Introduction
The Wnt signaling pathway plays a critical role in
proliferation, cell mobility and development (1–3). Wnt
signaling stabilizes cytoplasmic β-catenin, and β-catenin is
subsequently translocated into the nucleus where it interacts with
the T-cell factor/lymphoid enhancer-binding factor (TCF/LEF)
transcription factors to stimulate the transcription of Wnt target
genes, such as such as cyclin D1, c-Myc and Axin2 (4–7). Aberrant
activation of Wnt signaling has been implicated in a variety of
human cancers, including colon cancer (8,9).
Specificity protein 5 (Sp5), a member of the Sp
transcription factor family, is a direct target of
β-catenin-TCF/LEF (10–13). Sp5 is expressed at high levels in
normal and tumor tissues where Wnt signaling is activated (14), suggesting that Sp5 is a universal Wnt
signaling target. Sp5 contains three conserved Cys2His2 zinc finger
domains at its C-terminus, which bind the GC-box (GGGCGG) or
closely associated sequences (15,16). Sp5
directly represses the transcription of the cell cycle inhibitor
p21 by binding to the GC-boxes located in its promoter region, and
exogenous expression promotes the growth of breast cancer MCF-7
cells, which express no detectable Sp5 protein (11,14,17). By
contrast, overexpression of Sp5 inhibits cell growth and
transformation mediated by oncogenic KRAS in NIH3T3 cells (18). In addition, Sp5-deficient mice do not
exhibit evident abnormalities, possibly due to functional
compensation by other members of the Sp family (19). Although the analysis of Sp5 could
provide novel insights into the mechanisms underlying the
tumorigenesis of colon cancer, the role of Sp5 in the regulation of
cell proliferation has not been clearly established.
The present study demonstrates that the expression
of Sp5 is not upregulated in the colon cancer cell line HCT116, in
which Wnt signaling is constitutively activated. Furthermore, Sp5
has the potential to inhibit the proliferation of HCT116 cells
through regulation of the expression of the cell cycle inhibitor
p27.
Materials and methods
Cell culture and transfection
HCT116 cells were obtained from ATCC (Manassas, VA,
USA) and cultured in McCoy's 5A medium (Sigma-Aldrich, St. Louis,
MO, USA) supplemented with 10% fetal bovine serum (FBS; Nichirei
Biosciences, Tokyo, Japan). HEK293 cells were obtained from ATCC
and cultured in Dulbecco's modified Eagle's medium (DMEM; Nissui,
Tokyo, Japan) supplemented with 10% FBS. An siRNA targeting p27
(5′-CAAGTGGAATTTCGATTTT-3′) was obtained from Ambion (s2837; Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Plasmids and siRNAs
were transfected into cells using Lipofectamine LTX and RNAiMAX
(both Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Silencer negative control 1 siRNA
(Ambion; Thermo Fisher Scientific, Inc.) was used as a negative
control.
Cloning of mouse Sp5 cDNA
Mouse embryonic fibroblasts (MEF) were sourced from
C57BL/6 mice. Total RNA was isolated from MEFs using TRIsure
(Bioline Reagents Limited, London, UK) and cDNA was synthesized
from 500 ng of RNA using Super Script III Reverse Transcriptase
(Invitrogen; Thermo Fisher Scientific, Inc.) with random hexamers.
A FLAG tag was added to the 5′ end of the mouse Sp5 (mSp5) cDNA by
polymerase chain reaction (PCR) using KOD-plus-Neo (Toyobo Co.,
Ltd., Osaka, Japan) and cloned into a pcDNA3.1 vector (Invitrogen;
Thermo Fisher Scientific, Inc.) with the forward primer
5′-AAAGAATTCATGGCCGCTGTGGCCGTCCT-3′ and reverse primer
5′-TTTTCTAGATCATAGGTCCCGCGGATTCT-3′.
Reverse transcription
(RT)-quantitative PCR (qPCR) analysis
Total RNA was isolated using TRIsure and cDNA was
synthesized from 500 ng of RNA using PrimeScript RT Master Mix
(Takara Bio, Inc., Otsu, Japan). Reactions were performed at 37°C
for 15 min and inactivated at 85°C for 5 sec. All cDNA samples were
diluted (1:9) with RNase-free water for use as templates in qPCR.
qPCR was performed in duplicate using SYBR Green I (Takara Bio,
Inc.), and detected on a LightCycler 480 Real-Time PCR System
(Roche Diagnostics GmbH, Mannheim, Germany). qPCR was performed in
a 10-µl mixture that included 5 µl 2X SYBR Premix Ex Taq (Takara
Bio, Inc.), 3µ l RNase-free water containing 1.6 pmol of each
primer and 2µl cDNA template. The PCR cycling conditions were as
follows: A denaturation step of 30 sec at 95°C, and then 40 cycles
of amplification at 95°C for 5 sec and 60°C for 30 sec. All mRNA
quantification data were normalized to glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) expression and compared as differences in
ΔΔCq (20). Primer sequences were as
follows: GAPDH forward, 5′-GCACCGTCAAGGCTGAGAAC-3′; GAPDH reverse,
5′-TGGTGAAGACGCCAGTGGA-3′; Axin2 forward,
5′-GCCAATGGCCAAGTGTCTC-3′; Axin2 reverse,
5′-GGCTCTCCAACTCCAGCTTC-3′; p27 forward,
5′-GGCCTCAGAAGACGTCAAAC-3′; p27 reverse,
5′-CATGTATATCTTCCTTGCTTCATCA-3′; p21 forward,
5′-CTGGAGACTCTCAGGGTCGAAA-3′; and p21 reverse,
5′-GATTAGGGCTTCCTCTTGGAGAA-3′.
Immunoblotting analysis
Cells were lysed in radioimmunoprecipitation assay
buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM
ethylenediaminetetraacetic acid, 1% Triton X-100, 0.1% sodium
dodecyl sulfate, 0.5 mM dithiothreitol, 1:1,000 protease inhibitor
cocktail). The lysates were subjected to immunoblotting analysis
with monoclonal mouse anti-FLAG (#F1804; Sigma-Aldrich; dilution,
1:1,000) or anti-tubulin (#CP06; EMD Millipore, Billerica, MA, USA;
dilution, 1:1,000) primary antibodies, and horseradish peroxidase
(HRP)-conjugated secondary antibodies (#405306; BioLegend, San
Diego, CA, USA; dilution, 1:5,000).. Visualization was performed
using Luminata Forte HRP substrate (EMD Millipore).
Cell proliferation assays
HCT116 cells were transfected with Flag-tagged mSp5
or empty vector (control) using Lipofectamine LTX. Cells were
cultured for 1 day and then plated in 96-well plates (Day 0), and
cell numbers were measured using the CellTiter-Glo 2.0 assay kit
(Promega Corporation, Madison, WI, USA).
Luciferase assays
HEK293 cells were transfected with a
luciferase-reporter plasmid and cultured for 24 h. Cells were lysed
and firefly luciferase activity was measured with the Luciferase
Reporter Assay System (Promega Corporation) according to the
manufacturer's instructions. pRL-CMV was used as an internal
control. Primer sequences were as follows: p27 promoter (−300)
forward, 5′-AAACTCGAGCGGTCCTCTGGTCCAGGTCC-3′; p27 promoter (−1,000)
forward, 5′-AAACTCGAGCGAGGGGAGTATTTTCACCC-3′; p27 promoter (TSS)
reverse, 5′-TTTAAGCTTGGTGGCCCCGGCGCGGTTCC-3′; p27 promoter (+100)
reverse, 5′-TTTAAGCTTAGAAAATGATTGACACGGCG-3′.
Chromatin immunoprecipitation (ChIP) assays. ChIP
assays were performed as described previously (21). Briefly, HCT116 cells were transfected
with Mock or FLAG-mSp5 expression constructs 24 h prior to the
assays. DNA fragments immunoprecipitated with anti-FLAG antibody
(#F1804; Sigma-Aldrich) or mouse IgG (2 µg) were analyzed by
RT-qPCR using primers directed against a region containing the
predicted Sp5-binding site in the p27 promoter region (p27-TSS).
The promoter regions of GAPDH and p21 were used as negative
and positive controls, respectively. Primer sequences were as
follows: GAPDH (TSS) forward, 5′-TGCGTGCCCAGTTGAACCAG-3′; GAPDH
(TSS) reverse, 5′-AACAGGAGGAGCAGAGAGCGAAGC-3′; p21 (TSS) forward,
5′-CTGGGGGAGGAGGGAAGTG-3′; p21 (TSS) reverse,
5′-CTGGCCGAGTTCCAGCAG-3′; p27 (−1,000) forward,
5′-CCTTCAGCCCTCTGAAAGCA-3′; p27 (−1,000) reverse,
5′-AGTCCAGCCGCCAAATACAA-3′; p27 (TSS) forward,
5′-ACCGCCATATTGGGCCAC-3′; and p27 (TSS) reverse,
5′-GAGTCGCAGAGCCGTGAG-3′.
Statistical analysis
Statistical analysis was performed using Student's
t-test with Microsoft Excel 2010 (Microsoft Corporation, Seattle,
WA, USA). P<0.01 was considered to indicate a statistically
significant difference.
Results
Sp5 is expressed at low levels in
HCT116 cells
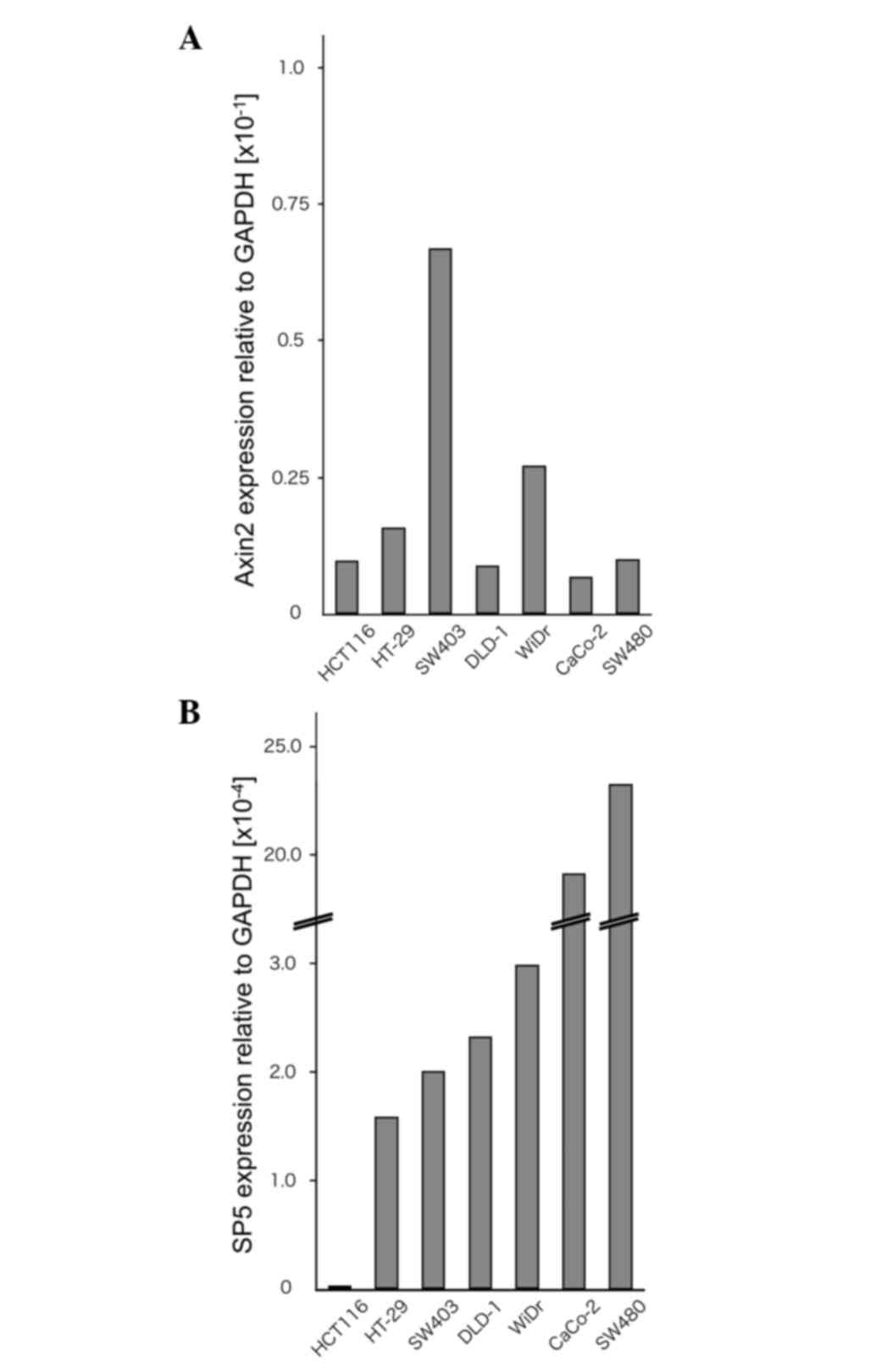
To examine the expression levels of Sp5 and the Wnt
target gene Axin2, RT-qPCR analyses were performed on the
colorectal cancer cell lines HCT116, HT-29, SW403, DLD-1, WiDr,
Caco-2 and SW480 (Fig. 1A and B).
HCT116 cells contain mutated β-catenin, whereas HT-29, SW403,
DLD-1, WiDr, Caco-2 and SW480 cells contain a truncated mutant of
the tumor suppressor adenomatous polyposis coli (22,23). In
the present study, Sp5 was found to be expressed at very low levels
in HCT116 cells compared with HT-29, SW403, DLD-1, WiDr, Caco-2 and
SW480 cells. By contrast, Axin2 expression was not downregulated in
HCT116 cells compared to the other cell lines.
mSp5 inhibited the growth of HCT116
cells
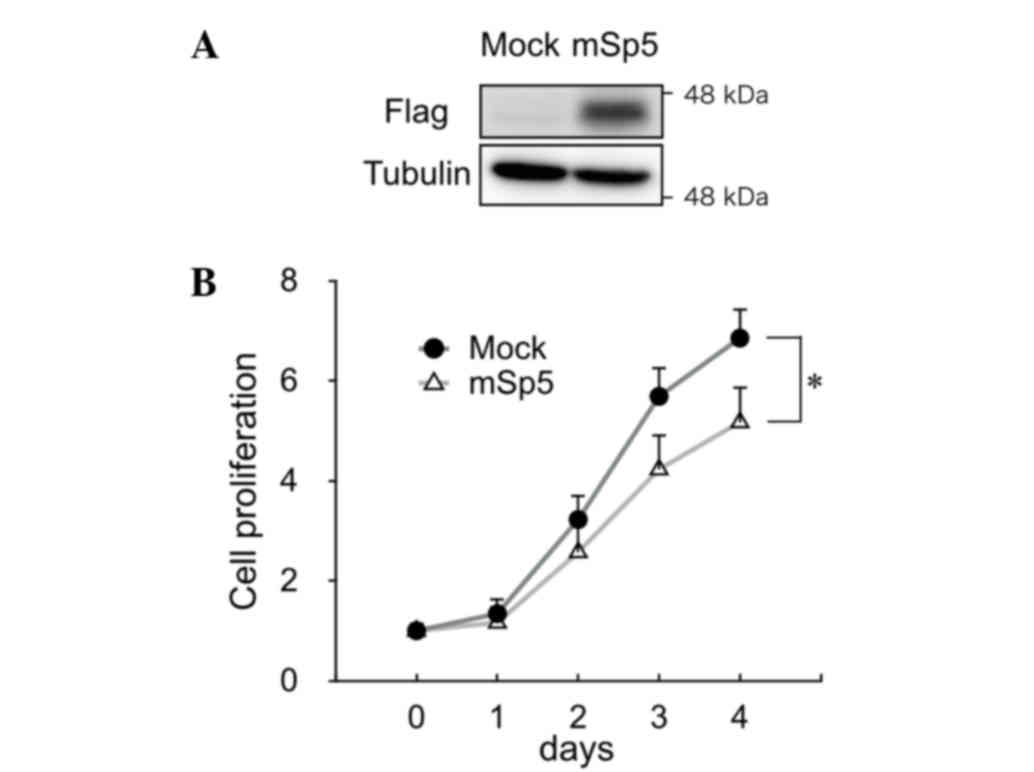
To study the effect of Sp5 on the proliferation of
colon cancer cells, mSp5 was overexpressed in HCT116 cells
(Fig. 2A). Overexpression of mSp5 was
found to result in a decrease in proliferation in these cells
(Fig. 2B).
mSp5 activates p27 transcription
It has been reported that overexpression of Sp5
suppresses p21 expression (11,14,17).
However, in the present study, RT-qPCR analysis revealed that
overexpression of mSp5 did not affect the expression levels of p21
in HCT116 cells (Fig. 3A). By
contrast, the overexpression of mSp5 resulted in the upregulation
of p27 (Fig. 3B). To clarify the
mechanisms underlying Sp5-mediated upregulation of p27, reporter
assays were performed using constructs in which various fragments
of the p27 promoter region were inserted upstream of the luciferase
gene (p27-P1, -P2, -P3 and -P4 in Fig.
3C). Overexpression of mSp5 in HEK293 cells enhanced the
activities of the p27-P2 and -P4, but not the p27-P1 and -P3
reporters (Fig. 3D). To examine
whether Sp5 transactivates p27 directly, ChIP assays were performed
on HCT116 cells transfected with FLAG-tagged mSp5 using an
anti-FLAG antibody. mSp5 binding to a DNA fragment (p27-TSS; shown
in Fig. 3C), which contains the
GC-box (Fig. 3E) was detected. The
promoter regions of GAPDH and p21 were used as negative and
positive controls, respectively. These results suggest that Sp5
directly upregulates the transcription of p27 by binding to the
GC-box in its promoter region.
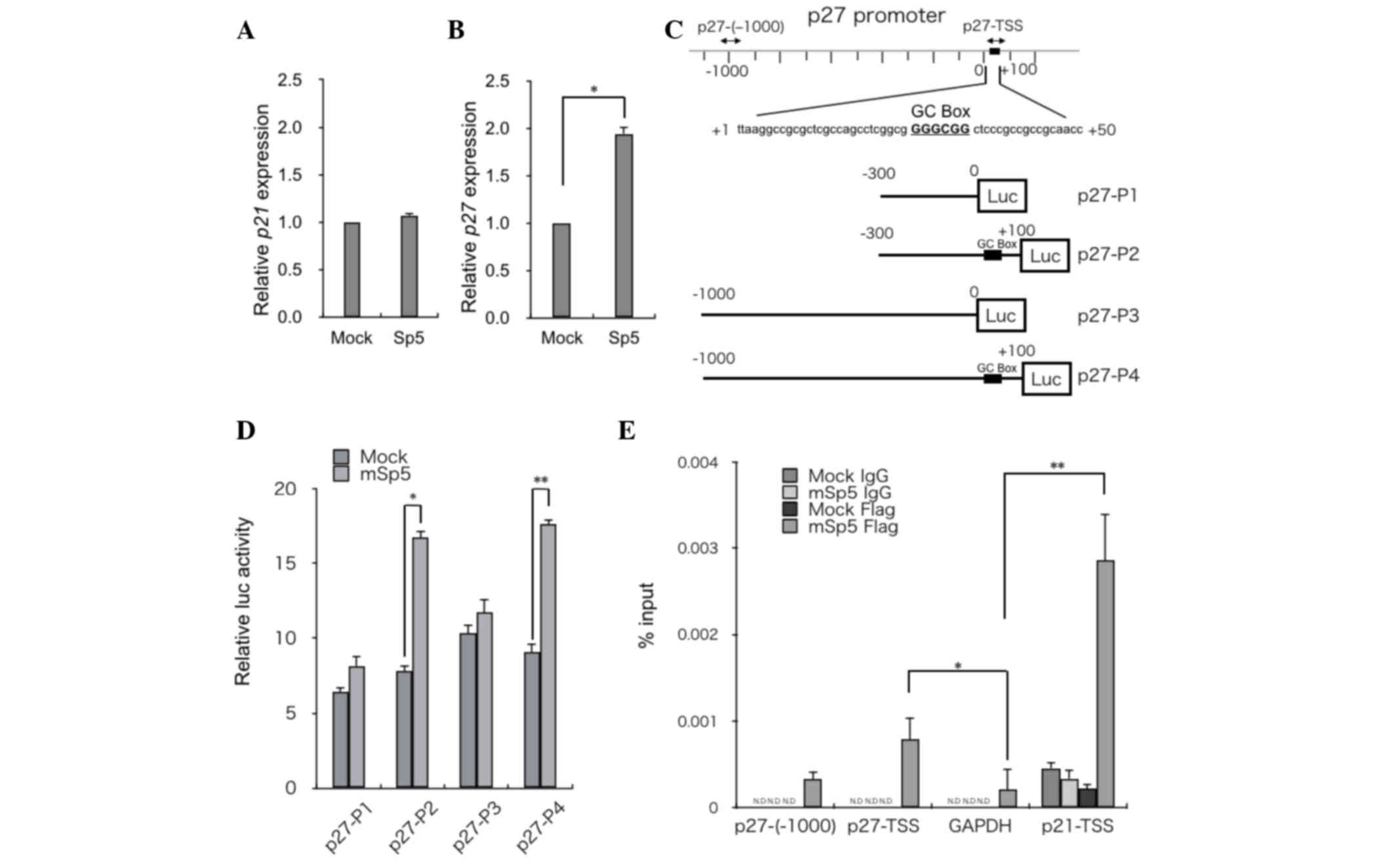 | Figure 3.mSp5 binds to the promoter region of
p27 and activates its transcription. Reverse
transcription-quantitative polymerase chain reaction analysis of
(A) p27 and (B) p21 in HCT116 cells transfected with mSp5. Results
are expressed as the means ± SD of three independent experiments
(*P=0.008, Student's t-test). (C) Schematic diagrams of reporter
constructs used for luciferase assays. Fragments of the p27
promoter were cloned upstream of the luciferase (Luc) gene: p27-P1
(from −300 to 0), p27-P2 (from −300 to +100), p27-P3 (from −1000 to
0), p27-P4 (from −1000 to +100). The potential Sp5 binding site
[GC-box (GGGCGG)] is indicated by black boxes. The positions of the
primers used for ChIP assays are indicated by arrows. (D) HEK293
cells were transfected with the indicated p27 reporter constructs
along with mSp5 and were subjected to luciferase assays. The
pRL-CMV vector was used as an internal control. Results are
expressed as the means ± SD of three independent experiments
(*P=0.014, **P=0.013, Student's t-test). (E) ChIP assays were
performed on HCT116 cells that had been transfected with mSp5 using
anti-Flag antibody or control IgG. The promoter regions of p27
[p27-(−1000) and p27-TSS] were amplified. The promoter regions of
GAPDH and p21-TSS were used as negative and positive controls,
respectively. Results are expressed as the means ± SD of three
independent experiments (*P<0.042, **P=0.036, Student's t-test).
mSp5, mouse specificity protein 5; SD, standard deviation; ChIP,
chromatin immunoprecipitation; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; N.D, not detected; TSS, transcription start
site. |
Knockdown of p27 partially restores
the growth of HCT116 cells overexpressing mSp5
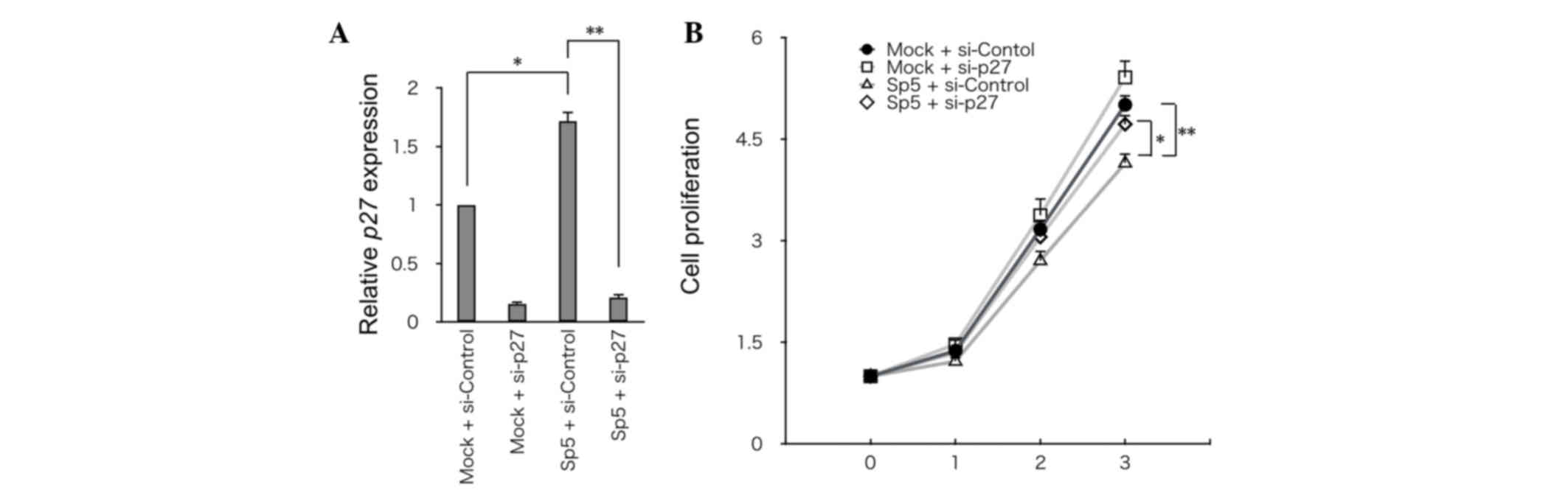
To address the significance of p27 in Sp5-mediated
suppression of cell growth, p27 was knocked down using siRNA in
HCT116 cells that had been transfected with mSp5 (Fig. 4A). Knockdown of p27 partially restored
the growth of HCT116 cells overexpressing mSp5 (Fig. 4B). Thus, Sp5-induced upregulation of
p27 may be required for inhibition of the growth of HCT116
cells.
Discussion
It has been shown that Sp5 is a direct target of Wnt
signaling and that its expression is commonly upregulated in
several colon cancer cell lines (24). The present study showed that the
expression of Sp5 is not upregulated in HCT116 cells, in which Wnt
signaling is constitutively activated. Although other studies have
reported that Sp5 represses the expression of p21 and promotes the
growth of MCF-7 cells (11,14,17), no
significant changes could be detected in the p21 expression in the
HCT116 cells transfected with Sp5 in the present study. This
discrepancy may be due to the differences in cell types and the
different expression levels of the Sp family of proteins.
Furthermore, the present results indicate that Sp5 has the
potential to inhibit cell proliferation by regulating the cell
cycle inhibitor p27 in HCT116 cells. We speculate that HCT116 cells
downregulate Sp5 in order to escape p27-mediated growth arrest. The
molecular mechanisms underlying the downregulation of Sp5 in HCT116
cells remain to be elucidated.
Acknowledgements
This study was supported by the Research Program of
Innovative Cell Biology by Innovative Technology (Integrated
Systems Analysis of Cellular Oncogenic Signaling Networks);
Grants-in-Aid for Scientific Research on Innovative Areas
(Integrative Research on Cancer Microenvironment Network); Project
for Development of Innovative Research on Cancer Therapeutics;
Takeda Science Foundation; and in part by the Global COE Program
(Integrative Life Science Based on the Study of Biosignaling
Mechanisms) and MEXT (Ministry of Education, Culture, Sports,
Science and Technology), Japan.
References
|
1
|
Logan CY and Nusse R: The Wnt signaling
pathway in development and disease. Annu Rev Cell Dev Biol.
20:781–810. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clevers H: Wnt/beta-catenin signaling in
development and disease. Cell. 127:469–480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eastman Q and Grosschedl R: Regulation of
LEF-1/TCF transcription factors by Wnt and other signals. Curr Opin
Cell Biol. 11:233–240. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beier F, Lee RJ, Taylor AC, Pestell RG and
LuValle P: Identification of the cyclin D1 gene as a target of
activating transcription factor 2 in chondrocytes. Proc Natl Acad
Sci USA. 96:1433–1438. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He TC, Sparks AB, Rago C, Hermeking H,
Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jho EH, Zhang T, Domon C, Joo CK, Freund
JN and Costantini F: Wnt/beta-catenin/Tcf signaling induces the
transcription of Axin2, a negative regulator of the signaling
pathway. Mol Cell Biol. 22:1172–1183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wodarz A and Nusse R: Mechanisms of Wnt
signaling in development. Annu Rev Cell Dev Biol. 14:59–88. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.PubMed/NCBI
|
|
10
|
Takahashi M, Nakamura Y, Obama K and
Furukawa Y: Identification of SP5 as a downstream gene of the
beta-catenin/Tcf pathway and its enhanced expression in human colon
cancer. Int J Oncol. 27:1483–1487. 2005.PubMed/NCBI
|
|
11
|
Fujimura N, Vacik T, Machon O, Vlcek C,
Scalabrin S, Speth M, Diep D, Krauss S and Kozmik Z: Wnt-mediated
down-regulation of Sp1 target genes by a transcriptional repressor
Sp5. J Biol Chem. 282:1225–1237. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wallmen B, Schrempp M and Hecht A:
Intrinsic properties of Tcf1 and Tcf4 splice variants determine
cell-type-specific Wnt/β-catenin target gene expression. Nucleic
Acids Res. 40:9455–9469. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fancy SP, Harrington EP, Baranzini SE,
Silbereis JC, Shiow LR, Yuen TJ, Huang EJ, Lomvardas S and Rowitch
DH: Parallel states of pathological Wnt signaling in neonatal brain
injury and colon cancer. Nat Neurosci. 17:506–512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Y, Guo Y, Ge X, Itoh H, Watanabe A,
Fujiwara T, Kodama T and Aburatani H: Elevated expression and
potential roles of human Sp5, a member of Sp transcription factor
family, in human cancers. Biochem Biophys Res Commun. 340:758–766.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kadonaga JT, Carner KR, Masiarz FR and
Tjian R: Isolation of cDNA encoding transcription factor Sp1 and
functional analysis of the DNA binding domain. Cell. 51:1079–1090.
1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kadonaga JT, Courey AJ, Ladika J and Tjian
R: Distinct regions of Sp1 modulate DNA binding and transcriptional
activation. Science. 242:1566–1570. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hoverter NP, Ting JH, Sundaresh S, Baldi P
and Waterman ML: A WNT/p21 circuit directed by the C-clamp, a
sequence-specific DNA binding domain in TCFs. Mol Cell Biol.
32:3648–3662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fernandez-Zapico ME, Lomberk GA, Tsuji S,
DeMars CJ, Bardsley MR, Lin YH, Almada LL, Han JJ, Mukhopadhyay D,
Ordog T, et al: A functional family-wide screening of SP/KLF
proteins identifies a subset of suppressors of KRAS-mediated cell
growth. Biochem J. 435:529–537. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harrison SM, Houzelstein D, Dunwoodie SL
and Beddington RS: Sp5, a new member of the Sp1 family, is
dynamically expressed during development and genetically interacts
with Brachyury. Dev Biol. 227:358–372. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsuura K, Jigami T, Taniue K, Morishita
Y, Adachi S, Senda T, Nonaka A, Aburatani H, Nakamura T and Akiyama
T: Identification of a link between Wnt/β-catenin signalling and
the cell fusion pathway. Nat Commun. 2:5482011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sparks AB, Morin PJ, Vogelstein B and
Kinzler KW: Mutational analysis of the APC/beta-catenin/Tcf pathway
in colorectal cancer. Cancer Res. 58:1130–1134. 1998.PubMed/NCBI
|
|
23
|
Ilyas M, Tomlinson IP, Rowan A, Pignatelli
M and Bodmer WF: Beta-catenin mutations in cell lines established
from human colorectal cancers. Proc Natl Acad Sci USA.
94:10330–10334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herbst A, Jurinovic V, Krebs S, Thieme SE,
Blum H, Göke B and Kolligs FT: Comprehensive analysis of β-catenin
target genes in colorectal carcinoma cell lines with deregulated
Wnt/β-catenin signaling. BMC Genomics. 15:742014. View Article : Google Scholar : PubMed/NCBI
|