). PEITC covalently modifies the
cysteine side chains of glutathione S-transferase, which
irreversibly inhibits its enzymatic activity (30). This irreversible inhibition may
contribute to effective cell-killing. The combination of PEITC and
TH588 may become a novel therapeutic strategy against cancers.
Acknowledgements
The present study was supported in part by the
SUIGAN project, Shimane University, Japan.
References
|
1
|
Conroy T, Desseigne F, Ychou M, Bouché O,
Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de
la Fouchardière C, et al: FOLFIRINOX versus gemcitabine for
metastatic pancreatic cancer. N Engl J Med. 364:1817–1825. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shaw AT, Winslow MM, Magendantz M, Ouyang
C, Dowdle J, Subramanian A, Lewis TA, Maglathin RL, Tolliday N and
Jacks T: Selective killing of K-ras mutant cancer cells by small
molecule inducers of oxidative stress. Proc Natl Acad Sci USA.
108:pp. 8773–8778. 2011; View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Trachootham D, Zhou Y, Zhang H, Demizu Y,
Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J and
Huang P: Selective killing of oncogenically transformed cells
through a ROS-mediated mechanism by beta-phenylethyl
isothiocyanate. Cancer Cell. 10:241–252. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Henzel AF, Kimmelman AC, Stanger BZ,
Bardeesy N and Depinho RA: Genetics and biology of pancreatic
ductal adenocarcinoma. Genes Dev. 20:1218–1249. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu X, Zhou QH and Xu K: Are
isothiocyanates potential anti-cancer drugs? Acta Pharmacol Sin.
30:501–512. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu X, Kassie F and Mersch-Sundermann V:
Induction of apoptosis in tumor cells by naturally occurring
sulfur-containing compounds. Mutat Res. 589:81–102. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kasukabe T, Honma Y, Okabe-Kado J, Higuchi
Y, Kato N and Kumakura S: Combined treatment with cotylenin A and
phenethyl isothiocyanate induces strong antitumor activity mainly
through the induction of ferroptotic cell death in human pancreatic
cancer cells. Oncol Rep. 36:968–976. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiao D, Powolny AA, Moura MB, Kelley EE,
Bommareddy A, Kim SH, Hahm ER, Normolle D, Van Houten B and Singh
SV: Phenethyl isothiocyanate inhibits oxidative phosphorylation to
trigger reactive oxygen species-mediated death of human prostate
cancer cells. J Biol Chem. 285:26558–26569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Topal MD and Baker MS: DNA precursor pool:
A significant target for N-methyl-N-nitrosourea in C3H/10T1/2 clone
8 cells. Proc Natl Acad Sci USA. 79:pp. 2211–2215. 1982; View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshimura D, Sakumi K, Ohno M, Sakai Y,
Furuichi M, Iwai S and Nakabeppu Y: An oxidized purine nucleoside
triphosphatase, MTH1, suppresses cell death caused by oxidative
stress. J Biol Chem. 278:37965–37973. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sakai Y, Furuichi M, Takahashi M, Mishima
M, Iwai S, Shirakawa M and Nakabeppu Y: A molecular basis for the
selective recognition of 2-hydroxy-dATP and 8-oxo-dGTP by human
MTH1. J Biol Chem. 277:8579–8587. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kennedy CH, Pass HI and Mitchell JB:
Expression of human MutT homologue (hMTH1) protein in primary
non-small-cell lung carcinomas and histologically normal
surrounding tissue. Free Radic Biol Med. 34:1447–1457. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Giribaldi MG, Munoz A, Halvorsen K, Patel
A and Rai P: MTH1 expression is required for effective
transformation by oncogenic HRAS. Oncotarget. 6:11519–11529. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patel A, Burton DG, Halvorsen K, Balkan W,
Reiner T, Perez-Stable C, Cohen A, Munoz A, Giribaldi MG, Singh S,
et al: MutT homolog 1 (MTH1) maintains multiple KRAS-driven
pro-malignant pathways. Oncogene. 34:2586–2596. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gad H, Koolmeister T, Jemth AS, Eshtad S,
Jacques SA, Ström CE, Svensson LM, Schultz N, Lundbäck T,
Einarsdottir BO, et al: MTH1 inhibition eradicates cancer by
preventing sanitation of the dNTP pool. Nature. 508:215–221. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huber KV, Salah E, Radic B, Gridling M,
Elkins JM, Stukalov A, Jemth AS, Göktürk C, Sanjiv K, Strömberg K,
et al: Stereospecific targeting of MTH1 by (S)-crizotinib as an
anticancer strategy. Nature. 508:222–227. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kawamura T, Kawatani M, Muroi M, Kondoh Y,
Futamura Y, Aono H, Tanaka M, Honda K and Osada H: Proteomic
profiling of small-molecule inhibitors reveals dispensability of
MTH1 for cancer cell survival. Sci Rep. 6:265212016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kettle JG, Alwan H, Bista M, Breed J,
Davies NL, Eckersley K, Fillery S, Foote KM, Goodwin L, Jones DR,
et al: Potent and selective inhibitors of MTH1 probe its role in
cancer cell survival. J Med Chem. 59:2346–2361. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kawakami K, Hattori M, Inoue T, Maruyama
Y, Ohkanda J, Kato N, Tongu M, Yamada T, Akimoto M, Takenaga K, et
al: A novel fusicoccin derivative preferentially targets hypoxic
tumor cells and inhibits tumor growth in xenografts. Anticancer
Agents Med Chem. 12:791–800. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang X, Song W, Zhou Y, Mao F, Lin Y,
Guan J and Sun Q: Expression and function of MutT homolog 1 in
distinct subtypes of breast cancer. Oncol Lett. 13:2161–2168. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Struthers L, Patel R, Clark J and Thomas
S: Direct detection of 8-oxodeoxyguanosine and 8-oxoguanine by
avidin and its analogues. Anal Biochem. 255:20–31. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Conners R, Hooley E, Clarke AR, Thomas S
and Brady R: Recognition of oxidatively modified bases within the
biotin-binding site by avidin. J Mol Biol. 357:263–274. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Goodman J and Hochstein P: Generation of
free radicals and lipid peroxidation by redox cycling of adriamycin
and daunomycin. Biochem Biophys Res Commun. 77:797–803. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hattori M, Kawakami K, Akimoto M, Takenaga
K, Suzumiya J and Honma Y: Antitumor effect of Japanese apricot
extract (MK615) on human cancer cells in vitro and in vivo through
a reactive oxygen species-dependent mechanism. Tumori. 99:239–248.
2013.PubMed/NCBI
|
|
25
|
Ishii Y, Hori Y, Sakai S and Honma Y:
Control of differentiation and apoptosis of human myeloid leukemia
cells by cytokinins and cytokinin nucleosides, plant
redifferentiation-inducing hormones. Cell Growth Differ. 13:19–26.
2002.PubMed/NCBI
|
|
26
|
Wang JY, Jin L, Yan XG, Sherwin S,
Farrelly M, Zhang YY, Liu F, Wang CY, Guo ST, Yari H, et al:
Reactive oxygen species dictate the apoptotic response of melanoma
cells to TH588. J Invest Dermatol. 136:2277–2286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tu Y, Wang Z, Wang X, Yang H, Zhang P,
Johnson M, Liu N, Liu H, Jin W, Zhang Y and Cui D4: Birth of MTH1
as a therapeutic target for glioblastoma: MTH1 is indispensable for
gliomatumorigenesis. Am J Transl Res. 8:2803–2811. 2016.PubMed/NCBI
|
|
28
|
Akiyama S, Saeki H, Nakashima Y, Iimori M,
Kitao H, Oki E, Oda Y, Nakabeppu Y, Kakeji Y and Maehara Y:
Prognostic impact of MutT homolog-1 expression on esophageal
squamous cell carcinoma. Cancer Med. 6:258–266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu C, Hu W, Wu H and Hu X: No evident
dose-response relationship between cellular ROS level and its
cytotoxicity-a paradoxical issue in ROS-based cancer therapy. Sci
Rep. 4:50292014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumari V, Dyba MA, Holland RJ, Liang YH,
Singh SV and Ji X: Irreversible inhibition of glutathione
S-transferase by phenethyl isothiocyanate (PEITC), a dietary cancer
chemopreventive phytochemical. PLoS One. 11:e01638212016.
View Article : Google Scholar : PubMed/NCBI
|