Introduction
Glioblastoma multiforme (GBM), classified as a grade
IV astrocytoma by the World Health Organization (1), is the most malignant form of
intracranial glioma tumor. The standard treatment for GBM is
surgery in combination with chemotherapy and/or radiotherapy.
Despite advances in therapeutic research, GBM patients have a
survival time of ~1 year post-diagnosis and <5% survive for 5
years (2,3). Highly invasive GBM cells may proliferate
extensively and infiltrate into the brain parenchyma, leading to
mortality (4,5).
The heterogeneity of GBM at the cellular level
hinders the improved understanding of the disease, making it
difficult to design and develop effective therapeutic strategies
(1,6).
To address this challenge, several studies (7,8) have
investigated the global gene expression profiles and genetic
alterations that drive tumor aggressiveness in GBM, and have
stratified GBM into four subtypes: Pro-neural, neural, classical
and mesenchymal. The majority of the established GBM cell lines
belong to the fourth subtype, which has a mesenchymal gene
signature (7).
In general, cadherin switching occurs during cancer
cell invasion: It is a process in which expression of epithelial
E-cadherin is switched to that of mesenchymal-associated cadherins,
including N-cadherin and vimentin, which are typical mesenchymal
markers that contribute toward the regulation of cell invasion
(9–13). It should be emphasized that, although
GBM cells are non-epithelial, these cells necessarily acquire their
invasive capability through parts of the epithelial-mesenchymal
transition (EMT) program, as exemplified by the upregulation of
N-cadherin in invasive glioma cells (14). Therefore, the use of several
epithelial or mesenchymal markers to determine the extent of glioma
cell invasion is a valid approach.
Elucidating the molecular basis of GBM invasion and
proliferation may contribute toward improving GBM therapy;
therefore, a number of studies (7,15) have
focused on using a variety of relevant genes in order to do so.
However, the knowledge obtained from studies into these known genes
is insufficient to elucidate the mechanism of GBM progression. As
such, the identification of novel molecular targets involved in
regulating invasion and proliferation in GBM is necessary.
Deubiquitinases (DUBs) have fundamental functions in
the ubiquitin system through their ability to specifically remove
ubiquitin from targeted proteins. The human genome encodes ~100
DUBs, which are stratified into 6 families (16). DUBs regulate the ubiquitinization
status and the degradation of proteins, affecting the activation
and localization of proteins, which in turn is essential for
numerous cellular processes and signaling pathways. Therefore, a
number of DUBs have been identified as pro- or anticancer factors
owing to their regulatory effects on key oncogenes or other
proteins involved in tumor progression (17–20). Of
these DUBs, the present study focused on ubiquitin-specific
protease 15 (USP15), a member of the USP family. A previous study
suggested that USP15 functions as a tumor suppressor and that it is
likely to be a key target for the inhibition of cancer cell
progression (21). For instance,
USP15 is downregulated in paclitaxel-resistant ovarian cancer
(22); however, other studies have
highlighted its pro-oncogenic functions. In metastatic breast
cancer MDA-MB-231 cells, USP15 is required for cell migration
(23). In a previous study by
Eichhorn et al (24), the
USP15 gene was revealed to be amplified in glioblastoma, breast
cancer and ovarian cancer, and the depletion of USP15 decreased the
oncogenic capacity of patient-derived glioma-initiating cells. The
present study primarily explores USP15 at the genetic level with a
focus on a particular glioma cell subpopulation. To the best of our
knowledge, there has been no previous research regarding the
general function of USP15 in glioma cells and therefore, the
present study was a preliminary investigation into the role of
USP15 in glioma cells. The U87-MG cell line was used as a general
model of glioma in order to elucidate the general gene functions
associated with glioma (25–27).
Materials and methods
Cell lines and cell culture
The glioma U251-MG and U87-MG cell lines were
obtained from the Cell Bank of Type Culture Collection of the
Chinese Academy of Sciences (Shanghai, China) and the American Type
Culture Collection (ATCC; Manassas, VA, USA), respectively. The two
human glioma cell lines were cultured at 37°C for 3 days in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) containing 2 mM glutamine, 10%
fetal bovine serum (FBS) (both from Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (both from
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells were
maintained in an incubator at 37°C in a 5% CO2
atmosphere. Cells (293) were cultured at 37°C for 2 days in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 2 mM glutamine,
10% FBS (both from Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin (both from Sigma-Aldrich;
Merck KGaA). HEB cells were cultured at 37°C for 3 days in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 2 mM glutamine,
10% FBS (both from Thermo Fisher Scientific, Inc.), 100 U/ml
penicillin and 100 µg/ml streptomycin (both from Sigma-Aldrich;
Merck KGaA).
Lentivirus production and
transduction
The transduction system was conducted as previously
described. Briefly, short hairpin RNA (shRNA) against USP15
(sequence, 5′-GCCAACTGAAGGTTGGAATAA-3′; USP15 KD) was generated and
another construct expressing shRNA against a non-mammalian
luciferase gene was used as a negative control (NC). These
constructs were co-transfected with packaging plasmids (15 µg;
Shanghai Hanyu Biotechnology Co., Ltd., Shanghai, China) into 293
cells using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions,
and viral particles were harvested 48 h later. U87-MG and U251-MG
cells were infected with a lentivirus containing 6 µg/ml polybrene
(Sigma-Aldrich; Merck KGaA).
Transwell invasion assay
For the Transwell invasion assay, 2×104
U87-MG and U251-MG cells stably expressing USP15 shRNAs were plated
into 24-well Boyden chambers (Sigma-Aldrich; Merck KGaA) with an
8-µm pore polycarbonate membrane, which was coated with 30 µg
Matrigel (BD Biosciences, San Jose, CA, USA). Cells
(1×104) were inserted into the upper chamber with 200 µl
serum-free DMEM (Thermo Fisher Scientific, Inc.), and DMEM
containing 20% FBS was added to the lower chamber to serve as a
chemoattractant. Following incubation for 24 h at 37°C in a 5%
CO2 atmosphere, cells were washed 3 times with phosphate
buffered saline. Cells on the top surface of the insert were
removed with a cotton swab. Cells adhering to the lower surface
were fixed with 100% methanol at 4°C for 15 min, stained with
Giemsa solution at room temperature for 30 sec and counted under a
light microscope in five predetermined fields (magnification,
×200).
Cell proliferation
Cells were counted using a Cell Counting Kit-8
(CCK-8) (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). A
total of 1×103 GBM (U87 and U251 cells) were plated onto
96-well culture plates in triplicate and maintained at 37°C, while
cell growth was determined daily for 5 days using a tetrazolium
salt-based colorimetric assay (Dojindo Molecular Technologies,
Inc.), according to the manufacturer's instructions. Absorbance was
measured at 450 nm. The corresponding cells expressing shRNA
against an irrelevant luciferase gene was used as a negative
control (NC). Three independent experiments were performed.
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay
buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA pH 8.0, 5 mM DTT and 2%
SDS) and protein concentration was determined using bicinchoninic
acid assay (Beyotime Institute of Biotechnology, Haimen, China).
Total protein (30 µg/lane) was resolved using a 10% SDS-PAGE gel
and was electro-transferred to polyvinylidene fluoride membranes
(Invitrogen; Thermo Fisher Scientific, Inc.), and blocked with 5%
skimmed dry milk in Tris-buffered saline (pH 7.5) at room
temperature for 1 h in Tris-buffered saline (pH 7.5). Membranes
were immunoblotted overnight at 4°C with the following primary
antibodies: N-cadherin (dilution 1:200; cat. no. 4061), vimentin
(dilution 1:200; cat. no. 3932), E-cadherin (dilution 1:500; cat.
no. 3195) (all from Cell Signaling Technology, Inc., Danvers, MA,
USA) and β-actin (dilution 1:3,000; cat. no. HRP60008; ProteinTech
Group, Inc., Chicago, IL, USA). A horseradish peroxidase-conjugated
IgG secondary antibody (dilution 1:2,000; cat. no. ab6721, Abcam,
Cambridge, MA, USA) was added and incubated at room temperature for
1 h. Bound antibodies were detected using the BeyoECL system (cat.
no. P0018; Beyotime Institute of Biotechnology), and densitometry
ImageQuant 5.2 software (GE Healthcare Life Sciences, Little
Chalfont, UK) was used for analysis.
Statistical analysis
Data are presented in the graphs as the mean ±
standard deviation of three independent experiments. The
differences among groups were determined using analysis of variance
with post hoc contrasts determined by Student-Newman-Keuls test and
comparisons between two groups were analyzed using the Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference. All statistical analyses were performed
using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla,
CA, USA).
Results
USP15 regulates glioma cell invasion
in vitro
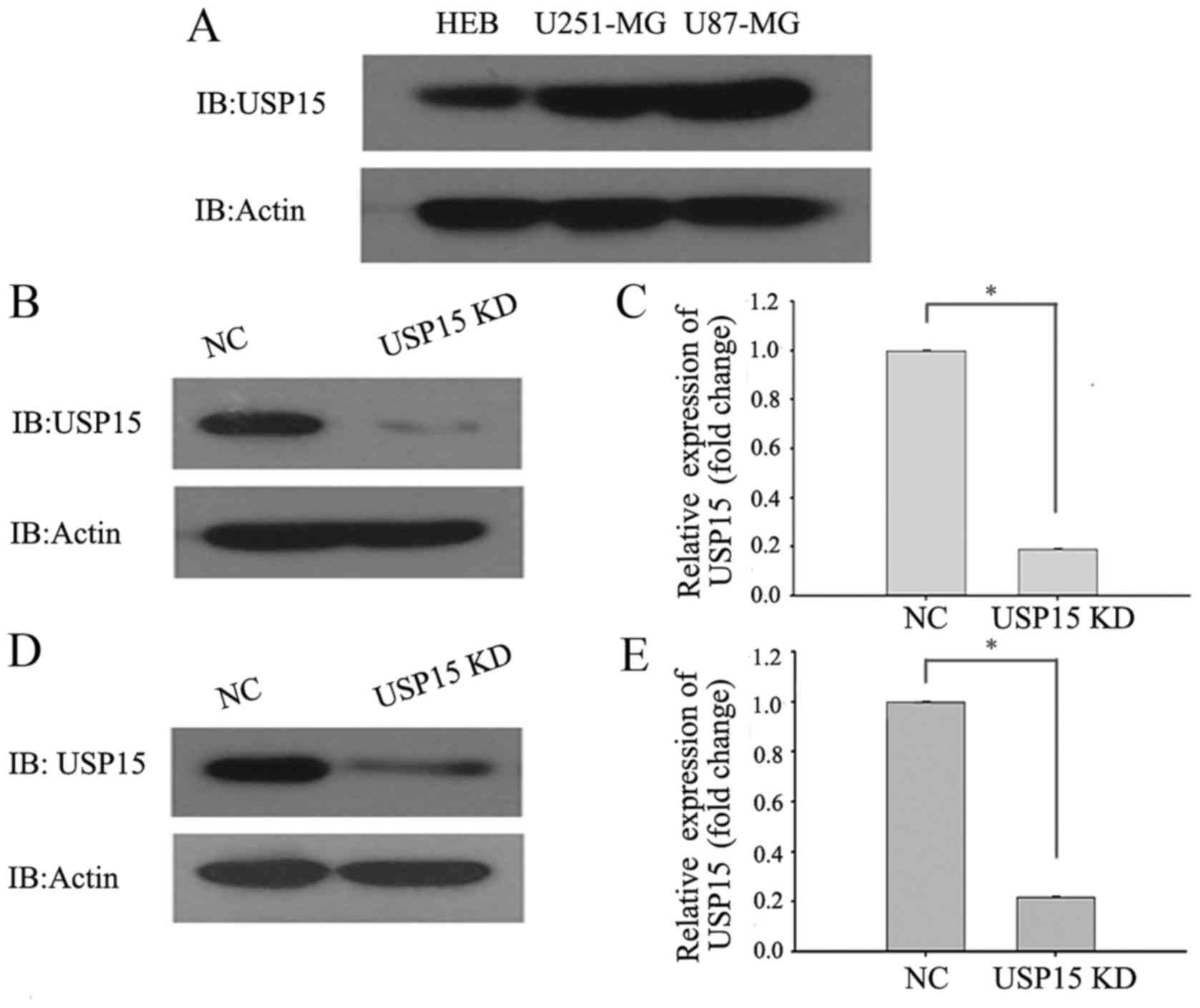
To directly investigate the role of USP15 in GBM
cells, the protein levels of USP15 in the HEB cell line and the
commonly used typical glioma cell lines (U87-MG and U251-MG) were
determined using western blot analysis. The results demonstrated
that the protein levels of USP15 in U87-MG and U251-MG cells were
higher in the glioma cells than in the HEB cell line (Fig. 1A). RNA interference technology was
subsequently used to deplete USP15 expression. U-87-MG and U251-MG
cells were transduced with a lentivirus-encoding control or USP15
shRNA. At 3 days after transduction, cell lysates were subjected to
western blot analysis to detect the protein level of USP15.
Densitometric analysis of the western blotting results, by
comparing USP15 protein levels in control shRNA-expressing cells
with those expressing USP15 shRNA, revealed significant reductions
in USP15 protein levels in the U87-MG (Fig. 1B and C) and U251-MG (Fig. 1D and E) glioma cells treated with
lentivirus-mediated USP15 shRNA. These findings indicated that
USP15 was successfully depleted in the two glioma cell lines.
Eichhorn et al (24) observed that neurospheres expressing
USP15 shRNA yielded fewer tumors than did the neurospheres
expressing control shRNA. This result indicated that expression of
USP15 may result in a loss of cell-cell contact and cell
scattering, a potential consequence of cell invasion. We therefore
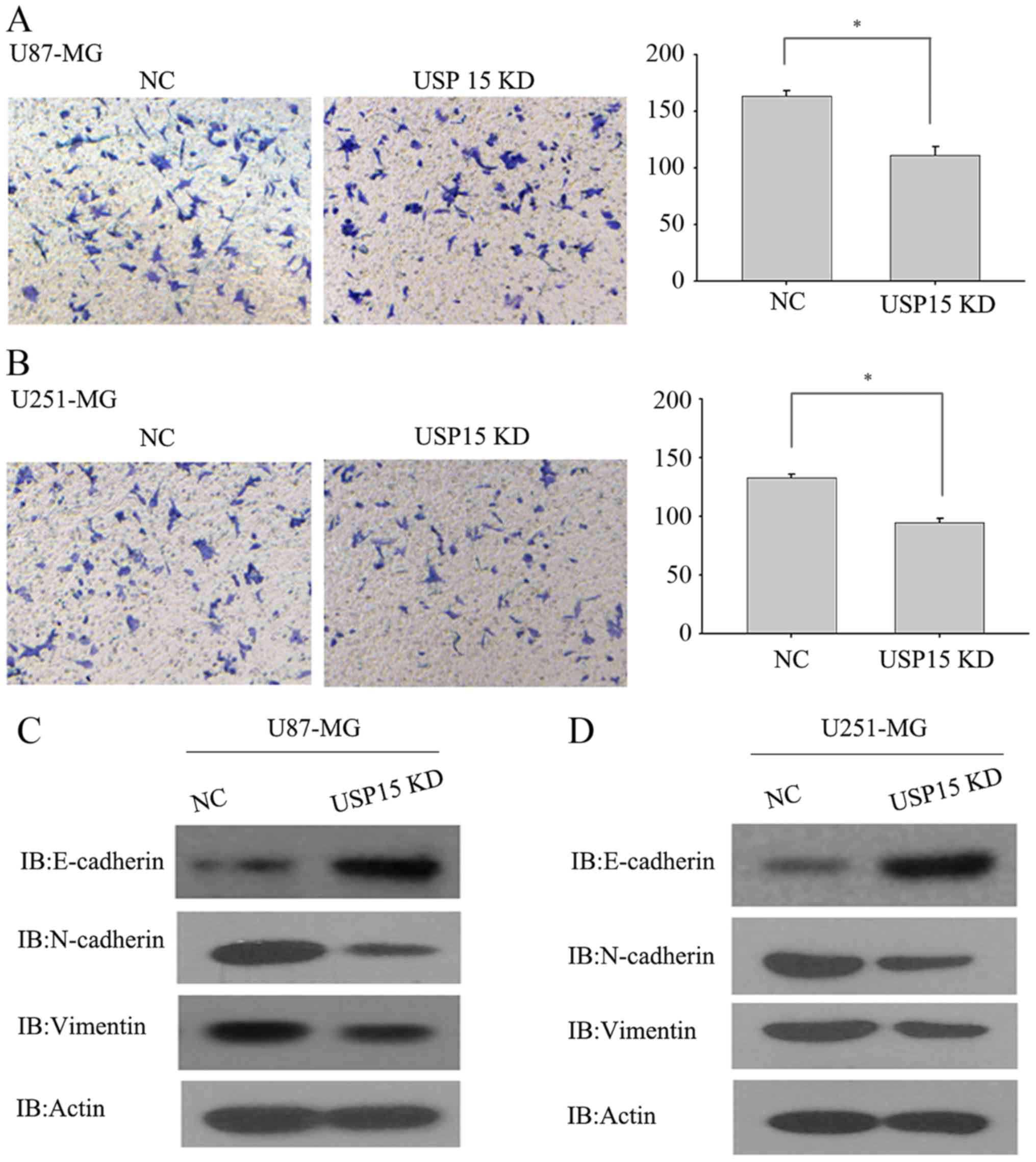
hypothesized that USP15 may promote glioma cell invasion. To test
this hypothesis, a Transwell invasion assay was conducted using
U87-MG and U251-MG cells, which are invasive in vitro, to
determine the invasive abilities of these cells. U87-MG and U251-MG
cells transduced with lentivirus-encoding control or USP15 shRNA
for 3 days were subjected to an invasion assay. Next, 24 h after
incubation, USP15 depletion was observed using lentivirus-mediated
USP15 shRNA in U87-MG cells, leading to a significant reduction in
cell invasion compared with that in the cells treated with control
shRNA (Fig. 2A; P=0.002).
Furthermore, depletion of USP15 in U251-MG cells resulted in a
significant reduction in invasion (P=0.012; Fig. 2B), albeit relatively moderate when
compared with the extent of difference in U87-MG cells. These data
indicate the role of USP15 in glioma cell invasion. Although the
USP15 depletion efficiency in the U87-MG and U251-MG cells was
virtually identical, as determined by western blot analysis
(Fig. 1B-E), the degree of impairment
of cell invasive capacities differed between the two cells types.
This may be attributable to the effect of USP15 on the invasive
capacity of different types of glioma cell.
As mesenchymal-associated genes are associated with
glioma cell invasion, USP15 expression was depleted in U87-MG and
U251-MG cells using lentivirus-encoding USP15-specific shRNA to
examine changes to the expression of N-cadherin, vimentin and
E-cadherin. As expected, USP15 depletion significantly diminished
the expression of N-cadherin and vimentin, while the protein level
of E-cadherin was increased upon USP15 depletion (Fig. 2C and D). These data provide further
support for the hypothesis that USP15 expression is required for
glioma cell invasion.
USP15 regulates glioma cell
proliferation in vitro
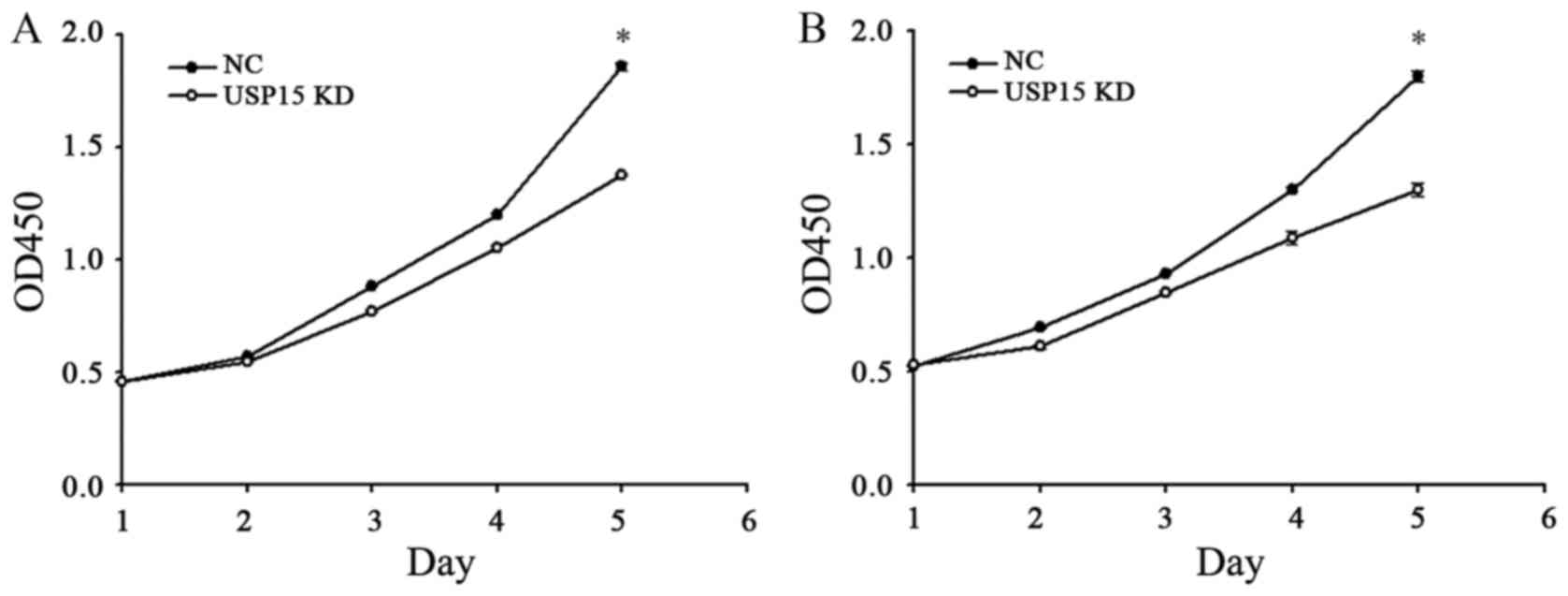
The fact that depletion of USP15 decreased the
oncogenic capacity of patient-derived glioma-initiating cells
indicated that cells with depleted expression of USP15 using
lentivirus-encoding USP15 shRNA generated smaller tumors than cells
expressing control shRNA. Therefore, further experimentation in the
present study aimed to determine whether USP15 may promote glioma
cell proliferation. To quantify cell proliferation, cell growth was
examined at 0-, 1-, 2-, 3-, 4- and 5-day time-points using a CCK-8
assay. The depletion of USP15 did not lead to a significant
reduction in U87-MG proliferation until day 4, with the peak
reduction occurring at day 5 (P<0.001; Fig. 3), indicating that USP15 promotes
glioma cell proliferation. A similar result was observed in U251-MG
cells, thereby corroborating the aforementioned conclusion
(Fig. 3B).
Discussion
During tumor progression, cancer cells acquire
capabilities that permit them to invade the neighboring tissues.
The steps involved in the dispersion of cancer cells include the
loss of cell-cell contact and increased invasiveness (28). In gliomas, tumor cells are known to
exhibit a diffusely infiltrative invasion pattern, in which the
tumor cells migrate as single cells, resulting in a reduced
requirement for surgical and radiological interventions (29). Therefore, the identification of potent
genes associated with glioma cell invasion is urgently required.
Considering the fact that targeting existing invasion-associated
gene targets has failed to significantly attenuate glioma cell
invasiveness, the present study instead focused on the
identification of novel targets that may exert suppressive effects
on glioma cell invasion. Owing to the fact that inhibition of USP15
(a member of the USP family involved in deubiquitinization) caused
a marked reduction in the number of glioma xenografts (24), it was speculated that the reduction
may be attributed to the loss of cell-cell contact and cell
scattering, a consequence of cancer cell invasion. The present
study demonstrated that depletion of USP15 expression resulted in a
reduction in the invasiveness of typical glioma cell models. These
findings indicated that USP15 promotes the invasive capacity of
glioma cells. Notably, a study by Eichhorn et al revealed
that downregulation of USP15 decreased the number of tumors
generated, indicating cancer cell scattering. However, cell
scattering alone is unable to recapitulate the invasive behavior of
cells. For instance, the overexpression of a truncated form of
E-cadherin that lacks the ectodomain of the wild-type protein may
result in loss of cell-cell contact and cell scattering. However,
the truncated product failed to elicit cancer cell invasion
(30). To the best of our knowledge,
the results of the present study demonstrate for the first time
that USP15 renders glioma cells capable of invasion and is involved
in glioma progression.
Cadherins are critical players in a number of
mechanisms with roles in tumor migration and invasion (9,31). These
genes were initially identified owing to their roles in metaplasia,
also known as EMT. During the EMT, upregulation of N-cadherin is
associated with the suppression of E-cadherin, in a process known
as the E- to N-cadherin switch (32).
Although glioma cells are non-epithelial, with limited or
non-existent E-cadherin expression, the cells with high invasive
ability may employ a program similar to EMT (33). Therefore, the relative expression of
E- and N-cadherin is regarded as a hallmark of examining glioma
cell invasiveness. Furthermore, upon USP15 depletion, changes in
the expression of E- and N-cadherin were associated with changes in
the invasiveness of glioma cells. The acquisition of invasive
ability prompts glioma cells to undergo a mesenchymal phenotypic
shift, upregulating N-cadherin and vimentin, which is consistent
with published findings that glioma cell invasion is associated
with the increased expression of N-cadherin (14). Nonetheless, the association of
E-cadherin expression with glioma cell invasiveness should be
interpreted with caution, given that a number of glioma cell lines
lack E-cadherin expression.
USP15 is known to be a tumor suppressor as it
promotes apoptotic processes and is downregulated in drug-resistant
ovarian cancer (22). Additionally,
regulation of the Wnt signaling pathway is essential for a number
of types of cancer, including glioma; USP15 acts as a negative
mediator of this pathway by deubiquitylating and stabilizing the
tumor suppressor adenomatous polyposis coli, thereby promoting the
degradation of β-catenin by the proteasome (34). Furthermore, USP15 negatively regulates
the nuclear factor NF-κB (NF-κB) signaling pathway, which is
required for cancer cell invasion, by stabilizing IκB kinase-α, an
inhibitor of NF-κB (35). However,
evidence indicates that USP15 serves an opposing role in cancer
cells, as exemplified by the USP15-dependent promotion of
glioblastoma oncogenesis (24) and
the USP-induced increase in MDA-MB-231 cell migration (23). With regards to the increase in
migration, the present study revealed that USP15 promoted cell
invasion and proliferation in glioma cells. The fact that USP15 was
identified to serve a dual function was expected, as certain DUBs
have already been documented to exhibit detrimental or protective
dual functions in cancer, depending upon the context and cell type
in which these specific DUBs function (21). For instance, USP2 rescues prostate
cancer cells from apoptosis by stabilizing fatty-acid synthase
(36) and deubiquitylates relevant
apoptosis-inducing factors, thereby promoting cell death (37). These data reveal that the pro-cancer
function of USP15 may contribute to an improved understanding of
the gene, and may rationally delineate its complex interaction with
key genes associated with cancer cell invasion and
proliferation.
In conclusion, the present study identified a
positive association between USP15 expression and cell invasion and
proliferation in U87-MG and U251-MG cells, providing an insight
into the importance of USP15 in a subset of high-grade gliomas.
Future studies should focus on identifying the detailed molecular
mechanisms underlying glioma invasion and proliferation. Since DUBs
have been demonstrated to be targetable by small organic molecules,
USP15 may be a potential therapeutic target and may contribute
toward novel therapeutic interventions in GBM.
Acknowledgements
The present study was funded by the National Natural
Science Foundation of China (grant no. 81660502).
References
|
1
|
Bonavia R, Inda MM, Cavenee WK and Furnari
FB: Heterogeneity maintenance in glioblastoma: A social network.
Cancer Res. 71:4055–4060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Persano L, Rampazzo E, Basso G and Viola
G: Glioblastoma cancer stem cells: Role of the microenvironment and
therapeutic targeting. Biochem Pharmacol. 85:612–622. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrom QT, Gittleman H, Farah P, Ondracek
A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the united states in 2006–2010. Neuro-Oncol. 15
Suppl 2:ii1–ii56. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Malzkorn B and Reifenberger G: Practical
implications of integrated glioma classification according to the
World Health Organization classification of tumors of the central
nervous system 2016. Curr Opin Oncol. 28:494–501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Colman H and Aldape K: Molecular
predictors in gliobla=stoma: Toward personalized therapy. Arch
Neurol. 65:877–883. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toki S, Kagaya S, Shinohara M, Matsumoto
T, Takahata Y, Saito H and Matsumoto K: Genome-wide gene expression
analysis of the human intestinal epithelial cells after stimulation
with lactic acid bacteria. J Allerg Clin Immunol. 117:S1532006.
View Article : Google Scholar
|
|
9
|
Hazan RB, Qiao R, Keren R, Badano I and
Suyama K: Cadherin switch in tumor progression. Ann N Y Acad Sci.
1014:155–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wheelock MJ, Shintani Y, Maeda M, Fukumoto
Y and Johnson KR: Cadherin switching. J Cell Sci. 121:727–735.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anastasiadis PZ: p120-ctn: A nexus for
contextual signaling via Rho GTPases. Biochim Biophys Acta.
1773:34–46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gumbiner BM: Cell adhesion: The molecular
basis of tissue architecture and morphogenesis. Cell. 84:345–357.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nelson WJ and Nusse R: Convergence of Wnt,
beta-catenin, and cadherin pathways. Science. 303:1483–1487. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu KV, Chang JP, Parachoniak CA, Pandika
MM, Aghi MK, Meyronet D, Isachenko N, Fouse SD, Phillips JJ,
Cheresh DA, et al: VEGF inhibits tumor cell invasion and
mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell.
22:21–35. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Newcomb EW, Cohen H, Lee SR, Bhalla SK,
Bloom J, Hayes RL and Miller DC: Survival of patients with
glioblastoma multiforme is not influenced by altered expression of
p16, p53, EGFR, MDM2 or Bcl-2 genes. Brain Pathol. 8:655–667. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sacco JJ, Coulson JM, Clague MJ and Urbé
S: Emerging roles of deubiquitinases in cancer-associated pathways.
IUBMB Life. 62:140–157. 2010.PubMed/NCBI
|
|
17
|
Stevenson LF, Sparks A, Allende-Vega N,
Xirodimas DP, Lane DP and Saville MK: The deubiquitinating enzyme
USP2a regulates the p53 pathway by targeting Mdm2. EMBO J.
26:976–986. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van der Horst A, de Vries-Smits AM,
Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM
and Burgering BM: FOXO4 transcriptional activity is regulated by
monoubiquitination and USP7/HAUSP. Nat Cell Biol. 8:1064–1073.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Colland F, Formstecher E, Jacq X, Reverdy
C, Planquette C, Conrath S, Trouplin V, Bianchi J, Aushev VN,
Camonis J, et al: Small-molecule inhibitor of USP7/HAUSP ubiquitin
protease stabilizes and activates p53 in cells. Mol Cancer Ther.
8:2286–2295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Allende-Vega N, Sparks A, Lane DP and
Saville MK: MdmX is a substrate for the deubiquitinating enzyme
USP2a. Oncogene. 29:432–441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fraile JM, Quesada V, Rodríguez D, Freije
JM and López-Otín C: Deubiquitinases in cancer: New functions and
therapeutic options. Oncogene. 31:2373–2388. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu M, Takanashi M, Oikawa K, Tanaka M,
Nishi H, Isaka K, Kudo M and Kuroda M: USP15 plays an essential
role for caspase-3 activation during Paclitaxel-induced apoptosis.
Biochem Biophys Res Commun. 388:366–371. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Inui M, Manfrin A, Mamidi A, Martello G,
Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S, et al: USP15
is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell
Biol. 13:1368–1375. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eichhorn PJ, Rodón L, Gonzàlez-Juncà A,
Dirac A, Gili M, Martínez-Sáez E, Aura C, Barba I, Peg V, Prat A,
et al: USP15 stabilizes TGF-β receptor I and promotes oncogenesis
through the activation of TGF-β signaling in glioblastoma. Nat Med.
18:429–435. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo Z, Li G, Bian E, Ma CC, Wan J and Zhao
B: TGF-β-mediated repression of MST1 by DNMT1 promotes glioma
malignancy. Biomed Pharmacother. 94:774–780. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao Y, Wang J, Yang J and Miao J:
Synergistic antitumor effect of ING4/PTEN double tumor suppressors
mediated by adenovirus modified with RGD on glioma. J Neurosurg
Sci. Apr 12–2017.(Epub ahead of print).
|
|
27
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gupta GP and Massagué J: Cancer
metastasis: Building a framework. Cell. 127:679–695. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Paw I, Carpenter RC, Watabe K, Debinski W
and Lo HW: Mechanisms regulating glioma invasion. Cancer Lett.
362:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takeichi M: Morphogenetic roles of classic
cadherins. Curr Opin Cell Biol. 7:619–627. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21 Suppl 7:vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hanahan D and Weinberg R: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang X, Langelotz C, Hetfeld-Pechoc BK,
Schwenk W and Dubiel W: The COP9 signalosome mediates beta-catenin
degradation by deneddylation and blocks adenomatous polyposis coli
destruction via USP15. J Mol Biol. 391:691–702. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schweitzer K, Bozko PM, Dubiel W and
Naumann M: CSN controls NF-kappaB by deubiquitinylation of
IkappaBalpha. EMBO J. 26:1532–1541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Graner E, Tang D, Rossi S, Baron A, Migita
T, Weinstein LJ, Lechpammer M, Huesken D, Zimmermann J, Signoretti
S and Loda M: The isopeptidase USP2a regulates the stability of
fatty acid synthase in prostate cancer. Cancer Cell. 5:253–261.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oh KH, Yang SW, Park JM, Seol JH, Iemura
S, Natsume T, Murata S, Tanaka K, Jeon YJ and Chung CH: Control of
AIF-mediated cell death by antagonistic functions of CHIP ubiquitin
E3 ligase and USP2 deubiquitinating enzyme. Cell Death Differ.
18:1326–1336. 2011. View Article : Google Scholar : PubMed/NCBI
|