Introduction
Human glioma is one of the most common and lethal
primary central nervous system (CNS) tumors. The broad category of
glioma represents ~27% of all CNS tumors according to the Central
Brain Tumor Registry of the United States (1) and 80% of malignant tumors (1). Gliomas vary widely in histology from
benign and potentially surgically curable grade I tumors (pilocytic
astrocytoma) to locally aggressive grade IV tumors (particularly
glioblastoma, GBM) with an increased risk of recurrence or
progression (2). The morbidity and
mortality rates for patients with glioma remain high, despite
undergoing aggressive multimodality therapy, which consists of
maximal surgical resection, adjuvant radiation and chemotherapy.
Despite optimal treatment, the median survival time is only 12–15
months for patients with GBM and 2–5 years for patients with
anaplastic gliomas (3). There are
several risk factors for which an association with brain tumors has
been established, including ionizing radiation, genetic
susceptibility, and allergic aspects (2). However, the molecular pathogenesis
underlying glioma tumorigenesis remains elusive. As the current
understanding of the molecular pathogenesis of these tumors
improves, it may be possible to select the most appropriate
therapies.
Centrosomal protein of 55 kDa (CEP55), also known as
FLJ10540, C10, f3 or URCC6, is an essential member of CEP family
proteins, which serve a vital function in mitotic exit and
cytokinesis (4). CEP55 localizes to
the mitotic spindle during prometaphase and metaphase, and
translocates to the spindle mid-zone and the mid-body during
anaphase and cytokinesis (5). During
cytokinesis, CEP55 may cooperate with members of endosomal sorting
complex required for transport (ESCRT) machinery, to constrict an
intracellular bridge to allow abscission (6–8). Apart
from cytokinesis, CEP55 is also associated with embryonic growth
(9). Additionally, CEP55 has been
associated with cancer progression by activating the
phosphoinositide 3-kinase (PI3K)/Akt signaling pathway (10), and is identified as a prognostic
marker for multiple types of cancer (10). A high expression of CEP55 expression
occurs in a diverse range of solid tumors, including prostate
cancer (11), bladder cancer
(12) and epithelial ovarian
carcinoma (13). Although the roles
of CEP55 in different types of cancer and the regulatory mechanisms
involved have been reported, the function and mechanism of CEP55 in
glioma tumorigenesis has not been fully elucidated.
Materials and methods
Analysis of the Oncomine database
Oncomine (www.oncomine.org) is a cancer microarray database and
web-based data-mining platform aimed at facilitating identification
from genome-wide expression analyses. To evaluate the mRNA
expression of CEP55 in glioma, data was extracted from the Oncomine
cancer microarray database. According to the standard procedures as
previously described (14), the
differential gene expression of CEP55 in normal brain tissues and
glioma tissues was obtained. French Brain Statistics (15), The Cancer Genome Atlas (TCGA;
www.cancergenome.nih.gov), Murat Brain
Statistics (16) and Sun Brain
Statistics (17) were used to compare
CEP55 mRNA expression levels between cancer and normal tissues.
Cell culture
The human glioma cell lines U251 and U343, normal
human astrocyte cells (HA) and the lentivirus vector package cell
line 293T used in the present study were obtained from Research
Center for Neurobiology of Xuzhou Medical University (Xuzhou,
China). All cells were cultured in Dulbecco's modified Eagle's
medium/high glucose (Hyclone; GE Healthcare, Chicago, IL, USA)
supplemented with 10% fetal bovine serum (FBS; Hyclone) and 1%
penicillin streptomycin solution (Vicmed, Xuzhou, China). The cells
were maintained at 37°C in a humidified incubator with 5%
CO2.
Establishment of stable U251 cell
lines which overexpress or downregulate CEP55
Three CEP55-targeted RNA interference (RNAi)
sequences were synthesized and inserted into a GV248 plasmid
[element sequence, hU6-multiple cloning site
(MCS)-ubiquitin-enhanced green fluorescent protein (EGFP)-internal
ribosome entry site (IRES)-puromycin] to construct the recombinant
plasmids. The sequences were as follows: RNAi-1,
5′-AGCGGGAAGTCTATGTAAA-3′; RNAi-2, 5′-AGGCATGTACTTTAGACTT-3′ and
RNAi-3, 5′-AAGCCTAGTAACTCCAAAT-3′. The GV358 lentivirus vector
(element sequence, Ubi-MCS-3FLAG-SV40-EGFP-IRES-puromycin;
Genechem, Shanghai, China) containing scrambled RNA (sequence,
5′-TTCTCCGAACGTGTCACGT-3′) was used as a negative control. The four
plasmids were co-transfected into 293T cells with packaging
plasmids, pHelper 1.0 and pHelper 2.0 using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's protocol. At ~48
h post-transfection, lentivirus was harvested by centrifugation at
4°C at 2,500 × g for 10 min, to dispose of cell debris and finally
collected by ultracentrifugation at 4°C at 50,000 × g for 2 h. The
transfected cells were selected by puromycin at 1.5 mg/ml for 2
weeks.
The full-length human CEP55 cDNA was cloned into the
lentiviral vector GV358 to construct the recombinant plasmid. A
blank GV358 lentiviral vector was used as negative control. Then,
the constructs were co-transfected into 293T cells with assistant
packaging plasmid using Lipofectamine® 2000, according
to the manufacturer's protocol. At 48 h post-transfection,
lentivirus was collected and infected into U251 cells. Transfected
cells were selected by puromycin for 2 weeks, and the cell line
with a stable overexpression of CEP55 was obtained. In all
experiments, untransfected cells were used as controls.
Western blot analysis
U251 and U343 cells were washed three times with
ice-cold PBS, and trypsinized and collected. Following lysis on ice
for 30 min using radioimmunoprecipitation assay lysis buffer (Merck
KGaA, Darmstadt, Germany) containing protease inhibitors and
phenylmethanesulfonyl fluoride (PMSF), the lysate was centrifuged
at 12,000 × g at 4°C for 20 min. Phosphatase inhibitors were added
when necessary. The concentration of the proteins was measured
using a BCA protein assay kit (Beyotime Institute of Biotechnology,
Haimen, China). Protein samples were loaded and separated by 12%
SDS-PAGE, and transferred onto nitrocellulose membranes. The
membranes were blocked in 5% non-fat milk suspended in washing
buffer (100 mmol/l NaCl, 10 mmol/l Tris/HCl and 0.1% Tween-20) for
1 h at room temperature. The membranes were incubated overnight at
4°C with primary antibodies against CEP55 (cat. no. 23891-1-AP;
dilution 1:500; Proteintech Group, Inc., Chicago, IL, USA), cyclin
B1 (cat. no. ab181593; dilution 1:11,000), cyclin A1 (cat. no.
ab133183; dilution 1:11,000), cyclin D1 (cat. no. ab134175;
dilution 1:11,000), p-Akt (cat. no. ab38449; dilution 1:11,000),
Akt (cat. no. ab182729; dilution 1:11,000), p21 (cat. no. ab109199;
dilution 1:11,000). GAPDH (cat. no. ab8245; dilution 1:1,000; all
Abcam, Cambridge, UK) was used as a loading control. The following
day, the membranes were probed using IRDye 800CW-conjugated goat
anti-rabbit secondary antibody (cat. no. 926-32211; dilution
1:5,000; LI-COR Biosciences, Lincoln, NE, USA) at room temperature
for 1 h. Protein bands were detected using an Odyssey infrared
imaging system (LI-COR Biosciences). The intensity of the bands was
quantified using ImageJ software (version 1.48; National Institutes
of Health, Bethesda, MD, USA), and the level of protein expression
was normalized to the expression of GAPDH.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from cells using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) in accordance
with the manufacturer's protocol. RNA was reverse transcribed into
cDNA using Vic qRT Super kit (Vicmed), according to the
manufacturer's protocol. qPCR was performed using SYBR qPCR mix
(Roche Diagnostics, Basel, Switzerland), run on a Light Cycler 480
Instrument II (Roche Diagnostics). The reactions were incubated in
a 96-well optical plate at 95°C for 10 min for 40 cycles of 95°C
for 10 sec, 60°C for 20 sec and 72°C for 30 sec. GAPDH was used as
an internal control, and the relative expression levels of the gene
of interest were calculated using the 2−ΔΔCq method
(18). The sequences of the primers
are as follows: CEP55 forward,
5′-GAGGATCCCCGGGTACCGGTCGCCACCATGTCTTCCAGAAGTACCAAAG-3′ and
reverse, 5′-TCCTTGTAGTCCATACCCTTTGAACAGTATTCCACATGGAC-3′; GAPDH
forward, 5′-CTCTCTGCTCCTCCTGTTCGAC-3′ and reverse,
5′-TGAGCGATGTGGCTCGGCT-3′. All RT-qPCRs were performed in
triplicate.
Cell Counting kit-8 (CCK-8) assay
Cell viability was determined by CCK-8 cell
viability assay (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). The U251 cells were seeded into 96-well plates at a density
of 3×103 cells/well. CCK-8 solution was added to each
well at 0, 24, 48 and 72 h. Following incubation for 1.5 h at 37°C,
the optical density was measured at a wavelength of 450 nm on a
microplate reader (BioTek China, Beijing, China). All experiments
were performed three times.
Cell cycle distribution analysis
For analysis of the cell cycle, Cell Cycle Detection
kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China) was used
according to manufacturer's protocol. The cells were trypsinized
and collected. After washing twice with ice-cold PBS, the U251
cells were fixed with 70% ethanol at 4°C overnight. Following
centrifugation at 1,000 × g for 5 min, at 4°C, the supernatant was
discarded. Subsequently, the cells were resuspended in PBS with 100
µl/ml RNase A in a 37°C water bath for 30 min. The cells were then
stained using propidium iodide (400 µl/ml) and incubated for 30 min
at room temperature in the dark. Finally, the cells were analyzed
using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA,
USA), and the data were analyzed using Multi Cycle software
(version AV; Phoenix Flow Systems, San Diego, CA, USA).
Cell apoptosis analysis
Cell apoptosis ratio was measured using an Annexin
V-allophycocyanin (APC)/7-aminoactinomycin D (AAD) Apoptosis
Detection kit (Nanjing KeyGen Biotech. Co., Ltd.), according to the
manufacturer's protocol. In brief, a total of 1×106 U251
cells were harvested, washed with PBS twice and resuspended in 500
µl binding buffer. Cells were subsequently dual stained with 5 µl
Annexin V-APC and 5 µl 7-AAD at room temperature in the dark.
Following 20 min incubation at room temperature, the cells were
immediately analyzed using a FACSCalibur flow cytometer (BD
Biosciences), and cell apoptosis ratio was determined using FlowJo
software (version X; FlowJo LLC, Ashland, OR, USA).
Statistical analysis
Quantitative data are presented as the mean ±
standard deviation of three independent experiments. SPSS software
(version 16.0; SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 5.0
software (GraphPad Software, Inc., La Jolla, CA, USA) were used for
all statistical analysis. Independent sample t-tests and paired
t-tests were used within groups for repeated measures. Multiple
comparisons between groups were performed using one-way analysis of
variance followed by Student-Newman-Keuls test for statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
CEP55 expression is upregulated in
glioma tissues and cell lines
To determine the expression pattern of CEP55 in
glioma, four independent microarray datasets from Oncomine database
were analyzed. When compared with normal brain tissues of patients
from the study of French Brain Statistics (15), The Cancer Genome Atlas (TCGA,
www.cancergenome.nih.gov), Murat Brain
Statistics (16) and Sun Brain
Statistics (17), CEP55 expression
was elevated in glioma tissues (data not shown). To validate this
observation, RT-qPCR and western blotting were further performed to
assess CEP55 expression levels in glioma cell lines, U343 and U251.
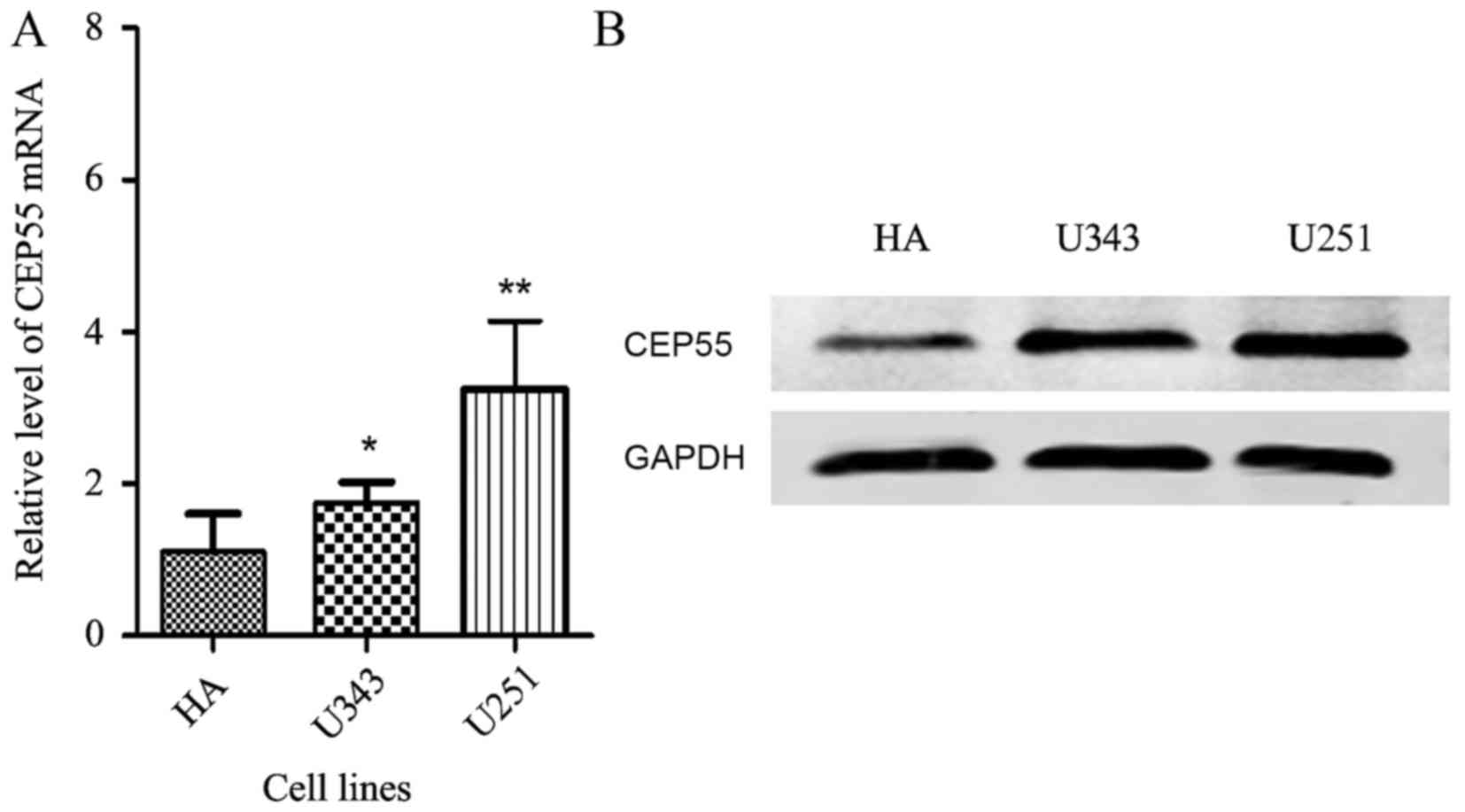
Compared with the normal human astrocytes (HA), the mRNA (Fig. 1A) and protein levels of CEP55 were
increased in these two glioma cell lines (Fig. 1B).
Successful stable RNAi knockdown and
overexpression of CEP55 in U251 cell lines
To investigate the effect of CEP55 on the behavior
of glioma cells, U251 cell lines with stable CEP55 knockdown and
overexpression were established using lentivirus-mediated
technology. Negative control and three CEP55 targeting RNA
interference 1, 2 and 3 (RNAi-1, 2 and 3) were infected into U251
cells, and the effects of CEP55 knockdown in these groups were
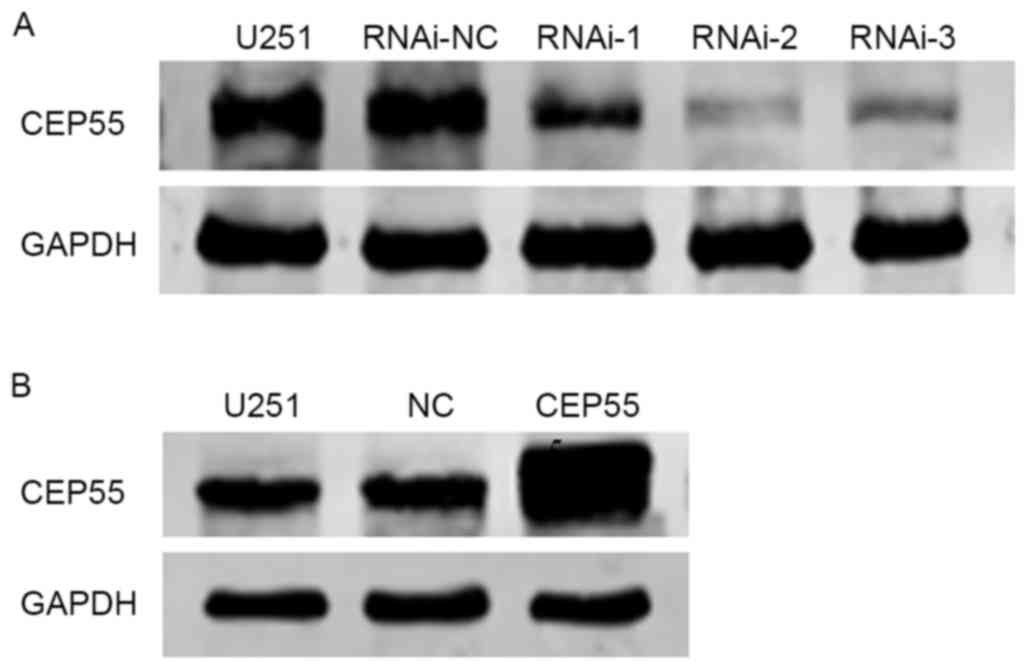
evaluated by western blot analysis. The expression of CEP55 was
decreased was most marked in response to RNAi-2 treatment in U251
cells compared with that in the negative control (untreated U251
cells; Fig. 2A), therefore RNAi-2 was
selected for subsequent experiments.
In addition, a CEP55-overexpressing lentivirus
vector was established and stably transfected into U251 cells.
Western blot analysis was used to evaluate the transfection
efficacy (Fig. 2B). As indicated in
Fig. 2B, the levels of CEP55 protein
expression in CEP55-overexpressing U251 cells were increased
compared with the empty vector transfected cells or untransfected
cells.
CEP55 is associated with viability of
U251 cells
To determine the function of CEP55 in regulating the
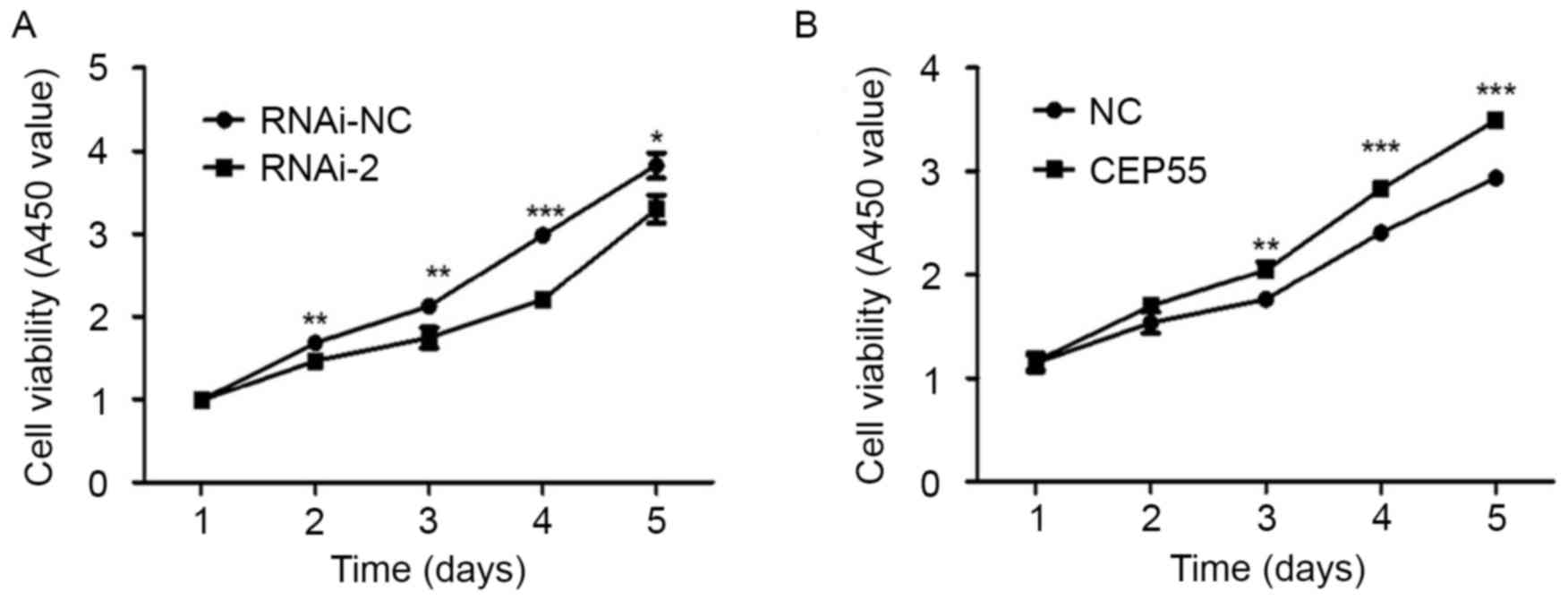
viability of U251 cells, a CCK-8 assay was used. The results
revealed that downregulating the expression of CEP55 significantly
inhibited the viability of U251 cells (Fig. 3A). Additionally, overexpression of
CEP55 promoted the viability of U251 cells (Fig. 3B). The results suggest that CEP55 may
serve an important function in regulating the viability of U251
cells.
Knockdown of CEP55 induces cell cycle
arrest and apoptosis
The regulation of cell viability may affect cell
cycle progression. To further investigate the mechanism of CEP55 in
proliferative ability of glioma cells, the cell cycle distribution
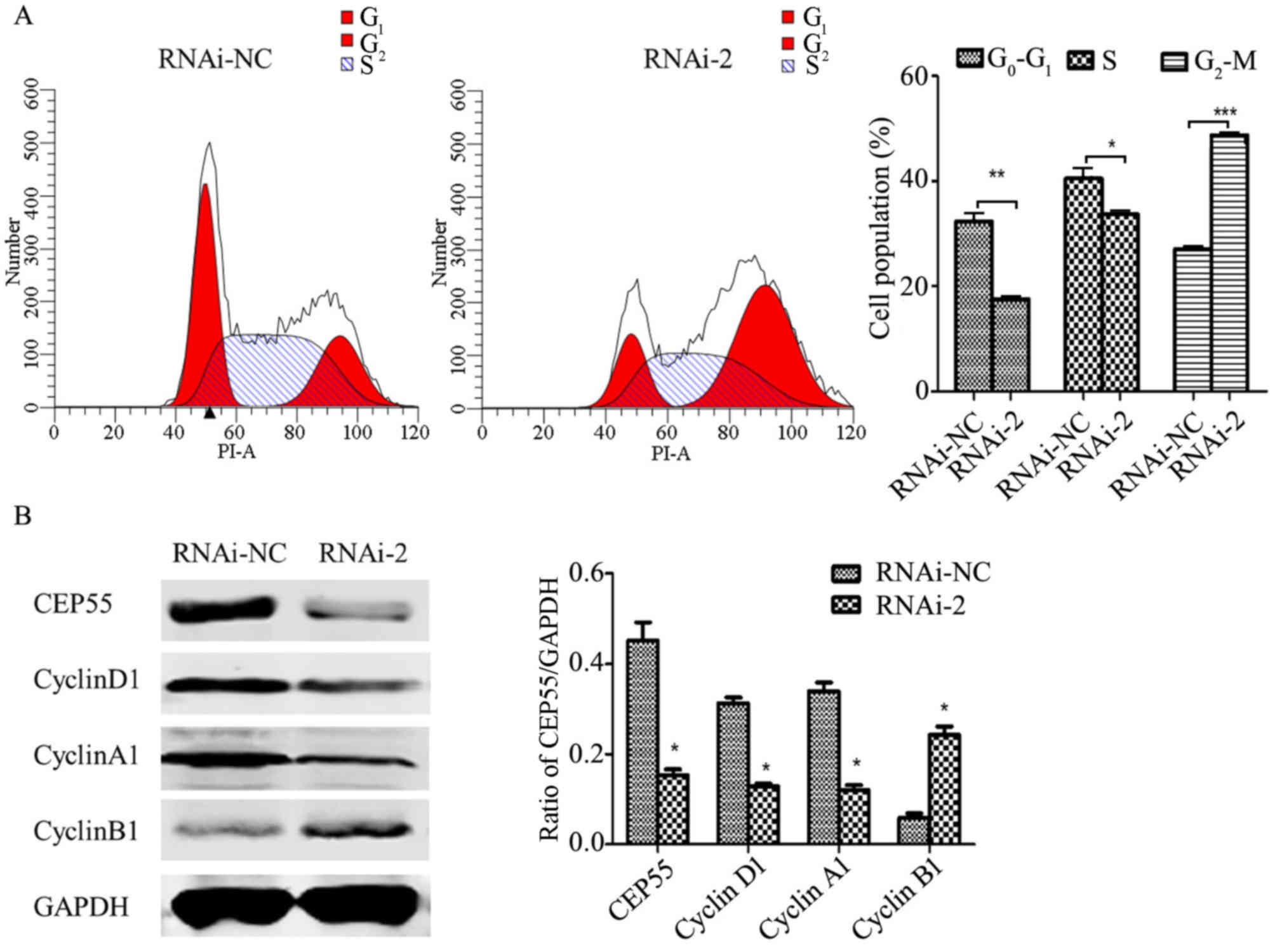
was detected by flow cytometric analysis. As presented in Fig. 4A, knockdown of CEP55 significantly
decreased the number of G0/G1 and S phase
cells, and increased the number of G2/M phase cells,
which indicates that an increased number of cells were arrested at
G2/M phase. The cell cycle distribution is associated
with the expression of cyclin-associated genes (19). Therefore, the level of cyclins was
detected by western blot analysis (Fig.
4B). The level of cyclin B1, a cyclin that is important for
G2 to M transition, was significantly increased in
CEP55-knockdown U251 cells compared with the RNAi-negative (NC)
group (Fig. 4B). However, the levels
of cyclin A1 and cyclin D1 were significantly reduced in
CEP55-knockdown U251 cells compared with the RNAi-NC group
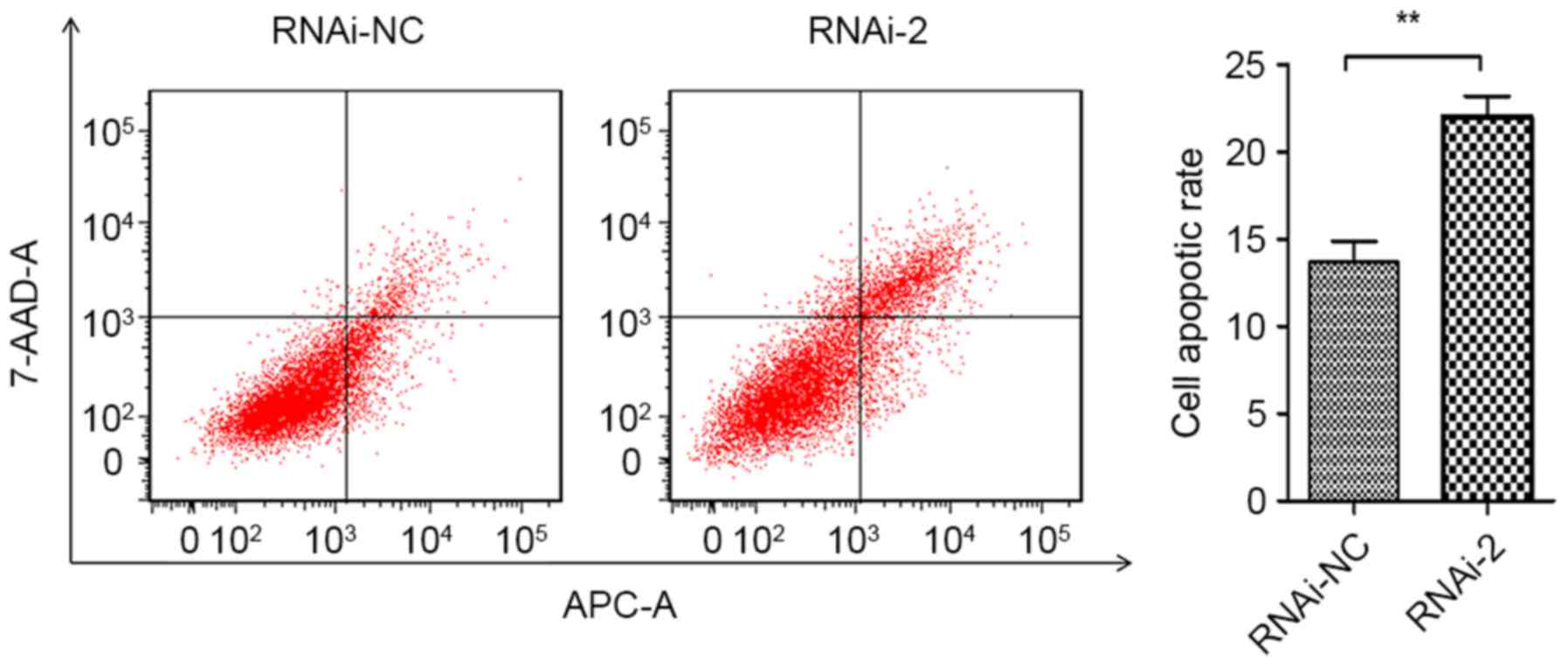
(Fig. 4B). Furthermore, Annexin
APC/7-AAD staining by flow cytometry was performed to evaluate the
function of CEP55 in apoptosis of U251 cells. The results
demonstrated that CEP55 knockdown significantly increased cell
apoptotic rates compared with the RNAi-NC group (13.67 to 22.01%;
Fig. 5). Taken together, these
results revealed that CEP55 may have a critical function in cell
cycle progression and apoptosis of U251 cells.
CEP55 regulate cell viability via the
PI3K/Akt/p21 signaling pathway
The cell cycle is regulated by a number of signaling
molecules and cyclins. CEP55 has been implicated to regulate the
PI3K/Akt signaling pathway via an interaction with the catalytic
subunit of PI3K (9,20). Previously, several molecular targets
of CEP55, including p21, cyclin-dependent kinase (CDK)4 and B-cell
lymphoma (Bcl)-2, associated with the cell cycle have been assessed
(21). An inhibitor of CDK, p21, was
a key cell cycle regulator and also a downstream target of the
PI3K/Akt signaling pathway (22). In
light of previous reports, PI3K/Akt/p21 may act as a downstream
target of CEP55. To investigate this hypothesis, a number of
proteins associated with the PI3K/Akt/p21 signaling pathway were
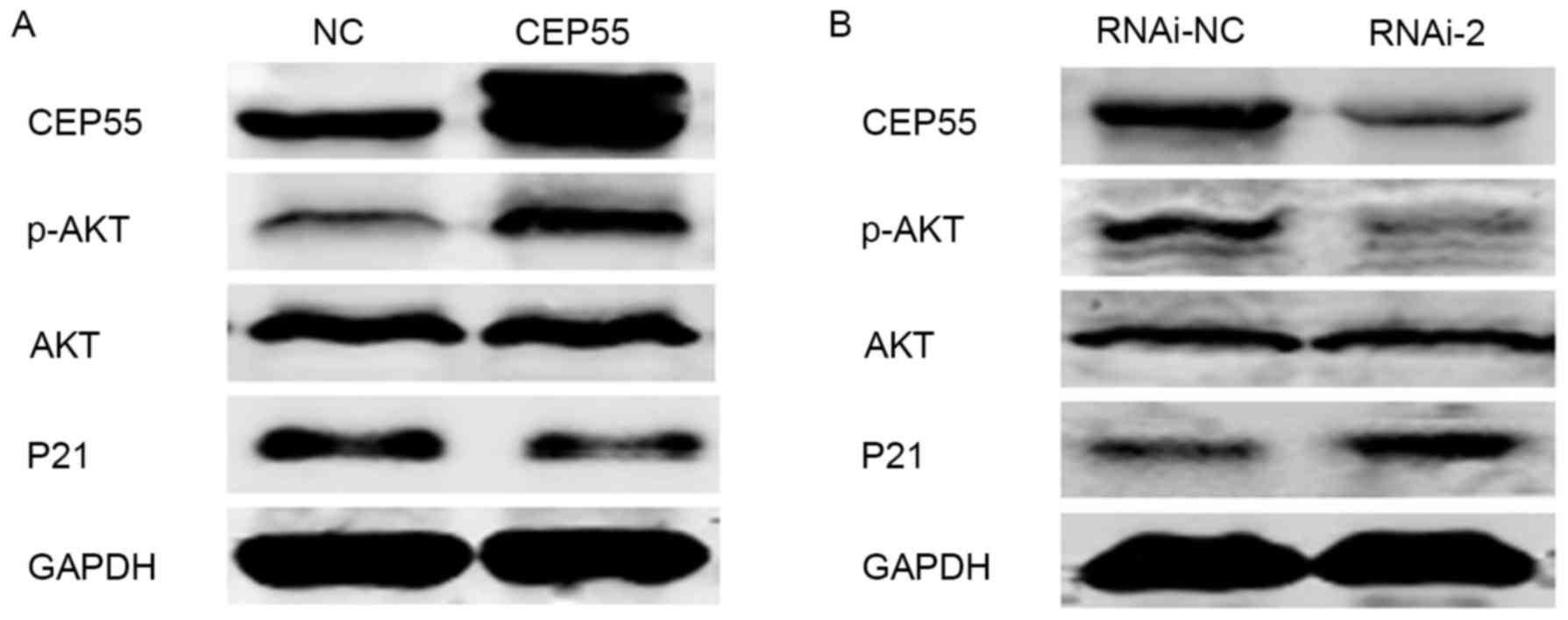
examined by western blot analysis. The results indicated that when
CEP55 was knocked down, the level of phospho-Akt was decreased, and
the level of p21 was increased (Fig.
6). Conversely, the level of phospho-Akt was increased, and the
level of p21 was decreased in response to overexpression of CEP55
in the U251 cell line (Fig. 6).
Collectively, the results of the present study suggested that CEP55
acts as a regulator of PI3K/Akt/p21 signaling, as overexpression of
CEP55 inhibited the activation of the PI3K/Akt signaling pathway,
further inhibiting cell viability.
Discussion
Gliomas are the second most common brain tumors in
adults, which account for ~24% of all adult brain tumors (2). Despite available therapies, including
radiotherapy, chemotherapy and adjuvant therapies, the relative
survival time for glioblastoma is short with only 5.1% of patients
who survive five years post diagnosis (1). Therefore, there is an urgent requirement
to discover more efficacious therapy methods and novel molecular
biomarkers for glioma tumorigenesis. It is well known that the
PI3K/Akt signaling pathway is one of the major pro-survival
pathways. CEP55 has been reported to be a regulator of the PI3K/Akt
signaling pathway (22–26), which may indicate that CEP55 may
participate in cell viability and tumor development, by regulating
this specific pathway. To date, the implication of CEP55 in cancer
cell viability has been frequently demonstrated in various types of
human cancer (23,25,27,28).
However, the function of CEP55 in glioma tumorigenesis remains
unclear.
In the present study, the expression of CEP55 in
glioma was detected. According to the results from the Oncomine
database, CEP55 expression was markedly increased in glioma
compared with normal brain tissues. In addition, the results of the
present study showed that CEP55 expression was also increased in
glioma cell lines compared with normal human astrocytes. All these
results indicated that CEP55 may be an interesting candidate for
its involvement in glioma tumorigenesis. In order to investigate
this hypothesis, the effect of overexpression and knockdown of
CEP55 on the cell viability of glioma U251 cells was evaluated. In
the present study, it was demonstrated that the knockdown of CEP55
expression led to decreased cell viability, apoptosis induction and
cell cycle arrest as well as the inactivation of the PI3K/Akt/p21
signaling pathway. By contrast, the overexpression of CEP55
promoted cell viability and the activation of the PI3K/Akt/p21
signaling pathway in the U251 glioma cell line. Therefore, it was
hypothesized that CEP55 is likely to regulate cell viability via
the PI3K/Akt/p21 signaling pathway in the glioma cell line.
The deregulation of cell viability is a key
characteristic of cancer cells, which is associated with cell cycle
regulation (29,30). CEP55 exerts a pivotal role in cell
cycle progression (31). It was
indicated in the present study that an increased number of cells
accumulated in G2/M phase following CEP55 knockdown.
This result was consistent with previous research on hepatocellular
and gastric carcinoma (25). In
addition, cyclin D1 (32), cyclin A
(33) and cyclin B1 (34) are the major cyclins that control the
G0/G1, S and G2/M phases of the
cell cycle, respectively. In the present study, it was demonstrated
that the knockdown of CEP55 expression promoted cyclin B1
expression and inhibited the expression of cyclin D1 and cyclin A1,
which further supported the results indicating G2/M
phase arrest. However, some other studies have data which differed
from the current results, where the knockdown of CEP55 induced
G0-G1 phase arrest in lung cancer cells
(21) and breast cancer (27). These differences in the results may be
due to the different mechanisms of cancer occurrence. Furthermore,
the potential mechanism underlying CEP55-associated tumor
phenotypes has not been elicited, and this maybe a key area for
future study.
The PI3K/Akt signaling pathway is tightly controlled
via a multistep process (20).
Activated PI3K/Akt signaling mediates numerous cellular functions,
including angiogenesis, metabolism, growth, cell viability,
survival, protein synthesis, transcription and apoptosis (20). Akt has diverse roles in cell survival,
cell cycle, angiogenesis, protein synthesis and metabolism
(35). Increased PI3K activation
results in an increased pool of phosphatidylinositol
3,4,5-trisphosphate (PIP3) and a subsequent increase in
Akt activation as marked by Ser473 phosphorylation
(10). Whereas mammalian target of
rapamycin (mTOR) and p21 are substrates for Akt, those two proteins
have been identified to exhibit opposite functions. The present
study identified an increase in those proteins in the U251 cell
line consistent with previous studies (10,32–35). CEP55
is confirmed to be overexpressed in a wide range of solid tumors,
including lung adenocarcinoma (23),
hepatocellular carcinoma (26),
glioma (24) and gastric carcinoma
(25), where the overexpression of
CEP55 promotes cell metastasis, invasion and cell viability via
upregulation of the PI3K/Akt signaling pathway. CEP55 directly
interacts with the PI3K catalytic subunit, PIK3CA (also known as
p110), which promotes its stability and activation (28). Consistent with previously reported
studies, the results of the present study demonstrated that the
overexpression of CEP55 resulted in the upregulation of the
PI3K/Akt signaling pathway, leading to dysregulated cell cycle via
inhibition of p21. However, Wang et al (24) implicated that CEP55 regulates glucose,
metabolism, cell viability and apoptosis of glioma cells via the
Akt/mTOR signaling pathway. Taken together, all these studies
demonstrate that CEP55 may promote tumor cell viability through
activation of the PI3K/Akt/p21 signaling pathway in glioma. It is
premature to draw any conclusions from the present studies with
CEP55, as several important questions remain unanswered, including
the underlying molecular signaling pathways of CEP55 in glioma.
Additional in vivo studies are required to confirm the
conclusions of the present study.
In conclusion, the results of the present study
suggested that CEP55 has important roles in regulating various
cellular processes, including cell viability, cell cycle and
apoptosis, by mediating PI3K/Akt/p21 signaling in glioma cell
lines. Combined with previous studies, the present study indicates
that CEP55 may be a potential therapeutic target for glioma.
Additional studies investigating the regulation and function of
CEP55 during cancer development and reoccurrence are required to
design therapeutic strategies for various human malignancies with
CEP55 overexpression.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant. no. 81402073),
Natural Science Foundation of Jiangsu Province (grant. no.
BK20130218), the Program of the China Postdoctoral Science
Foundation (grant. no. 2014M551663), Jiangsu Province Universities
(grant no. 17KJB310016) and the Foundation of Jiangsu Province Six
Talents Peak (grant. no. JY-061).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FL and DJ contributed equally to the present study
by designing and conducting experiments, analyzing data and writing
the paper. FL, DJ and CXT conducted experiments and collected data.
DSG conceived of the project and experiments and analyzed data. All
authors critically revised the manuscript and provided final
approval.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GBM
|
glioblastoma multiforme
|
|
HA
|
astrocyte cell
|
|
FBS
|
fetal bovine serum
|
|
PMSF
|
phenylmethanesulfonyl fluoride
|
|
PI
|
propidium iodide
|
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the united states in 2008–2012. Neuro Oncol. 17
Suppl 4:iv1–iv62. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
McNeill KA: Epidemiology of brain tumors.
Neurol Clin. 34:981–998. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fabbro M, Zhou BB, Takahashi M, Sarcevic
B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y and
Khanna KK: Cdk1/Erk2- and Plk1-dependent phosphorylation of a
centrosome protein, Cep55, is required for its recruitment to
midbody and cytokinesis. Dev Cell. 9:477–488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao WM, Seki A and Fang G: Cep55, a
microtubule-bundling protein, associates with centralspindlin to
control the midbody integrity and cell abscission during
cytokinesis. Mol Biol Cell. 17:3881–3896. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee HH, Elia N, Ghirlando R,
Lippincott-Schwartz J and Hurley JH: Midbody targeting of the ESCRT
machinery by a noncanonical coiled coil in CEP55. Science.
322:576–580. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carlton JG, Agromayor M and Martin-Serrano
J: Differential requirements for Alix and ESCRT-III in cytokinesis
and HIV-1 release. Proc Natl Acad Sci USA. 105:pp. 10541–10546.
2008; View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carlton JG and Martin-Serrano J: Parallels
between cytokinesis and retroviral budding: A role for the ESCRT
machinery. Science. 316:1908–1912. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jeffery J, Neyt C, Moore W, Paterson S,
Bower NI, Chenevix-Trench G, Verkade H, Hogan BM and Khanna KK:
Cep55 regulates embryonic growth and development by promoting Akt
stability in zebrafish. FASEB J. 29:1999–2009. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jeffery J, Sinha D, Srihari S, Kalimutho M
and Khanna KK: Beyond cytokinesis: The emerging roles of CEP55 in
tumorigenesis. Oncogene. 35:683–690. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shiraishi T, Terada N, Zeng Y, Suyama T,
Luo J, Trock B, Kulkarni P and Getzenberg RH: Cancer/Testis
Antigens as potential predictors of biochemical recurrence of
prostate cancer following radical prostatectomy. J Transl Med.
9:1532011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh PK, Srivastava AK, Rath SK, Dalela
D, Goel MM and Bhatt ML: Expression and clinical significance of
Centrosomal protein 55 (CEP55) in human urinary bladder
transitional cell carcinoma. Immunobiology. 220:103–108. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang W, Niu C, He W, Hou T, Sun X, Xu L
and Zhang Y: Upregulation of centrosomal protein 55 is associated
with unfavorable prognosis and tumor invasion in epithelial ovarian
carcinoma. Tumour Biol. 37:6239–6254. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rhodes DR, Kalyana-Sundaram S, Mahavisno
V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ,
Kincead-Beal C, Kulkarni P, et al: Oncomine 3.0: Genes, pathways,
and networks in a collection of 18,000 cancer gene expression
profiles. Neoplasia. 9:166–180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
French PJ, Swagemakers SM, Nagel JH,
Kouwenhoven MC, Brouwer E, van der Spek P, Luider TM, Kros JM, van
den Bent MJ and Sillevis Smitt PA: Gene expression profiles
associated with treatment response in oligodendrogliomas. Cancer
Res. 65:11335–11344. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murat A, Migliavacca E, Gorlia T, Lambiv
WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven
MC, et al: Stem cell-related ‘self-renewal’ signature and high
epidermal growth factor receptor expression associated with
resistance to concomitant chemoradiotherapy in glioblastoma. J Clin
Oncol. 26:3015–3024. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun L, Hui AM, Su Q, Vortmeyer A,
Kotliarov Y, Pastorino S, Passaniti A, Menon J, Walling J, Bailey
R, et al: Neuronal and glioma-derived stem cell factor induces
angiogenesis within the brain. Cancer Cell. 9:287–300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Casimiro MC, Crosariol M, Loro E, Li Z and
Pestell RG: Cyclins and cell cycle control in cancer and disease.
Genes Cancer. 3:649–657. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hemmings BA and Restuccia DF: PI3K-PKB/Akt
pathway. Cold Spring Harb Perspect Biol. 4:a0111892012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu L, Mei Q, Zhao J, Dai Y and Fu Q:
Suppression of CEP55 reduces cell viability and induces apoptosis
in human lung cancer. Oncol Rep. 36:1939–1945. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa
JR and Walsh K: Cell cycle withdrawal promotes myogenic induction
of Akt, a positive modulator of myocyte survival. Mol Cell Biol.
19:5073–5082. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen CH, Lai JM, Chou TY, Chen CY, Su LJ,
Lee YC, Cheng TS, Hong YR, Chou CK, Whang-Peng J, et al: VEGFA
upregulates FLJ10540 and modulates migration and invasion of lung
cancer via PI3K/AKT pathway. PLoS One. 4:e50522009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang G, Liu M, Wang H, Yu S, Jiang Z, Sun
J, Han K, Shen J, Zhu M, Lin Z, et al: Centrosomal protein of 55
regulates glucose metabolism, proliferation and apoptosis of glioma
cells via the Akt/mTOR signaling pathway. J Cancer. 7:1431–1440.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tao J, Zhi X, Tian Y, Li Z, Zhu Y, Wang W,
Xie K, Tang J, Zhang X, Wang L and Xu Z: CEP55 contributes to human
gastric carcinoma by regulating cell proliferation. Tumour Biol.
35:4389–4399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen CH, Lu PJ, Chen YC, Fu SL, Wu KJ,
Tsou AP, Lee YC, Lin TC, Hsu SL, Lin WJ, et al: FLJ10540-elicited
cell transformation is through the activation of PI3-kinase/AKT
pathway. Oncogene. 26:4272–4283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Jin T, Dai X and Xu J:
Lentivirus-mediated knockdown of CEP55 suppresses cell
proliferation of breast cancer cells. Biosci Trends. 10:67–73.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen CH, Shiu LY, Su LJ, Huang CY, Huang
SC, Huang CC, Yin YF, Wang WS, Tsai HT, Fang FM, et al: FLJ10540 is
associated with tumor progression in nasopharyngeal carcinomas and
contributes to nasopharyngeal cell proliferation, and metastasis
via osteopontin/CD44 pathway. J Transl Med. 10:932012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nguyen-Ba G and Vasseur P: Epigenetic
events during the process of cell transformation induced by
carcinogens (review). Oncol Rep. 6:925–932. 1999.PubMed/NCBI
|
|
30
|
Vermeulen K, Van Bockstaele DR and
Berneman ZN: The cell cycle: A review of regulation, deregulation
and therapeutic targets in cancer. Cell Prolif. 36:131–149. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hui L, Yang N, Yang H, Guo X and Jang X:
Identification of biomarkers with a tumor stage-dependent
expression and exploration of the mechanism involved in laryngeal
squamous cell carcinoma. Oncol Rep. 34:2627–2635. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alt JR, Gladden AB and Diehl JA: p21(Cip1)
Promotes cyclin D1 nuclear accumulation via direct inhibition of
nuclear export. J Biol Chem. 277:8517–8523. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brehm A, Miska EA, McCance DJ, Reid JL,
Bannister AJ and Kouzarides T: Retinoblastoma protein recruits
histone deacetylase to repress transcription. Nature. 391:597–601.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Archer SY, Johnson J, Kim HJ, Ma Q, Mou H,
Daesety V, Meng S and Hodin RA: The histone deacetylase inhibitor
butyrate downregulates cyclin B1 gene expression via a
p21/WAF-1-dependent mechanism in human colon cancer cells. Am J
Physiol Gastrointest Liver Physiol. 289:G696–G703. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|