Introduction
Gastric cancer (GC) is the third leading cause of
cancer-associated mortality worldwide (1). Statistics from the National Central
Cancer Registry of China revealed that GC, with ~498,000 associated
mortalities in 2015, is the second most common cause of
cancer-associated mortality in China (2). Due to the fact that >80% of GC cases
are diagnosed in the middle-late stages, there is a <25% 5-year
survival rate in these patients (3).
The efficacy of adjuvant chemotherapy, the main treatment for
advanced GC, is limited due to its non-specific anticancer
activity. Therefore, cellular antitumor strategies, including
activated dendritic cells, tumor-infiltrating lymphocytes and,
particularly, genetically engineered T lymphocytes, have
demonstrated potential for the treatment of solid tumors (4).
Chimeric antigen receptors (CARs), which are
responsible for the success of T-cell immunotherapy, contain three
main elements; an extracellular domain, a single-chain
antigen-recognition domain (usually an scFv) and a transmembrane
domain. ScFv recognizes and binds to a specific tumor antigen
independently of major histocompatibility complex molecules, while
the transmembrane domain is an intracellular signaling domain
including a signal-transduction molecule of the T cell receptor
[usually cluster of differentiation (CD)3ζ] and costimulatory
receptors (e.g., CD137, CD28 or OX40) (5). For hematological tumors, CD19-, CD20-
and CD22-targeted CAR-T cell treatments have completed clinical
trials and have displayed notable antitumor activity (4). Previous studies have reported that
CD19-specific CAR-T-cell treatment had been the most effective
therapy with complete response (CR) rates of 70–90% (6). For solid tumors, a series of CARs
targeting 23 different antigens, including prostate-specific
membrane antigen, fibroblast activation protein and GD2, had been
constructed and validated in clinical trials (4). As for GC, CARs targeting
carcinoembryonic antigen, mucin 1 and receptor tyrosine-protein
kinase erbB-2 remain in clinical trials (4). Considering the heterogeneity of GC
cells, the identification of novel antigen-based CAR-T cells for
the treatment of GC is urgently required.
Previously, a monoclonal antibody, mAb-3H11, was
obtained using the hybridoma technique with spleen cells from mice
immunized with five human GC cell lines (7). Briefly, Balb/c mice were immunized with
one of the following human GC cell lines: M85, SGC7901, Kato III or
MGC-803 once a week, and were mice were injected with MKN45 cells
at the middle of each week by intra-splenic injection. Spleen cells
from mice immunized with GC cells were were harvested and fused
(via membrane fusion) with the murine myeloma SP2/0 cells using the
cell fusion hybridoma technique. Candidate hybridoma cells
producing mAbs were obtained by selective culture and ELISA
screening in GC cells. Among all candidate antibodies, mAb-3H11 was
confirmed to exhibit a high specific binding capacity to GC cells
and a reaction rate of ~93.5% in human GC tissues; however, it did
not react with normal cells (7).
Furthermore, it was also determined whether mAb 3H11 reacted with
normal human organ tissue, including heart, liver, spleen, lung,
kidney, stomach, colon, brain, bone, muscle, skin and nerve
(7). As a result, there was only a
weak positive reaction rate for mAb-3H11 in 1/15 normal gastric
mucosa and a week cross-reaction with salivary gland, sweat gland,
bronchus and intestinal content (7).
The above results suggested that mAb 3H11 reacted with most GC
tissues, but reacted weakly with the few of normal tissues,
indicating that mAb 3H11 has potential for GC diagnosis or target
therapy.
Furthermore, I131-labeled or
I125-labeled mAb 3H11 indicated a high sensitivity and
specificity for intraoperative detection of the lymphatic
micrometastasis in two clinical trials for radioimmunoguided
surgery (8,9), suggesting its potential application as a
radioiodination reagent to detect metastatic cancer in clinical
practice. In addition, our previous study also confirmed the
sequences of variable region heavy chain (VH) and light
chain (VL) for mAb-3H11 (10). The antigen of mAb-3H11 was previously
reported to be centrosomal protein 290 (CEP290) by cDNA library
screening (11); however, the
intracellular location of CEP290 and predominant membrane location
of the antigen of mAb 3H11 remain unclear.
The present study not only designed
lentivirus-mediated scFv-3H11 CAR-harboring CD28, CD137 and CD3ζ
signaling domains, but also evaluated the antitumor activity of
CAR-T cells against GC cells both in vitro and in
vivo. The results of the present study provide information for
future clinical trials testing the use of this CAR-T cell
immunotherapy for patients with GC.
Materials and methods
Cell culture
The human GC cell lines, NCI-N87, MKN45, AGS, NUGC3,
SGC7901, MGC803 and BGC823, and Jurkat cells, were obtained from
the Cell Center of Peking Union Medical University (Beijing,
China). All GC cells and Jurkat cells were cultured in RPMI-1640
medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.) and incubated at 37°C in a humidified incubator.
Isolation of primary GC cells
Two cases of primary human GC cells (name as −1 and
PGCC-2) were surgically obtained via tumor resection from a 62 year
old male and a 64-year-old male in October 2013 (Peking University
Cancer Hospital and Institute, Beijing, China). All samples were
mechanically cut into 1 mm pieces using scissors, and digested with
1 mg/ml collagenase I and 1 mg/ml collagenase IV (Thermo Fisher
Scientific, Inc.) at 37°C for 1 h. Subsequently, PGCCs were
cultured in F-12 nutrient mixture (Ham)-Dulbecco's modified Eagle's
medium (Invitrogen; Thermo Fisher Scientific, Inc.) for conditional
reprogramming and immortalization of epithelial cells as previously
described (12). Written informed
consent had obtained from all patients enrolled in the present
study and all protocols were approved by the Peking University
Cancer Hospital and Institute Ethical Committee.
RNA extraction and overlap
extension-polymerase chain reaction (OE-PCR)
Total RNA was extracted from hybridoma-3H11 cells
using an RNeasy Mini kit (Qiagen, Inc., Valencia, CA, USA),
according to the manufacturer's protocols. A total of 2 µg RNA was
reverse transcribed using TransScript® All-in-One
First-Strand cDNA Synthesis SuperMix (Beijing TransGen Biotech Co.,
Ltd., Beijing, China) according to the manufacturer's protocol. The
annealing temperature was designed on the basis of our previous
study (10). VH and
VL were amplified by PCR using the following
thermocycling conditions: Pre-denaturation at 94°C for 5 min,
followed by 94°C 30 sec, 45°C for 60 sec, 72°C for 90 sec for 30
cycles and finally, 72°C for 10 min and 4°C for 1 min. The
sequences for primers were VH-forward, 5′-GAATTCCAGGTTCAGCTGTG-3′
VH-reverse-1, 5′-TGAGGAGACGGTGACTGAGG-3′ and VH-reverse-2,
5′-CGATCCGCCACCGCCAGAGCCACCTCCGCCTGAACCGCCTCCACCTGAGGAGACGGTGACTGAGG-3′;
VL-forward-1, 5′-GGTGGAGGCGGTTCAGGCGGAGGTGGCTCTGGCGGTGGCGGATCGCAAATTGTACTCACCCAGTC-3′,
VL-forward-2, 5′-CAAATTGTACTCACCCAGTC-3′ and VL-reverse,
5′-CTCGAGTTTTATTTCCAGCTTG-3′. Underlined sequences indicate the
overlap linker (Gly4Ser)3 sequence.
CDR analysis by IgBLAST
VH and VL OE-PCR products were
cloned into PCR-blunt vectors (Invitrogen; Thermo Fisher
Scientific, Inc.), sequenced by SinoGenoMax Co., Ltd. (Beijing,
China) and analyzed by the IgBLAST program (13) using ImMunoGeneTics databases
(http://www.imgt.org/).
Constructs and lentiviral
packaging
The scFv-3H11 sequence, connecting the VH
and VL using a (Gly4Ser)3 polypeptide linker,
was obtained by OE-PCR and was subsequently constructed in pEGFPN1
and plenti6-TR expression vectors. The structures of recombinant
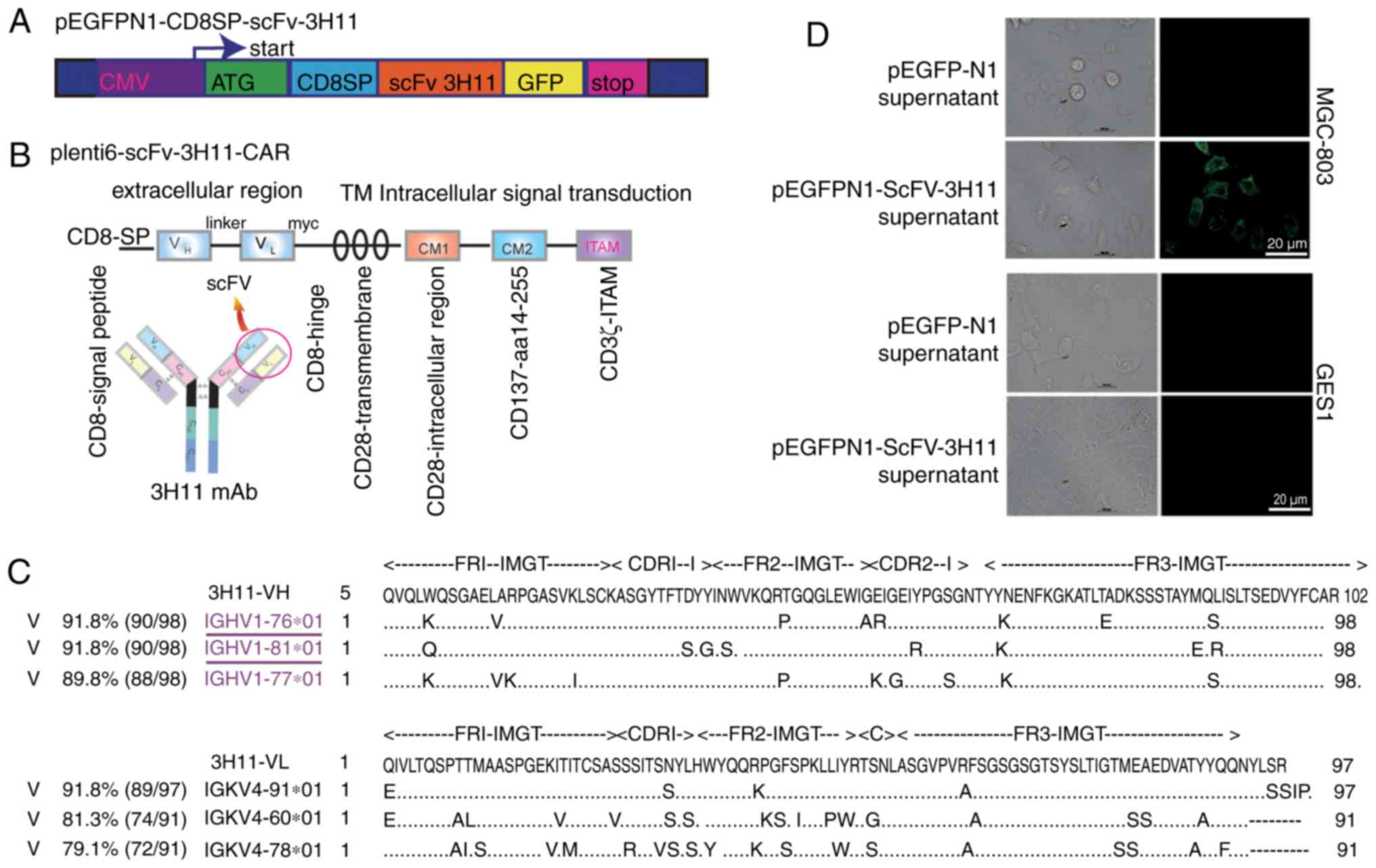
plasmids are presented in Fig. 1A and
B. A signal peptide from the CD8 family, CD8SP, was added prior
to the scFv-3H11 sequence. The entire DNA sequence of 3H11-CAR
contained scFv-3H11, human CD8 hinge, a transmembrane domain, and
CD28 and CD137 co-stimulatory and CD3ζ signaling domains (Fig. 1B). In order to verify transduction
efficiency, a tag of Myc was added immediately after the carboxyl
terminus of the scFv-3H11. A lentiviral shuttle plasmid containing
luciferase reporter, pELNS-Luciferase-IRES-Neo, was used to
establish stable luciferase expression in GC cell lines in order to
detect cytotoxicity. Viral particles were produced using
transfection into package HEK-293FT cells with a four-plasmid
system and were purified using PEG-it™ Virus Precipitation Solution
(Stratech Scientific Ltd., Newmarket, UK). All the recombinant
constructions were confirmed by sequencing (data not shown).
Secretory expression of
scFv-3H11-green fluorescent protein (GFP)
The pEGFPN1-CD8SP-ScFv-3H11 plasmid was transfected
into COS-7 cells at 80–90% confluence using Lipofectamine reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to
manufacturer's protocols. Two days later, the culture medium
containing secretory expression of recombinant scFv-3H11 was
collected for analysis of activity.
Generation and expansion of 3H11-CAR T
cells from peripheral blood mononuclear cells (PBMCs)
PBMCs from healthy donors were purified using
lymphocyte separation medium (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) by density gradient
centrifugation at 1,360 × g for 20 min in room temperature.
CD3+ T cells in PBMCs were enriched using the
EasySep™ Human T Cell Isolation kit (Stemcell
Technologies, Inc., Vancouver, BC, Canada), activated using
Dynabeads® Human T-Expander CD3/CD28 (Thermo Fisher
Scientific, Inc.) and expanded in OpTmizer™ T-Cell Expansion medium
(Thermo Fisher Scientific, Inc.), containing interleukin (IL)-2
(300 U/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany).
Lentivirus infection efficiency of the CAR-T cells was evaluated by
fluorescence-activated cell sorting (FACS) analysis on day 10 prior
to the first treatment.
Flow cytometry analysis
Live cells were incubated with mAb-3H11 (made in our
laboratory by affinity-purification from mouse ascites using
sepharose protein A; 1 µg/µl; 1:100 dilution) or an anti-Myc (1:100
dilution; cat. no. ab32; Abcam, Cambridge, UK) antibody at 37°C for
20 min, prior to being incubated with fluorescein isothiocyanate
(FITC)-labeled goat anti-mouse secondary antibodies (cat no.
115-095-003; 1:100 dilution; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA) at 37°C for 20 min, followed by analysis
on a BD Accuri C6 Cytometer using C6 software (version 1.0.264.21;
BD Biosciences, Franklin Lakes, NJ, USA).
Cytotoxicity assays and cytokine
secretion assays
BGC-823 and NCI-N87 cells were mixed directly with
lentivirus-containing luciferase reporter for 48 h (Shanghai
GeneChem Co., Ltd., Shanghai, China) and then screened by G418 for
~2 weeks to establish stable firefly luciferase-expressing cancer
cells in order to detect cytotoxicity. Firefly luciferase activity
in the cells (representing the live cell numbers) were measured
using a Luciferase Reporter assay (Promega Corporation, Madison,
WI, USA) according to the manufacturer's protocol. Mock T cells
were used as the control group to normalize luciferase data for
CAR-T cells, and cell lysis was calculated using the following
formula: Cell lysis (%) = (1-luciferase experiment/luciferase
control) ×100. The specific in vitro antitumor activity of
CAR-T cells was performed. Briefly, different types of target cells
(with luciferase reporter gene) were seeded onto triplicate 96-well
plates, at a density of 103 cells/well, with 50 µl
RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.). Subsequently, an
equal volume of effector cells or control medium was added to each
well to ensure an effector-target ratio (E/T ratio) of 25:1,
12.5:1, 6.2:1, 3.1:1, 1.6:1, or 0.8:1. Following an 8-h incubation,
cell supernatants were obtained by centrifugation at 800 × g for 10
min at room temperature and collected for cytokine measurements of
IL-2 and interferon (IFN)-γ concentrations using an ELISA kit
(BioLegend, Inc., San Diego, CA, USA), according to the
manufacturer's protocols.
Xenograft mouse model of GC
Ten NOD/SCID mice (male; 7 weeks old; 18–20 g) were
purchased from Vital River Laboratories Co., Ltd. and were housed
in a pathogen-free animal housing facility at Beijing University
Cancer Hospital and Institute at 23±3°C, a relative humidity of
~50%, 12 h light/dark cycle and a standard sterilized rodent diet
from Vital River Laboratories Co., Ltd (Bejing, China) and
sterilized water ad libitum. The animal experiments were approved
by the Animal Ethics Committee of Peking University Cancer Hospital
and Institute. A total of 100 µl 1×106
3H11-antigen-positive MGC-803 cells, were injected subcutaneously
into NOD/SCID mice on day 0. Tumor-bearing mice were randomly
assigned to the CAR-T cell and control T cell groups prior to
treatment, with five mice in each group. The tumor volume (TV) of
each mouse was measured twice weekly using a vernier caliper and
was calculated according to the following formula: TV = 1/2 ×
length × width2. On day 14, when the mean TV reached
~100 mm3, 200 µl 2×107 CAR-T cells or control
T cells, were infused into the tumors of the mice twice weekly by
multipoint injection.
Immunohistochemical examinations
Humane endpoints were used in accordance with
Peiking University Cancer Hospital and Institute standard operating
protocols. Tumor tissue samples from sacrificed mice on the 35th
day, according to the humane endpoints of diameter of the tumor
mass (i.e., greater than 1.5 cm diameter in mice) were fixed in 10%
formaldehyde solution for 24 h, dehydrated in ethyl alcohol, and
embedded in paraffin, prior to being cut into 6 µm thick sections
using a microtome. Immunohistochemical (IHC) staining was performed
according to standard procedures. Briefly, slides were immersed in
xylene to remove paraffin, washed in a graded series of ethanol,
immersed in citrate buffer at pH 6.0 and then incubated in a
high-pressure sterilization oven for antigen retrieval at 100°C for
3 min. Endogenous peroxidase activity was blocked in a blocking
solution with 3% H2O2 in PBS for 10 min at
room temperature, and then the slides were incubated with PBS
containing 1% bovine serum albumin (Amresco, Solon, OH, USA) for 10
min at room temperature to block non-specific binding. The tissue
sections were incubated at room temperature for 1 h with a primary
rabbit anti-human CD3 antibody (1:200; cat. no. ab5690; Abcam),
followed by incubation with horseradish peroxidase-conjugated goat
anti-mouse IgG (1:500; cat no. A4416; Sigma-Aldrich; Merck KGaA)
for 1 h at room temperature. Then, the slides were visualized with
0.1% 3,3-diaminobenzidine (Sigma-Aldrich; Merck KGaA) for 2 min,
and counterstained with one drop of 1% hematoxylin for 10 min at
room temperature.
Statistical analysis
Statistical analyses were performed using Prism
V5.00 for Windows (GraphPad Software, Inc., La Jolla, CA, USA). The
differences between two groups were assessed using independent
samples t-test. Dunnett's multiple comparison tests were used to
compare differences between treatment groups and the control group
following one-way analysis of variance. P<0.05 was considered to
indicate a statistically significant difference.
Results
Plasmid construction and analysis for
CDR of VH and VL of mAb-3H11 using the
IgBLAST database
The structures of recombinant plasmids are presented
in Fig. 1A and B. In order to
determine the secretory expression of scFv-3H11, a signal peptide
from the CD8, named CD8SP, was added before the scFv-3H11 sequence.
The entire DNA sequence of 3H11-CAR contained scFv-3H11, human CD8
hinge, a transmembrane domain, and CD28 and CD137 co-stimulatory
and CD3ζ signaling domains (Fig. 1B).
In order to verify transduction efficiency, a Myc tag was added
immediately after the carboxyl terminus of scFv-3H11. The deduced
amino acid sequences of VH and VL were
submitted to the online IgBLAST database, and were aligned to
maximize the homology with the IGHV1-76 and
IGKV4-91, respectively (Fig.
1C). The complementarity determining regions (CDRs) for the
heavy and light chains of mAb-3H11 are presented in Fig. 1C.
Secretory scFv-3H11 indicated good
binding activity
The binding activity of scFv-3H11 in the cultured
medium of COS-7 cells was detected by live cell staining. As
demonstrated in Fig. 1D, the
secretory scFv-3H11 antibody in the harvested medium from COS-7
cells clearly stained the protein on the membrane of MGC-803 cells,
but not of GES-1 cells. This result suggested that recombinant
scFv-3H11 protein had good binding activity, similar to that of the
natural antibody.
The antigens of mAb-3H11 were highly
expressed in GC cells
FACS was used to assess the surface expression of
3H11-antigen proteins in a series of human GC cell lines (GCCLs),
including NCI-N87, MKN45, AGS, NUGC3, SGC7901, MGC803 and BGC823
cells, and in PGCCs obtained from two patients with GC. The results
of the present study suggested that 3H11-antigen was highly
expressed in all the GCCLs, with the positive percentage ranging
between 56.6 and 99.6%, and in the PGCCs from patient 1 (PGCC-1)
with a positive percentage of 87.0% and patient 2 (PGCC-2) with a
positive percentage of 50.8% (Fig.
2), while the positive percentage was only 0.9% in normal
gastric epithelial GES1 cells. Therefore, mAb-3H11 demonstrated a
50.8 to 99.6% positive reaction with GC cells. NCI-N87 with a 56.6%
positive rate and a BGC823 positive rate of 98.9% were selected for
further study.
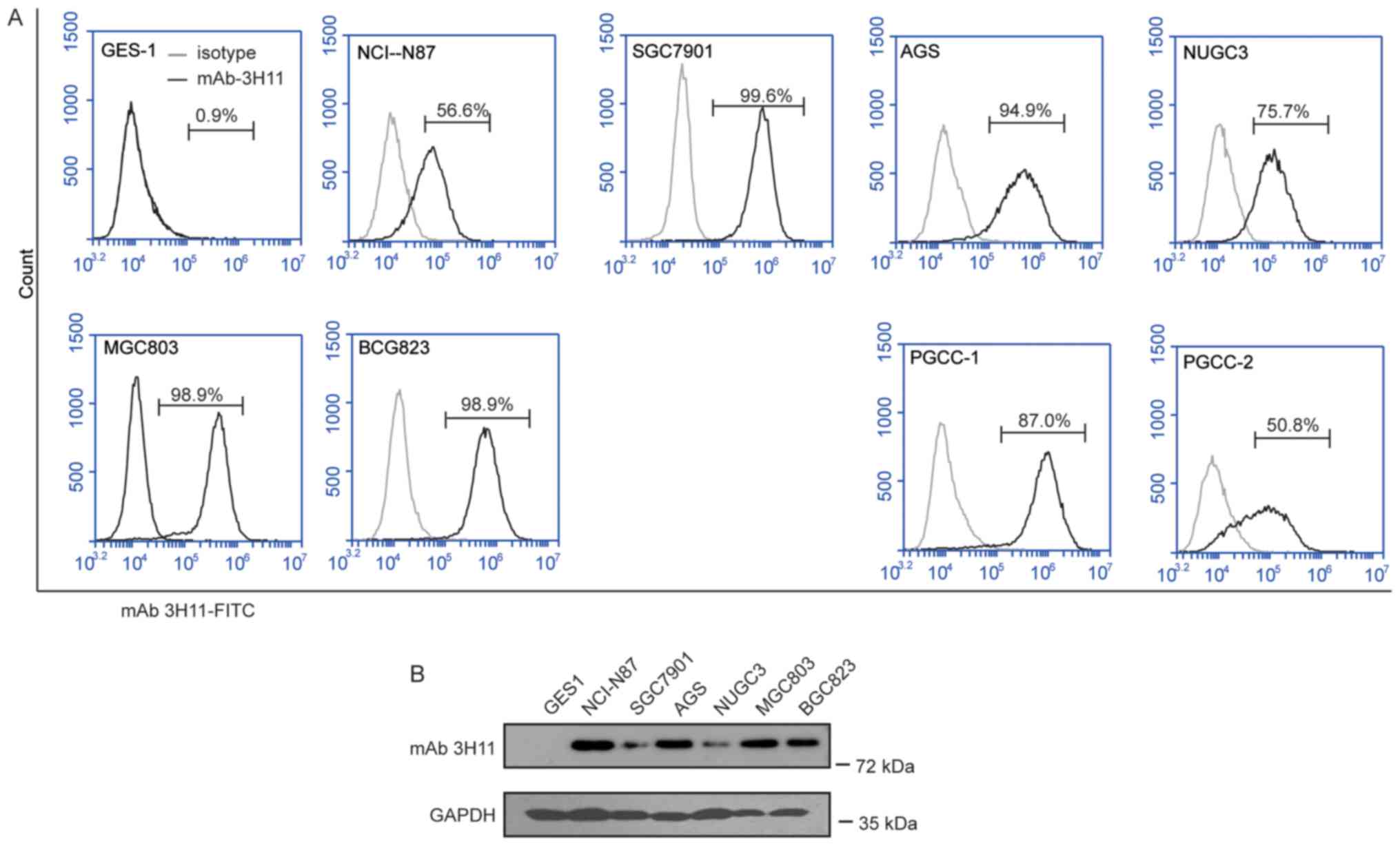 | Figure 2.Analysis of the antigen expression of
mAb-3H11 in GC cell lines. (A) Flow cytometry histogram plots of
the surface antigen expression of mAb-3H11 in GES1 cells, MGC803,
NUGC3, MKN45, NCI-N87, BCG823, MGC7901 and AGS cell lines, and two
patient-derived GC cell lines. (B) Western blot analysis of the
antigen expression of mAb-3H11 in GES1, NCI-N87, MGC7901, AGS,
NUGC3, MGC803 and BCG823 cells. mAb, monoclonal antibody; GC,
gastric cancer; PGCC, primary human GC cells. |
scFV-3H11-CAR T cells killed GC cells
accompanied with an increased expression of IL-2 and IFN-γ ex
vivo
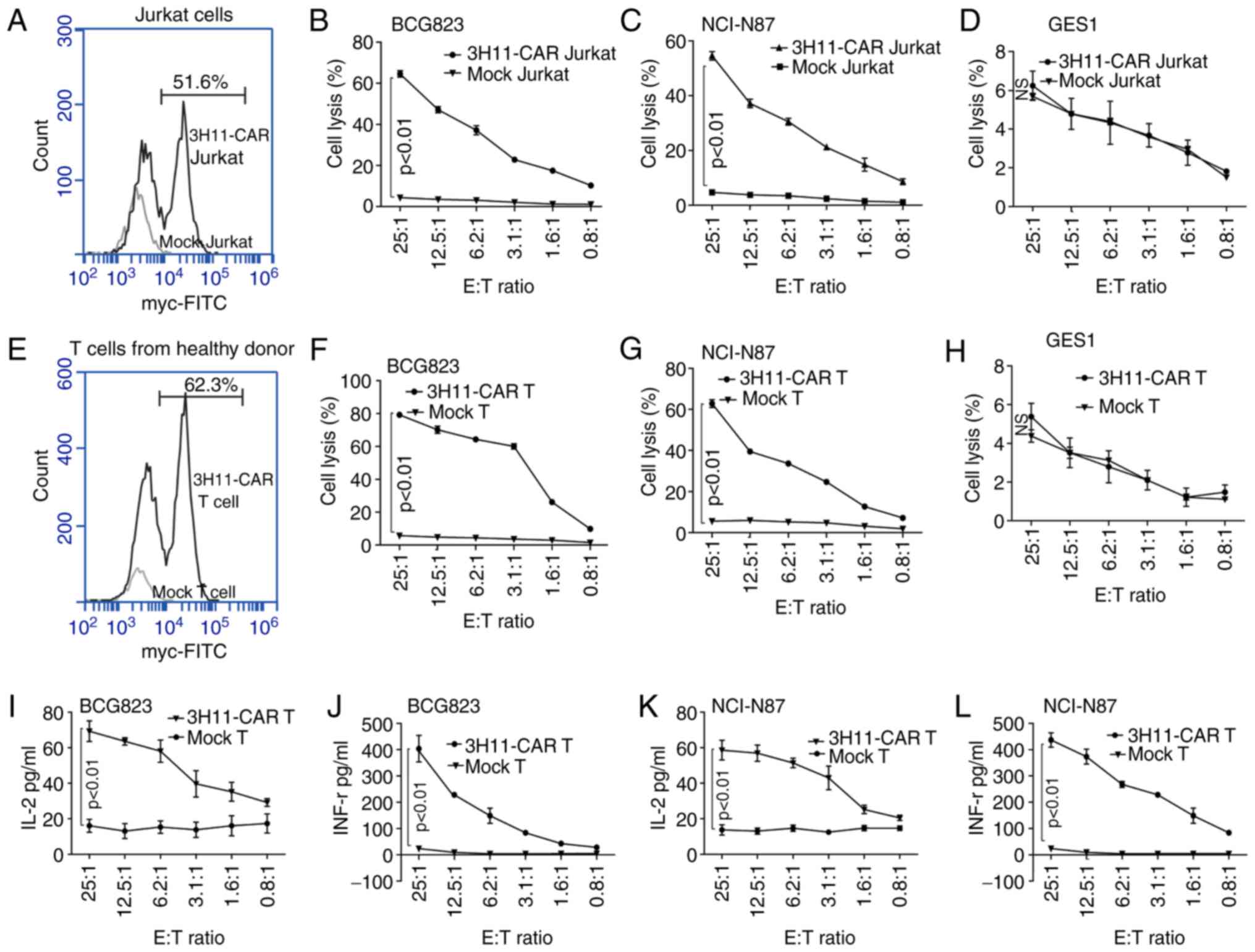
Following lentivirus infection, the transduction
efficiency of scFV-3H11-CAR in Jurkat cells was 51.6% (Fig. 3A), and the cytotoxicity of CAR-Jurkat
cells exhibited higher average killing activity against BCG823
cells (64.6±2.7%; Fig. 3B) and
NCI-N87 cells (54.6±2.7%; Fig. 3C)
than did mock Jurkat cells with a low rate of lysis activity at a
ratio of 25:1 E/T against BCG823 cells and NCI-N87 cells.
Additionally, a dose-dependent cell killing was also performed, as
demonstrated in Fig. 3B and C, and it
was revealed that the cytotoxicity of scFV-3H11-CAR Jurkat cells
increased as the E/T ratio increased. As for the human normal
gastric epithelial GES1 cell line, there was no significant killing
effect for the CAR Jurkat cells compared with the mock Jurkat cells
(Fig. 3D).
Similarly, following lentivirus infection, the
transduction efficiency of 3H11-CAR-T cells from healthy donors was
62.3% (Fig. 3E). The cytotoxicity of
3H11-CAR-T cells exhibited higher average killing activity against
BCG823 cells (79.2±1.5%; Fig. 3F) and
NCI-N87 cells (Fig. 3G, 62.8±3.3%)
than did mock T cells with a low rate of cell lysis at a ratio of
25:1 E/T, and the cytotoxic effects displayed a dose-dependent
pattern (P<0.05; Fig. 3F and G).
As for the GES1 cells, the CAR-T cells had no significant killing
effect, compared with the mock T cells (Fig. 3H). Furthermore, following the
incubation, the levels of cytokines released by scFV-3H11-CAR T
cells, including IL-2 and IFN-γ, were significantly elevated in the
supernatants of BGC823 and NCI-N87 cells compared with those of the
mock T cells, and the cytokine level also exhibited a
dose-dependent pattern (Fig. 3I and K
for IL-2 expression; Fig. 3J and L
for IFN-γ expression).
scFV-3H11-CAR-T cells exhibited
effective antitumor activity against xenografts
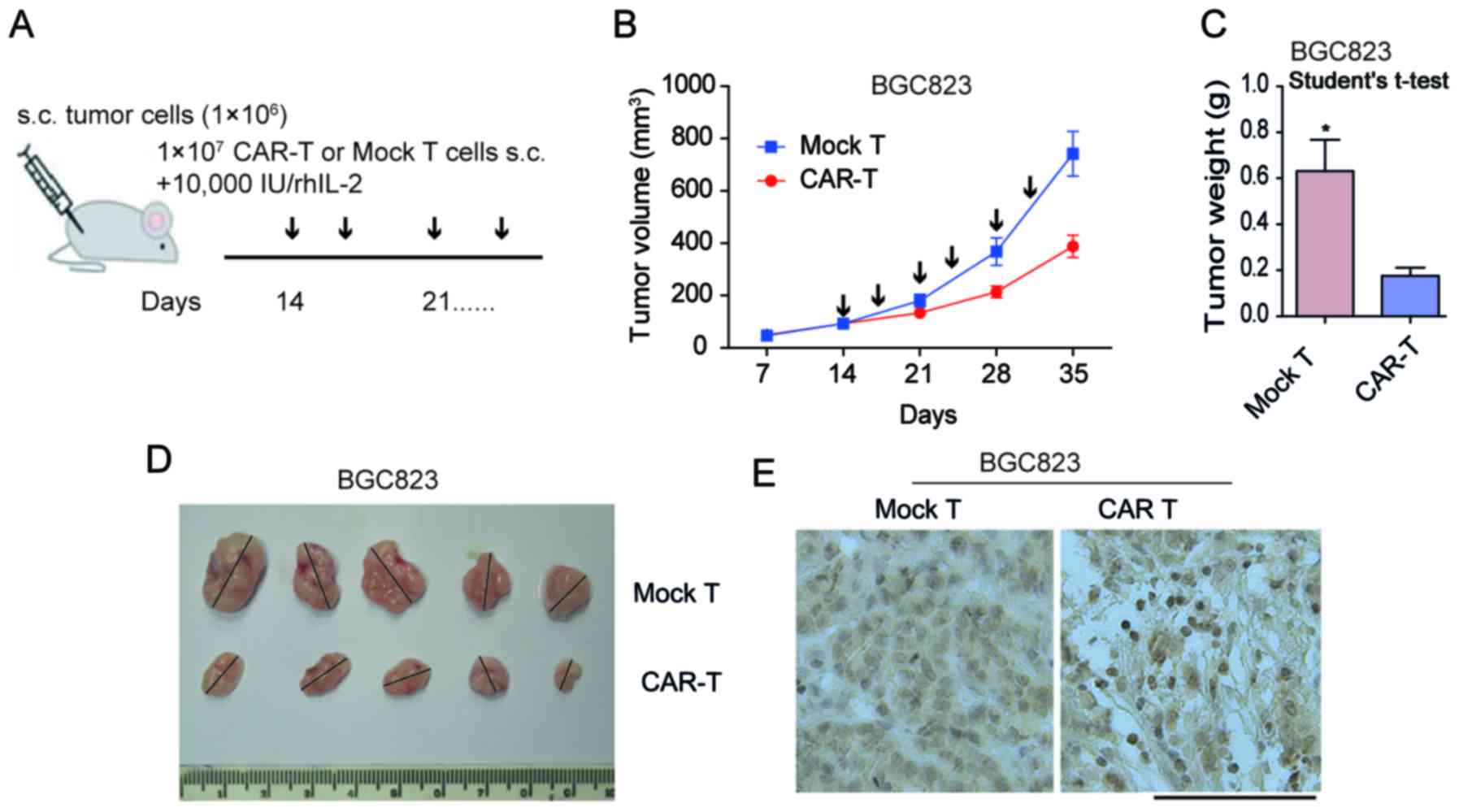
The subcutaneous xenotransplanted tumor model of GC
BGC823 cells were established in NOD/SCID mice in order to
determine the antitumor capacity of scFV-3H11-CAR-T cells in
vivo. As demonstrated in Fig. 4A.
treatment with CAR-T cells was performed on day 14, when the mean
tumor volume (TV) had reached ~100 mm3 by multipoint
intra-tumor injection with effector cells twice a week. In the
present study, all animals presented with only one tumor. In the
mouse model, the tumor weight was considerably inhibited by
treatment with 3H11-CAR-T cells, while tumors in the control group
continued to grow rapidly following injection with mock T cells
(Fig. 4B). When the mice were
sacrificed at 35 days, the mean BGC823 tumor weight in the
3H11-CART group was 176 mg, while that in the mock T group was 631
mg (P=0.01; Fig. 4C and D),
suggesting a 72.1% suppression in tumor weight following 3H11-CAR-T
therapy in BGC823 cells. In order to assess the infiltration of
3H11-CAR-T cells, IHC labeling for CD3 T cells was performed on
tumor samples initiated from BGC823 cells. The results of the
present study demonstrated a considerable increase in human
CD3+ T cells in the experimental group, while only a
small number of CD3+ T cells were observed in the mock T
group (Fig. 4E). These findings
suggested that 3H11-CAR-T cells may effectively inhibit tumor
growth. The results for the PGCC-1 cells were similar (data not
shown).
Discussion
The high effectiveness and low toxicity of CAR-T
cell therapy is primarily dependent upon the specificity of the
target antigen. In our previous study, mAb-3H11 exhibited a
positive reaction with five immunized GC cell lines and a negative
reaction with lymphocytes, red blood cells, bone marrow cells and
diploid fibroblasts. Furthermore, it also exhibited a high positive
reaction rate of ~93.5% in tissues of all histopathological types
of GC, including well-differentiated, poorly differentiated,
mucosal and metastatic GC (7). The
aforementioned study also detected the the reactivity of mAb 3H11
in 32 types of normal human tissue, including heart, liver, spleen,
lung, kidney, stomach, colon, brain, bone, muscle, skin and nerve,
and the results demonstrated that there were weak positive reaction
rates for mAb-3H11 with 1/15 normal gastric mucosa and a week
cross-reaction with salivary gland, sweat gland, bronchus and
intestinal content (7). These
previous results suggested that mAb-3H11-based CAR-T cell therapy
was feasible because it had little on-target, off-tumor toxicity
resulting from the shared antigens in normal tissues. Therefore, in
the present study, CAR-T cells were created using a single-chain
variable fragment (scFv) of mAb-3H11 in order to kill GC. The
results of the present study suggested that 3H11-CAR-T cells
induced robust T cell cytotoxicity and elicited high levels of IL-2
and IFN-γ production compared with control CAR T cells.
The signaling region of the first-generation (1st-G)
CAR mimics T cell receptor (TcR) signaling by fusing the
antigen-binding region to the CD3-ζ chain, and the
second-generation (2nd-G) CAR mimics TcR and costimulatory
signaling by adding, for example, CD28 or CD137 domains to the
intracellular region, while the third-generation (3rd-G) CAR has
two costimulatory domains fused with the TcR CD3-ζ chain (14). The results of a previous study
demonstrated that the 3rd-G CAR-T cells exhibited superior
activation and proliferation capacity compared with the 2nd-G CAR-T
cells carrying only one costimulatory domain (15). In the present study, in order to
obtain the optimal antitumor effect, lentivirus-expressing
scFV-3H11 3rd-G CAR was constructed. With regards to the killing
mechanism of effector T cells on target tumor cells, a previous
study suggested that CARs were hybrid proteins consisting of an
antigen specific binding domain (usually scFV) fused to
intracellular T-cell activation domains (CD28 and CD137/CD3ζ
receptor), and CAR-expressing engineered T lymphocytes may directly
recognize and kill tumor cells expressing its antigen in an human
leukocyte antigen-independent manner (16). Another previous study suggested that
it was possible that antigen-targeted CAR-T cells may not only
efficiently kill single tumor targets, but may also kill multiple
tumor targets in a sequential manner (17).
Jurkat cells are an immortalized line of human T
lymphocyte cells established in the late 1970s from the peripheral
blood of a 14-year-old boy (18).
They have been successfully used in the evaluation of in
vitro effectiveness of certain CAR-T cell therapies (19–22). To
the best of our knowledge, the present study was the first to
examine the cytotoxicity of scFV-3H11 CAR Jurkat cells in
vitro, and it was revealed that scFV-3H11 CAR Jurkat cells
killed more than half of the GC cells at a ratio of 25:1 E/T.
Subsequently, 3H11-CAR were transduced into primary T cells and
their antitumor effects both in vitro and in vivo
were detected. The results of the present study suggested that
scFV-3H11 CAR-T cells not only killed 62.8% NCI-N87 cells and 72.9%
BGC823 cells at a ratio of 25:1 E/T in vitro, but also
inhibited 72.1% tumor growth of BGC823 and 57.1% tumor growth of
PGCC-1 cells in vivo.
The ideal delivery method of CAR-T cell therapy in
solid tumors was by intravenous infusion. For the treatment of
xenografts in the mouse model used in the present study, CAR-T
cells were first administered by tail intravenous injection.
However, little therapeutic activity was detected following
treatment (data not shown). Considering that unsuccessful
intravenous injection was possibly due to the poor ability of
infused CAR-T cells to reach tumor-specific sites, the
tumor-bearing mice were administered with an intra-tumor injection
of CAR-T cells or mock T cells, and revealed that the therapeutic
efficacy was improved significantly. These results were consistent
with those of previous studies for T cell therapy in animal models
(23–25), which had exhibited no effect by
systematic infusion. Although scFv-3H11 CAR-T cells did not
overcome the current barriers of CAR-T cell therapy in GC in the
present study, the changes in protein levels in cultured CAR-T
lymphocytes, including increased expression of chemokine receptors
(26) or heparanase (27), may promote tumor infiltration and
antitumor activity. Therefore, the development of 3H11-CAR-T cells
co-expressed with heparanase or chemokine receptors may be a way to
improve the efficacy of this therapeutic tool in the treatment of
GC.
Acknowledgements
Not applicable.
Funding
This study was supported by the Beijing Committee of
Science and Technology in China (grant no. D131100005313010), the
National High Technology Research and Development Program of China
(863 Program; grant no. 2014AA020603), the National Natural Science
Foundation of China (grant nos. 81201964, 81772632 and 81773144),
Peking University (PKU) 985 Special Funding for Collaborative
Research with PKU Hospitals (2013-11-20) and the interdisciplinary
medicine Seed Fund of Peking University (grant no.
BMU2018MX019).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HH, SW, YH, and ZL performed most experiments. WY
and YL participated in the in vitro study. LW, LZ and JJ
designed the experiments and coordinated the project.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients enrolled in the present study and all protocols were
approved by the Peking University Cancer Hospital and Institute
Ethical Committee.
Consent for publication
Patients provided written informed consent for the
publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zong L, Abe M, Seto Y and Ji J: The
challenge of screening for early gastric cancer in China. Lancet.
388:26062016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson LA and June CH: Driving
gene-engineered T cell immunotherapy of cancer. Cell Res. 27:38–58.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Essand M and Loskog AS: Genetically
engineered T cells for the treatment of cancer. J Intern Med.
273:166–181. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Batlevi CL, Matsuki E, Brentjens RJ and
Younes A: Novel immunotherapies in lymphoid malignancies. Nat Rev
Clin Oncol. 13:25–40. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei SM: Monoclonal antibodies against
gastric cancer and their selective reaction on various tissues.
Zhonghua Zhong Liu Za Zhi. 11:162–164. 1989.(In Chinese).
PubMed/NCBI
|
|
8
|
Xu G, Zhang M, Liu B, Li Z, Lin B, Xu X,
Jin M, Li J, Wu J, Dong Z, et al: Radioimmunoguided surgery in
gastric cancer using 131-I labeled monoclonal antibody 3H11. Semin
Surg Oncol. 10:88–94. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang C, Wang Y, Su X, Lin B, Xu X, Zhang
M, Li J and Xu G: Iodine-125 labeled monoclonal antibody 3H11: In
radioimmunoguided surgery for primary gastric cancer. Zhonghua Wai
Ke Za Zhi. 38:507–509. 2000.(In Chinese). PubMed/NCBI
|
|
10
|
Li J, Wang Y, Li QX, Wang YM, Xu JJ and
Dong ZW: Cloning of 3H11 mAb variable region gene and expression of
3H11 human-mouse chimeric light Chain. World J Gastroenterol.
4:41–44. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen D and Shou C: Molecular cloning of a
tumor-associated antigen recognized by monoclonal antibody 3H11.
Biochem Biophys Res Commun. 280:99–103. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Ory V, Chapman S, Yuan H, Albanese
C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al:
ROCK inhibitor and feeder cells induce the conditional
reprogramming of epithelial cells. Am J Pathol. 180:599–607. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ye J, Ma N, Madden TL and Ostell JM:
IgBLAST: An immunoglobulin variable domain sequence analysis tool.
Nucleic Acids Res. 41:(Web Server Issue). W34–W40. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Enblad G, Karlsson H and Loskog AS: CAR
T-Cell therapy: The role of physical barriers and immunosuppression
in lymphoma. Hum Gene Ther. 26:498–505. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang XY, Sun Y, Zhang A, Hu GL, Cao W,
Wang DH, Zhang B and Chen H: Third-generation CD28/4-1BB chimeric
antigen receptor T cells for chemotherapy relapsed or refractory
acute lymphoblastic leukaemia: A non-randomised, open-label phase I
trial protocol. BMJ Open. 6:e0139042016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozawa K: Current status and future
development of CAR-T gene therapy. Rinsho Ketsueki. 56:2180–2185.
2015.(In Japanese). PubMed/NCBI
|
|
17
|
Davenport AJ, Jenkins MR, Ritchie DS,
Prince HM, Trapani JA, Kershaw MH, Darcy PK and Neeson PJ: CAR-T
cells are serial killers. Oncoimmunology. 4:e10536842015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schneider U, Schwenk HU and Bornkamm G:
Characterization of EBV-genome negative ‘null’ and ‘T’ cell lines
derived from children with acute lymphoblastic leukemia and
leukemic transformed non-Hodgkin lymphoma. Int J Cancer.
19:621–626. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kobayashi E, Kishi H, Ozawa T, Hamana H,
Nakagawa H, Jin A, Lin Z and Muraguchi A: A chimeric antigen
receptor for TRAIL-receptor 1 induces apoptosis in various types of
tumor cells. Biochem Biophys Res Commun. 453:798–803. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shirasu N, Shibaguci H and Kuroki M,
Yamada H and Kuroki M: Construction and molecular characterization
of human chimeric T-cell antigen receptors specific for
carcinoembryonic antigen. Anticancer Res. 30:2731–2738.
2010.PubMed/NCBI
|
|
21
|
Jamnani FR, Rahbarizadeh F, Shokrgozar MA,
Mahboudi F, Ahmadvand D, Sharifzadeh Z, Parhamifar L and Moghimi
SM: T cells expressing VHH-directed oligoclonal chimeric HER2
antigen receptors: Towards tumor-directed oligoclonal T cell
therapy. Biochim Biophys Acta. 1840:378–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Khaleghi S, Rahbarizadeh F, Ahmadvand D,
Rasaee MJ and Pognonec P: A caspase 8-based suicide switch induces
apoptosis in nanobody-directed chimeric receptor expressing T
cells. Int J Hematol. 95:434–444. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Q, Wang H, Li H, Xu J, Tian K, Yang
J, Lu Z and Zheng J: Chimeric antigen receptor-modified T Cells
inhibit the growth and metastases of established tissue
factor-positive tumors in NOG mice. Oncotarget. 8:9488–9499.
2017.PubMed/NCBI
|
|
24
|
Pule MA, Savoldo B, Myers GD, Rossig C,
Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z, et al:
Virus-specific T cells engineered to coexpress tumor-specific
receptors: Persistence and antitumor activity in individuals with
neuroblastoma. Nat Med. 14:1264–1270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zuccolotto G, Fracasso G, Merlo A,
Montagner IM, Rondina M, Bobisse S, Figini M, Cingarlini S,
Colombatti M, Zanovello P and Rosato A: PSMA-specific
CAR-engineered T cells eradicate disseminated prostate cancer in
preclinical models. PLoS One. 9:e1094272014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Craddock JA, Lu A, Bear A, Pule M, Brenner
MK, Rooney CM and Foster AE: Enhanced tumor trafficking of GD2
chimeric antigen receptor T cells by expression of the chemokine
receptor CCR2b. J Immunother. 33:780–788. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Caruana I, Savoldo B, Hoyos V, Weber G,
Liu H, Kim ES, Ittmann MM, Marchetti D and Dotti G: Heparanase
promotes tumor infiltration and antitumor activity of
CAR-redirected T lymphocytes. Nat Med. 21:524–529. 2015. View Article : Google Scholar : PubMed/NCBI
|