Introduction
Colorectal cancer (CRC) is one of the most common
types of malignancies in Western countries, causing ~500,000
cancer-associated mortalities annually worldwide (1). Despite undergoing surgical resection,
nearly 50% of patients do not respond to chemotherapy due to drug
resistance (2). Previous studies have
revealed that this may be due, at least in part, to anti-apoptotic
responses (3–5).
The inhibitor of apoptosis protein (IAP) family is
crucial to the anti-apoptotic responses of tumor cells (6). Livin, an important member of IAP, is
considered to negatively regulate apoptosis-associated proteins and
prevent apoptosis. It is strongly expressed in numerous common
human neoplasms including CRC and lung cancer (7,8).
Overexpression of Livin in tumors is associated with resistance to
chemotherapy and radiotherapy, increased tumor recurrence and
decreased patient overall survival (9–11). In CRC
tissues, Livin expression levels are significantly increased and
considered to be an independent prognostic factor (12).
In the past three decades, 5-fluorouracil (5-FU)
chemotherapy has been the primary choice for the adjuvant treatment
of CRC, although its resistance in the course of treatment has
become a common problem. Briefly, 5-FU is an analog of uracil with
a fluorine atom at the C-5 position in place of hydrogen. It
inhibits thymidylate synthase and influences uracil metabolism to
induce apoptosis in cancer cells (13,14).
Previous studies have revealed that Livin serves a role in 5-FU
resistance, and that the knockdown of Livin expression in cancer
cell lines facilitates 5-FU-induced cell apoptosis (15,16).
However, the mechanisms underlying Livin-mediated reversal of 5-FU
chemoresistance have not yet been fully elucidated.
Apoptosis, also known as type I cell death, is a
well-researched form of programmed cell death that involves the
rapid demolition of cellular structures and organelles via the
activation of catabolic enzymes (17). Autophagy is a conserved catabolic
process in which cellular contents are degraded in lysosomes
(18). Autophagy is considered to
serve an important role in tumor development and chemoresistance
(19). Previous studies have
demonstrated that autophagy may serve a beneficial or detrimental
effect on cancer cells, depending on the response to environmental
stressors (20). With regard to
benefits, autophagy provides the metabolites and ATP required for
cell survival upon exposure to diverse environmental stressors,
including treatment with chemotherapeutic agents, endoplasmic
reticulum stress and hypoxia (21).
Conversely, in certain cases, autophagy can lead to cell death by
lysosome-mediate cell degradation (a distinct form of cell death
compared with apoptosis) (22).
Autophagy and apoptosis serve important roles in oncogenesis and
tumor progression in mammals; they may be triggered by common
upstream signals, resulting in combined autophagy and apoptosis
(23,24). In other instances, their occurrence
may be mutually exclusive. For example, researchers have
demonstrated autophagy-mediated removal of protein of aggregates
and harmful organelles, which induced inhibition of cell apoptosis
(25). Despite efforts, the crosstalk
between autophagy and apoptosis remains unclear. Previous studies
have suggested the involvement of B-cell lymphoma-2
(Bcl-2)-beclin-1 interaction (25).
In more recent studies, protein kinase B (Akt) was hypothesized to
be involved in the crosstalk between apoptosis and autophagy
(26). The present study revealed
that apoptosis and autophagy were triggered by two common upstream
signals, specifically Bcl-2 and the Akt signaling pathway.
Materials and methods
Antibodies and drugs
Anti-Livin, anti-Schizont membrane-associated
cytoadherence protein (SMAC), anti-light chain 3 (LC3), anti-actin,
anti-P62, anti-Bcl-2 and anti-caspase-3 antibodies were purchased
from Abcam (Cambridge, UK); anti-phosphorylated (p)-Akt, anti-total
(T)-Akt were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The 5-FU, autophagy inhibitor 3-methyladenine
(3-MA) and apoptosis inhibitor benzyloxycarbonyl-Val-Ala-Asp
fluoromethylketone (z-VAD-FMK) were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany).
Cell lines and cell culture
HCT116 and SW620 human colon carcinoma cell lines
were obtained from the Cell Bank of the Chinese Academy of Sciences
(Shanghai, China) and maintained in RPMI-1640 (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) culture medium supplemented
with 10% fetal bovine serum and 1% penicillin/streptomycin. All
cells were grown in a humidified incubator at 37°C with 5%
CO2.
Short hairpin (sh)RNA synthesis and
construction of Livin shRNA lentiviral vectors
The double stranded RNA sequences, simultaneously
aimed at both Livin mRNA isomers (GenBank accession nos. NM_139317
and NM_022161), were designed as previously described (27,28). The
sense strand was 5′-CAGGAGAGAGGTCCAGTCCAGTCTGA-3′ and the antisense
strand was 5′-TCAGACTGGACCTCTCTCCTG-3′. They were synthesized by
Shanghai GeneChem Co., Ltd. (Shanghai, China). Their connection to
the lentiviral vector pGCL-GFP (Shanghai GeneChem Co., Ltd.) was
verified by Shanghai GeneChem Co., Ltd. using polymerase chain
reaction (PCR) and DNA sequencing (data not shown). Briefly, the
products were transformed to fresh component bacteria E.
coli (Shanghai GeneChem Co., Ltd.) and 10 clones were selected
for each plasmid. PureLink™ Genomic DNA Purification kit
(Thermo Fisher Scientific, Inc.) was used for DNA extraction and
purification prior to being subjected to PCR amplification (forward
primer, 5′-CGCACGGCACAAAGACGA-3′ and reverse primer,
5′-GTCAGTTCCTGCTCCGGTCAA-3′). Taq DNA Polymerase (Thermo
Fisher Scientific, Inc.) was used for DNA amplification. The
thermocycling conditions were as follows: 95°C for 5 min, followed
by 25 cycles of 95°C for 30 sec, 55°C for 60 sec, 72°C for 60 sec,
and a final extension at 72°C for 7 min. The products were
identified by 2% agarose gel electrophoresis and further determined
by DNA sequencing (Shanghai GeneChem Co., Ltd.).
Transfection
The verified lentiviral vector was packaged by 293T
packaging cells (Shanghai Gefan Biotechnology Co., Ltd., Shanghai
China), and vector particles were concentrated and purified to
reduce toxicity. These lentiviral vectors infected HCT116 and SW620
cells when the cells achieved a confluence of ~80% following
incubation at 37°C for 48 h. Expression of the green fluorescent
protein reporter gene on the lentivirus was observed 4–5 days after
infection (multiplicity of infection =50). Cells were collected
with a transfection efficiency >80%. Further experiments were
performed 6–10 days after transfection. The cells into which the
lentivirus-shLivin was transfected were named the shLivin group,
the cells transfected with the negative control (NC) shRNA were
named the NC group, and the untransfected HCT116 and SW620 cells
were named the control group.
Reverse transcription-quantitative
(RT-q)PCR analysis
Livin expression levels in HCT116 and SW620 cell
lines were determined by RT-qPCR analysis, using the standard
methods previously described (29).
Total RNA was extracted from HCT116 and SW620 cell pellets prepared
by centrifugation at 500 × g for 5 min at room temperature using
TRIzol® reagent (Takara Biotechnology Co., Ltd., Dalian,
China). Reverse transcription was performed using the Prime Script
Strand cDNA Synthesis kit (Takara Biotechnology Co., Ltd.)
according to the manufacturer's protocol. cDNA were amplified using
SYBR Premix Ex Taq (Takara Biotechnology Co., Ltd.). The PCR primer
sequences were as follows: Livin forward, 5′-CGCACGGCACAAAGACGA-3′
and reverse, 5′-GTCAGTTCCTGCTCCGGTCAA-3′; β-actin forward,
5′-AGCCATGTACGTAGCCATCC-3′ and reverse, 5′-CTCTCAGCTGTGGTGGTGAA-3′.
PCR thermocycling conditions were set as follows: Pre-denaturing at
95°C for 30 sec, denaturing at 95°C for 5 sec, annealing at 60°C
for 34 sec with 40 cycles, denaturing at 95°C for 15 sec, annealing
at 60°C for 60 sec, and 95°C for a final 15 sec. PCR was performed
using the Mastercycler nexus (Eppendorf, Hamburg, Germany). Data
were analyzed using the comparative Cq method (2−ΔΔCq)
(30). Three independent experiments
were performed for each clone.
Western blot analysis
Western blot analysis was performed as previously
described (31). The cells were
homogenized in radioimmunoprecipitation assay lysis buffer (Thermo
Fisher Scientific, Inc.) and heated to 100°C for 10 min prior to
analysis. Protein concentration is performed using the Bradford
assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). In total, 30
µg of protein mixture from cells was loaded per lane and separated
by electrophoresis on an SDS-PAGE (10% gel). Proteins were then
transferred to nitrocellulose blotting membranes. Membranes were
blocked for 1 h with 5% milk. The membranes were then blotted with
anti-Livin (cat. no. ab97350; 1:1,000), anti-LC3 (cat. no. ab51520;
1:1,000), anti-p62 (cat. no. ab91526; 1:1,000), anti-caspase-3
(cat. no. ab13847; 1:1,000), anti-SMAC (cat. no. ab8115; 1:1,000),
anti-p-Akt (cat. no. ab81283; 1:1,000), anti-Akt (cat. no.
ab179463; 1:1,000), anti-Bcl-2 (cat. no. ab59348; 1:1,000 diluted)
and anti-actin (cat. no. ab1801 1:1,000 diluted) which were all
purchased from Abcam (Cambridge, UK) at 4°C over night. After
washing three times with TBST (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), membranes were incubated with corresponding
horseradish peroxidase-conjugated secondary antibodies (goat
anti-rabbit; cat. no. ab6721; 1,5,000; Abcam) for 2 h at room
temperature. Protein densitometry values were performed using an
open source image-processing program ImageJ 1.46r (National
Institutes of Health, Bethesda, MD, USA) according to a protocol
described previously (32). The
relative protein expression levels in different cell lines were
normalized to β-actin. Three independent experiments were performed
for each clone.
Cell viability assay
Cell viability was assessed using a Cell Counting
Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) assay, as described previously (33). Cells were seeded (5×103
cells/well) in 96-well microplates and grown in humidified
incubator (at 37°C and 5% CO2) overnight. Various
concentrations of 5-FU (10, 20, 40, 80 and 100 µM) were used to
determine the concentration that produced the optimal HCT116 and
SW620 cell response, following incubation for 24 h. The
concentrations of 5-FU that promoted a half-maximal growth
inhibition (IC50) are presented as the mean ± standard
deviation (SD), derived from triplicate samples of three
independent experiments. The roles of autophagy and apoptosis in
the Livin knockdown-induced recovery of 5-FU sensitivity were also
further defined. In this regard, following Lv-sh Livin
transfection, HCT-116 and SW620 cells were pretreated with an
autophagy inhibitor (3-MA, 3 mM) at 37°C for 2 h, or an apoptosis
inhibitor (Z-VAD-FMK, 40 µM) at 37°C for 1 h prior to 5-FU (20 µM)
treatment, as previously described (34,35).
Acridine orange (AO)/ethidium bromide
(EB) double staining
The DNA binding dyes AO and EB can be used for the
morphological detection of cell death (36). A cocktail of EB and AO (100 µg/ml) was
prepared in PBS. Following treatment with 5-FU (20 µM) at 37°C for
24 h, the cells were washed twice with PBS, followed by replacement
with fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.).
Following 30 min incubation at 37°C, the cells were washed again
with PBS, stained with AO/EB, and incubated at 37°C for 30 min in
the dark. The cells were washed with PBS and analyzed by
fluorescent microscopy (magnification, ×200) (36). The cell death rate (%) was calculated
as the percentage of positively stained cells, namely the number of
cells undergoing programmed cell death (per 100 cells). Three
independent experiments were performed for each clone.
Lactate dehydrogenase (LDH) assay
To further investigate the effect of Livin-knockdown
on the response of colon cancer cells to 5-FU, an LDH assay was
performed. LDH release from HCT116 and SW620 cells was determined
using the LDH Cytotoxicity Detection kit-PLUS (Roche Applied
Science, Penzberg, Germany), according to the manufacturer's
protocol. Briefly, cells were seeded in 96-well plates and cultured
at 37°C for 24 h with 20 µM 5-FU. The percentage of LDH released
from the treated cells was calculated by comparing it with the
maximum release obtained with 2% Triton X-100 treatment (positive
control) and spontaneous LDH release (mock-treated cells considered
as a negative control), as follows: Cytotoxicity (%) =
(experimental value - low control)/(positive control - low control)
×100.
Transmission electron microscopy
The colon cancer cells were seeded into 6-well
plates at 37°C for 48 h and then fixed in 3% glutaraldehyde in 0.1
M cacodylate buffer (Electron Microscope Sciences, Hatfield, PA,
USA) at 37°C for 1 h. Following fixation, the samples were
post-fixed in 1% osmium tetraoxide in the same buffer at 37°C for
30 min. Ultra-thin sections were then observed by transmission
electron microscopy (magnification, ×12,000).
Statistical analysis
Statistical analyses were performed using the
GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA,
USA). One-way analysis of variance (ANOVA) or two-way ANOVA, with
Tukey's or Bonferroni post hoc tests, were utilized to analyze the
differences between groups. Data are presented as the mean ±
standard error. P<0.05 was considered to indicate a
statistically significant difference.
Results
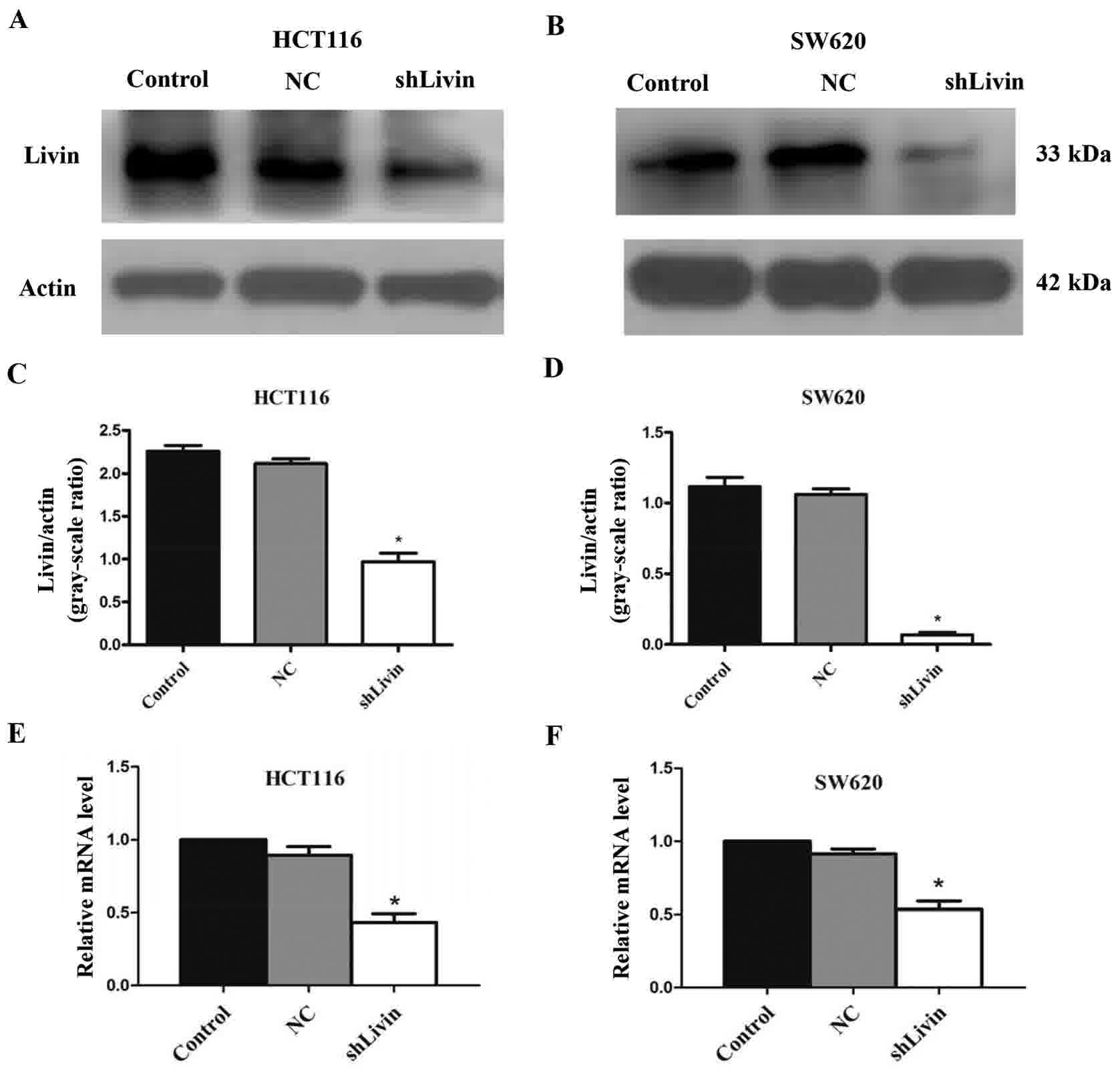
Protein and mRNA expression levels of
Livin are markedly decreased in Livin-knockdown HCT116 and SW620
cell lines
To confirm the effect of Livin knockdown in HCT116
and SW620 cell lines, the protein and mRNA expression levels of
Livin were evaluated by western blot analysis and RT-qPCR,
respectively. Western blot analysis indicated that Livin was
significantly down regulated in the shLivin group, compared with in
the NC and control groups, in HCT116 and SW620 cells. β-actin was
used as the loading control. All western blotting results are
representative of at least three independent experiments (Fig. 1A and B). Ratios between Livin to actin
in the shLivin group were significantly lower compared with the
control or NC groups (Fig. 1C and D).
Band densitometries were evaluated using ImageJ as aforementioned,
expression level comparisons of Livin and actin were compared using
two-way analysis of variance (P<0.05, shLivin group vs. control
or NC groups). In addition, a marked inhibition of Livin mRNA
expression was observed in the shLivin group compared with in the
NC and control groups (P<0.05, shLivin group vs. control or NC
groups; Fig. 1E and F). Each bar
represents the mean ± SD (n=3).
Silencing Livin increases the
chemotherapeutic sensitivity of colon cancer cells to 5-FU
AO-EB staining was performed to confirm that
silencing Livin increased the chemotherapeutic sensitivity of colon
cancer cells to 5-FU. Following treatment with 5-FU (20 µM) for 24
h, higher level of granular yellow-green nuclear AO staining and
concentrated orange nuclear EB staining, were detected in the
shLivin group compared with the control or NC groups (Fig. 2A). This indicated a higher level of
apoptosis. The cell death rate of shLivin group was significantly
higher compared with those of the control and NC groups (P<0.05;
Fig. 2B). These results confirmed
that silencing Livin enhanced 5-FU sensitivity in HCT116 and SW620
cell lines.
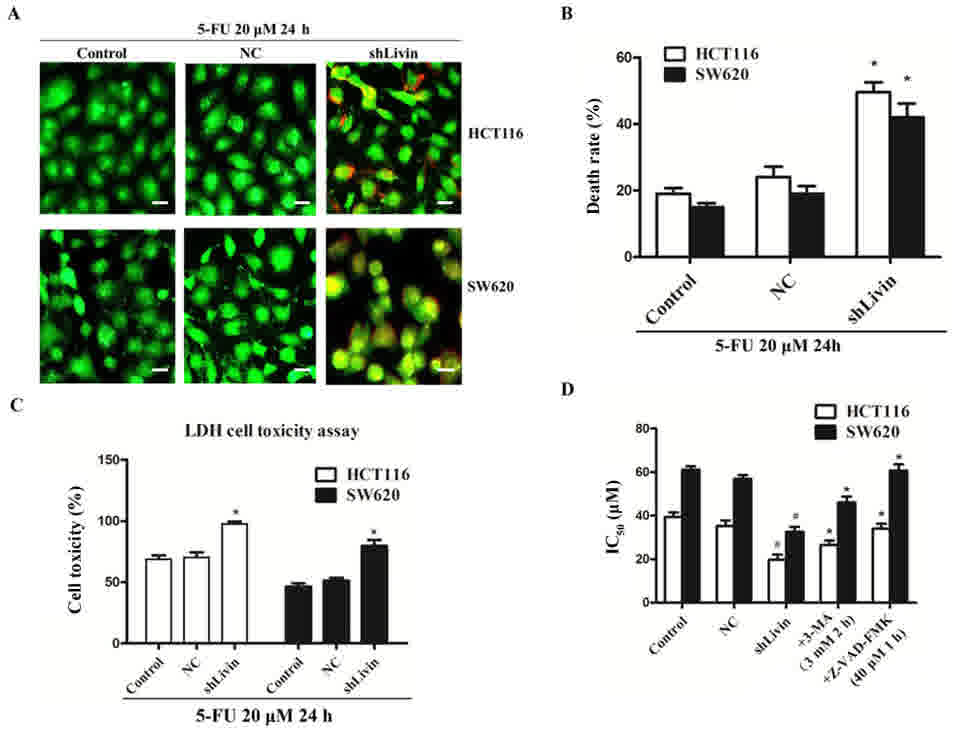 | Figure 2.Silencing Livin increased the 5-FU
sensitivity of colon cancer cells. (A) Acridine orange-ethidium
bromide staining was performed following treatment with 5-FU (20
µM) for 24 h. A greater number of stress granules were observed in
the cytoplasm of the shLivin group compared with in the control and
NC groups. Images are obtained fluorescence microscopy
(magnification, ×200). Scale bar, 100 µm. The results are
representative of ≥3 independent experiments. (B) The cell death
rate (%) was determined as the percentage of positively stained
cells. The cell death rate in the shLivin group was significantly
high, compared with those of the control and NC groups, following
treatment with 5-FU (20 µM) for 24 h (*P<0.05, shLivin group vs.
control or NC groups). (C) Results of the LDH assay for the shLivin
group compared with the control and NC groups. Silencing Livin
improved 5-FU chemotherapy sensitivities of HCT-116 and SW0620 cell
lines (*P<0.05, shLivin group vs. control or NC groups). (D) The
IC50 of the shLivin group was significantly lower
compared with the control and NC groups (#P<0.05,
shLivin group vs. control or NC groups). The IC50 of
3-MA and Z-VAD-FMK pretreated groups were significantly increased
compared with the non-pretreated shLivin group due to the
inhibition of autophagy and apoptosis, respectively (*P<0.05).
All data are presented as the mean ± standard deviation derived
from triplicate samples from three independent experiments. 5-FU,
5-fluorouracil; IC50, half-maximal inhibitory
concentration; NC, negative control; shLivin, lentivirus-short
hairpin Livin. |
To further investigate the effect of Livin knockdown
on the response of colon cancer cells to 5-FU, the LDH assay was
performed. The present study revealed that the shLivin group had a
significantly higher cell toxicity compared with the control and NC
groups, which indicated participation of Livin in 5-FU-induced cell
death (P<0.05; Fig. 2C).
Cell viability was evaluated by a CCK-8 assay.
Concentrations of 5-FU that resulted in a 50% growth inhibition are
indicated as the IC50 values (Fig. 2D). In the HCT116 cells, the
IC50 of the shLivin, NC and control groups after 24 h
were 19.733±4.127, 35.323±4.356 and 39.380±3.628 µM, respectively.
In the SW620 cell line, the IC50 of the shLivin, NC and
control groups after 24 h were 32.537±3.925, 56.880±3.082 and
61.120±2.661 µM, respectively. The IC50 of the shLivin
group was significantly lower, compared with those of the control
and NC groups (P<0.05; Fig. 2D).
Data are presented as the mean ± SD of three experiments.
Blockade of autophagy or apoptosis
inhibits Livin knockdown-induced recovery of 5-FU sensitivity in
HCT116 and SW620 cells
To further confirm the involvement of autophagy and
apoptosis in Livin knockdown-induced HCT116 and SW620 cell death,
the autophagy inhibitor 3-MA, and the apoptosis inhibitor
Z-VAD-FMK, were used to investigate whether Livin knockdown-induced
cell death may be attributed to autophagy and apoptosis. Following
Lv-sh Livin transfection into HCT-116 and SW620 cells, 3 mM 3-MA
was added 2 h prior to 24 h of 5-FU (20 µM) treatment. Cell
viability was evaluated with a CCK-8 assay; the IC50
values of pretreated groups were significantly increased to
26.583±3.387 and 46.027±4.832 µM in HCT-116 and SW620 cell lines,
respectively, which was significantly increased compared with the
non-pretreated shLivin group (P<0.05; Fig. 2D). Similarly, Z-VAD-FMK (40 µM) was
added to the culture for 1 h prior to 5-FU treatment; the
IC50 of pretreated groups were raised to 34.053±3.913
and 60.703±5.001 µM in the HCT-116 and SW620 cells, respectively,
which was significantly increased compared with in the
non-pretreated shLivin group (P<0.05; Fig. 2D). Data are presented as the mean ± SD
from three repeated experiments. Inhibitor pretreatments markedly
abrogated Livin knockdown-induced cell death, which indicated that
autophagy and apoptosis contributed to Livin knockdown-induced 5-FU
sensitivity recovery in HCT-116 and SW620 cells.
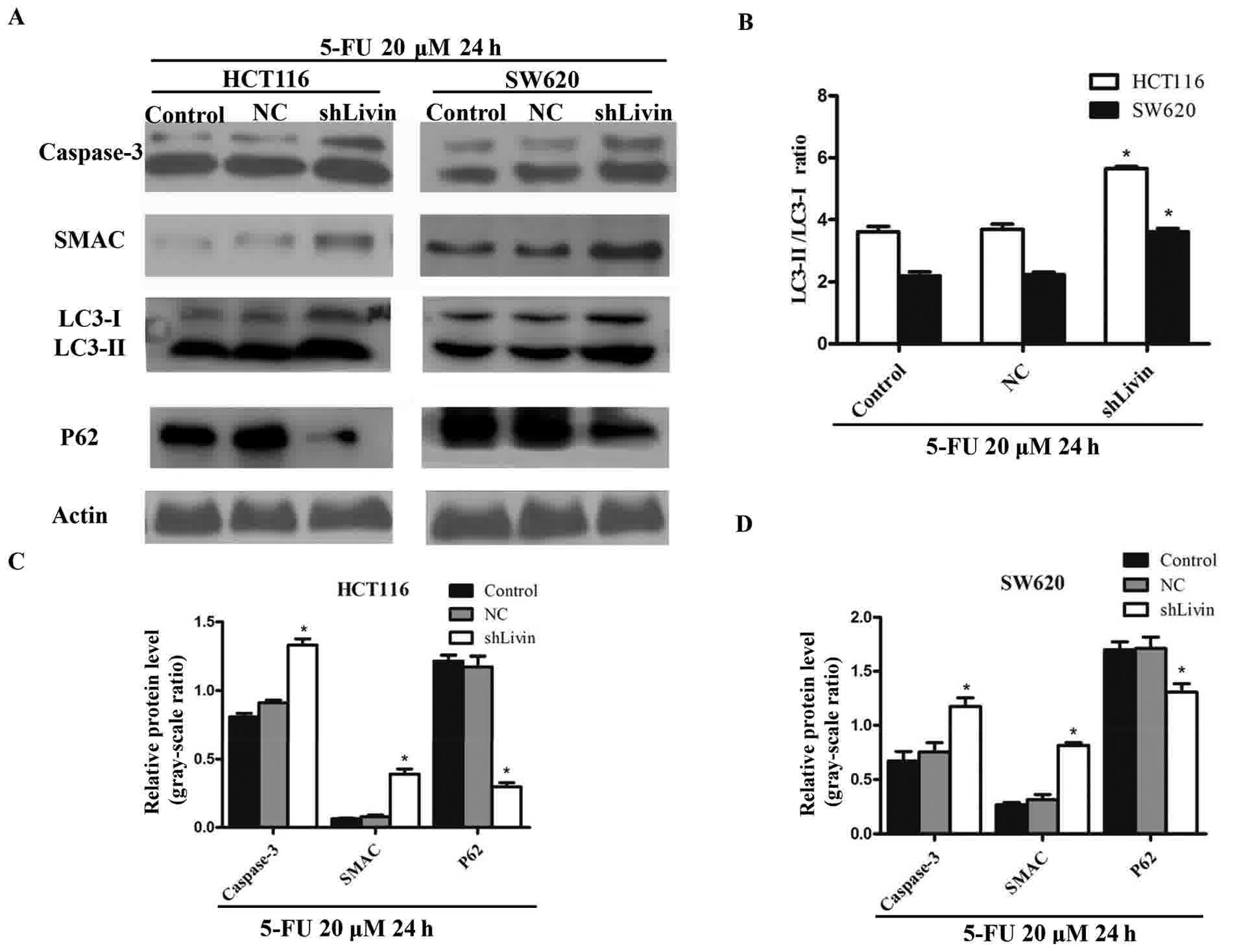
Silencing Livin induces the
upregulation of apoptosis and autophagy in HCT116 and SW620
cells
Western blot analysis was performed to determine the
expression levels of autophagic and apoptotic markers in the
control, NC and shLivin groups (Fig.
3A). The ratio of LC3-II to LC3-I, an established indicator of
autophagy, was markedly enhanced in the shLivin group, compared
with in the control and NC groups. Conversely, P62 remained at a
lower level in the shLivin group compared with in the control and
NC groups (Fig. 3B-D). Caspase-3 and
SMAC, crucial executioners of apoptosis, were highly upregulated
following Livin silencing, which indicated that apoptosis is
upregulated by Livin down regulation (P<0.05; Fig. 3C and D). These results indicated an
induction of apoptosis and autophagy mediated by Livin silencing.
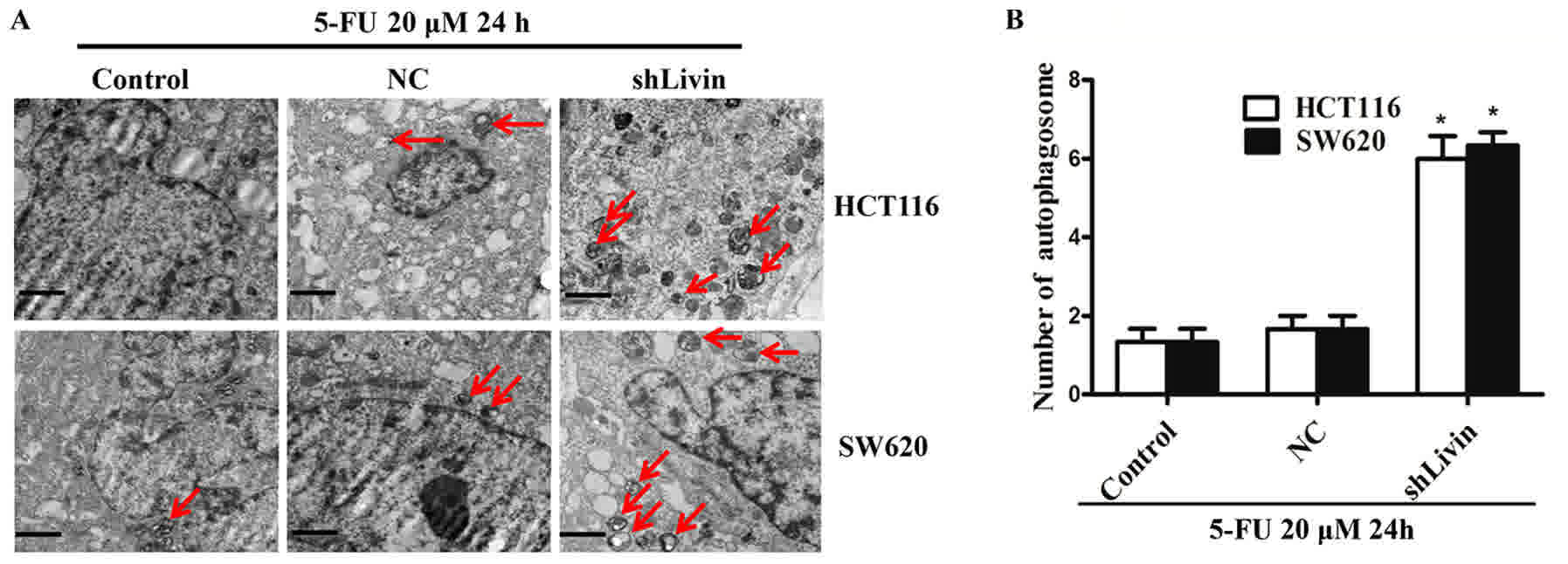
To confirm the activation of autophagy, the ultrastructure of cells
was observed with electron microscopy (Fig. 4A and B). A markedly higher number of
autophagosomes were observed in the shLivin group compared with in
the control and NC groups (Fig. 4A).
Quantification of autophagosomes from 12 random fields of view in
three independent experiments revealed that a significantly higher
number of autophagosomes in shLivin group compared with the control
and NC groups (P<0.05; Fig.
4B).
Inhibition of the Bcl-2 and Akt
signaling pathways is involved in cell death induced by Livin
silencing
Previous studies have demonstrated that Bcl-2 can
regulate autophagy and apoptosis due to cooperation with a number
of substances, including Ca2+ and autophagy protein 5
(Atg5) (23). The Search Tool for the
Retrieval of Interacting Genes/Proteins analysis revealed Livin and
associated protein-protein interactions in the literature
(confidence mode; http://string-db.org/). Livin, and the
autophagy/apoptosis markers Bcl-2 and Akt, were input into STRING
and a main cluster was formed. Blue lines represent interactions
between proteins, and line thickness denotes the confidence level
associated with each interaction. Within this cluster, Bcl-2 and
Akt, located in the key nodes and interact with each other, were
selected for further analysis (Fig.
5A). Western blot analysis was performed to detect the
expression levels of Bcl-2, p-Akt and T-Akt in the control, NC and
shLivin groups (Fig. 5B). The results
indicated that silencing Livin decreased the expression levels of
Bcl-2 and Akt (P<0.05; Fig. 5C and
D). These results suggested the involvement of Bcl-2 and the
Akt signaling pathway in cell death induced by Livin silencing.
Bcl-2 and the Akt signaling pathway may serve key roles in
regulating the crosstalk involved in silenced-Livin-induced
apoptosis and autophagy.
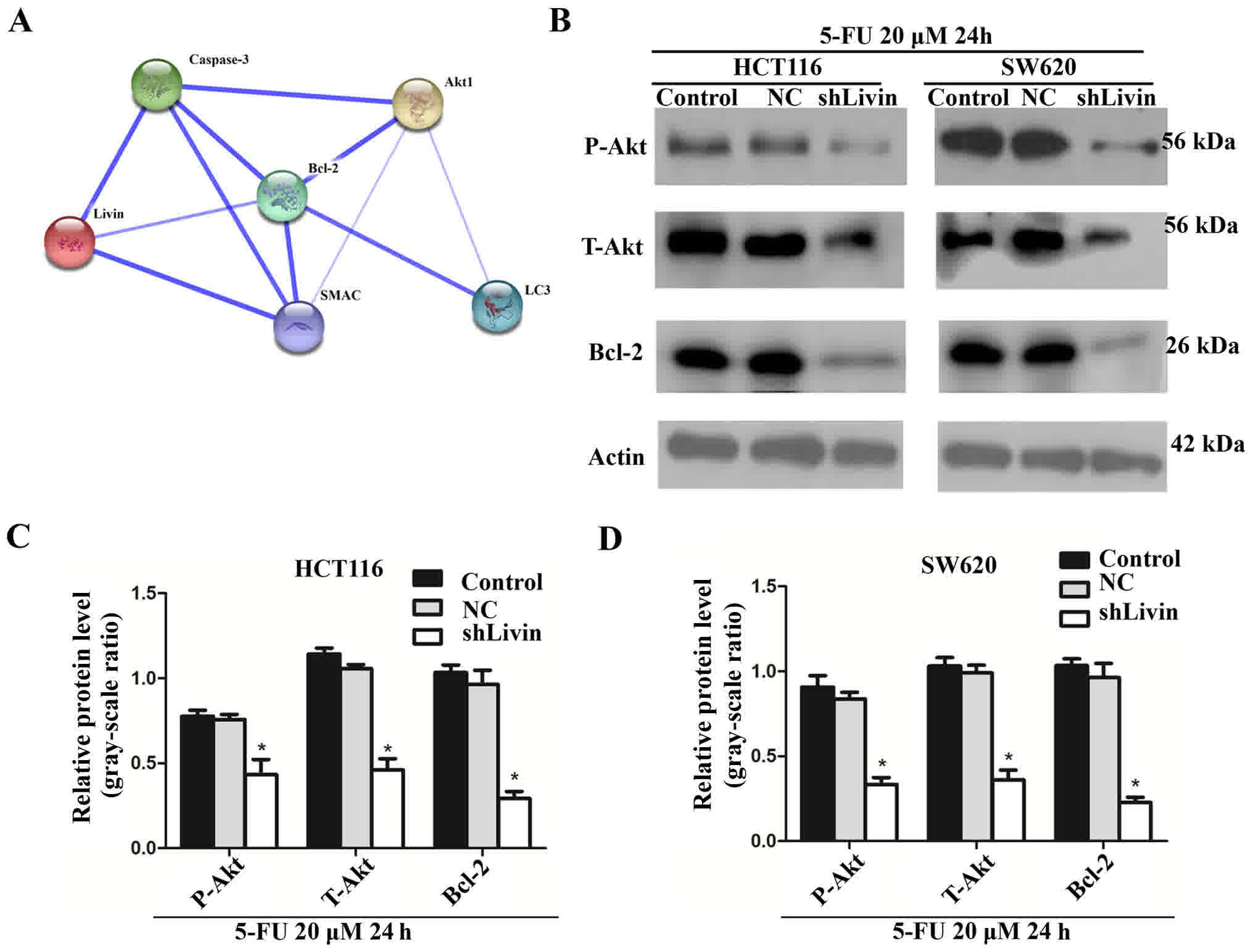 | Figure 5.Inhibition of Bcl-2 and the Akt
signaling pathway were involved in silenced-Livin-induced cell
death. (A) STRING analysis revealed Livin and associated
protein-protein interactions (confidence mode; http://string-db.org/). Within this cluster, Bcl-2
and Akt, which were located in the key nodes and mutually
interacted, were selected for further analysis. (B) HCT116 and
SW620 cells were incubated with 20 µM 5-FU for 24 h, harvested and
subjected to western blot analysis to detect the protein expression
level of Bcl-2, p-Akt and T-Akt from the control, NC and shLivin
groups. Images are representative of three independent experiments.
Histograms represent p-Akt, T-Akt and Bcl-2 protein expression
levels quantified by western blotting and the optical analysis
software ImageJ in (C) HCT116 and (D) SW620 cells (*P<0.05,
shLivin group vs. control or NC groups). Data are presented as the
mean ± standard deviation of three independent experiments. Bcl-2,
B-cell lymphoma-2; LC3, light chain 3; SMAC, Schizont
membrane-associated cytoadherence protein; Akt1, protein kinase B
1; p, phosphorylated; T, total; NC, negative control; shLivin,
lentivirus-short hairpin Livin; STRING, Search Tool for the
Retrieval of Interacting Genes/Proteins; 5-FU, 5-fluorouracil. |
Discussion
Livin is a crucial factor that contributes to
therapeutic resistance in various tumor types (37). Previous studies have proposed Livin as
a potential therapeutic target, owing to its high expression level
and association with tumor pathology and outcome (38). A previous study investigated the
expression levels of Livin in four colon cancer cell lines and
revealed that the overexpression of Livin inhibited the activation
of apoptosis in the chemotherapy-resistant cells (39). The present study confirmed that
silencing Livin may improve sensitivity to 5-FU chemotherapy. The
downregulation of Livin induced a high level of cell death, as
revealed by AO-EB staining. Furthermore, it was demonstrated that
this cell death was induced by apoptosis and autophagy. To the best
of our knowledge, no previous experiments have been performed to
analyze the association between Livin and the autophagic cell death
of colon cancer cells when treated with 5-FU.
Self-destructive cell processes, namely autophagy
and apoptosis, have been the focus of numerous researchers. Cell
apoptosis serves an important role in suppressing cancer cell
proliferation, which is involved in the activation of catabolic
enzymes in particular proteases in the signaling cascade and leads
to the rapid destruction of cellular structures and organelles
(40). Previous studies have revealed
that the apoptosis-associated gene Livin can regulate tumor cell
apoptosis induced by chemotherapy (41). The present study demonstrated the
upregulation of caspase-3 following Livin knockdown, which revealed
that silencing Livin enhanced the sensitivity of colon cancer cells
to 5-FU, leading to apoptosis. The use of the IAP (Z-VAD-FMK)
indicated that the IC50 of SVI-shLivin cells
significantly increased following inhibition of apoptosis by
Z-VAD-FMK. These results indicated that silencing Livin improved
the sensitivity of colon cancer cells to 5-FU, resulting in
apoptosis.
Autophagy is initially induced to allow cells to
adapt to stress; however, it also induces cell death in certain
cases (42). Previous studies
indicated that autophagy may be stimulated by numerous forms of
cellular stresses, including nutrient or growth factor deprivation
and hypoxia (43,44). Conversely, certain studies suggested
that autophagy may kill cancer cells via cellular digestion caused
by lysosomal enzymes (45,46). The results of the present study are
consistent with the latter hypothesis. The expression levels of
autophagy-associated proteins were detected via western blotting.
The results indicated an increase in LC3-II, and a decrease in
LC3-I, P62 and Bcl-2 expression levels in shLivin group compared
with control and NC groups. Electron microscopy was used to observe
the appearance of autophagosomes. The results indicated that there
was a markedly increased number of autophagosomes in the shLivin
group, compared with in the control and NC groups, which indicated
that autophagy was promoted by silencing Livin. To further confirm
the involvement of autophagic cell death, the inhibitor of
autophagy, 3-MA, was added. The results demonstrated that the
IC50 of SVI-shLivin cells was significantly increased
following the inhibition of autophagy. These results implied that
silencing Livin enhanced the sensitivity of colon cancer cells to
5-FU, leading to autophagic cell death.
The mechanisms underlying apoptosis and autophagy
vary, and the functional association/crosstalk between them is
complex (25). A previous study
revealed that Bcl-2 can regulate autophagy and apoptosis due to
cooperation with numerous substances, including Ca2+ and
Atg5, which may be involved in oncogenesis and tumor progression
(23). Based on the aforementioned
information, and on the co-participation of autophagic cell death
and apoptosis in the silenced-Livin-induced 5-FU sensitivity
improvement, Bcl-2 expression was evaluated in the Livin-silenced
cells. The result demonstrated a significant decrease in Bcl-2
protein expression level. Certain previous studies confirmed the
involvement of the Akt signaling pathway in the induction of
autophagy (47,48). The expression level of Akt was
detected by western blotting to verify whether it was involved in
the cell death induced by Livin silencing. The results indicated
that Akt expression was significantly decreased due to Livin
knockdown. The present study investigated the involvement of Bcl-2
and the Akt signaling pathway. Future studies are required to
investigate the specific mechanism underlying the decrease in Bcl-2
and Akt expression induced by Livin silencing, and to explore the
crosstalk between autophagic cell death and apoptosis processes
regulated by Bcl-2 and Akt.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shandong Key
Research and Development Project (grant nos. 2014GSF118134 and
2015GSF118055), the Medicine and Healthcare Technology Development
Project of Shandong Province (grant no. 2014WS0341), the Natural
Science Foundation of Shandong Province (grant nos. BS2010YY060,
ZR2014HM111 and ZR2014HP105) and the National Natural Science
Foundation of China (grant no. 81602227).
Availability of data and materials
The datasets generated and analyzed in the present
study are included in this published article.
Authors' contributions
CSL designed the experiments; SL and XL performed
the experiments and analyzed the data. QL, HJL, YLS, HQZ and HJZ
gave technical support and conceptional advice for the experiment
and data analysis. SL and XL wrote the manuscript in consultation
with QL, HJL, YLS, HQZ and HJZ. All authors approved the final
manuscript.
Ethics and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Greenlee RT, Hill-Harmon MB, Murray T and
Thun M: Cancer statistics, 2001. CA Cancer J Clin. 51:15–36. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Saunders M and Iveson T: Management of
advanced colorectal cancer: State of the art. Br J Cancer.
95:131–138. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu M, Liao M, Dai C, Chen JF, Yang CJ,
Liu M, Chen ZG and Yao MC: Sanguisorba officinalis L
synergistically enhanced 5-fluorouracil cytotoxicity in colorectal
cancer cells by promoting a reactive oxygen species-mediated,
mitochondria-caspase-dependent apoptotic pathway. Sci Rep.
6:342452016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
He S, Zhao Z, Yang Y, O'Connell D, Zhang
X, Oh S, Ma B, Lee JH, Zhang T, Varghese B, et al: Truncating
mutation in the autophagy gene UVRAG confers oncogenic properties
and chemosensitivity in colorectal cancers. Nat Commun. 6:78392015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang H, Tang J, Li C, Kong J, Wang J, Wu
Y, Xu E and Lai M: MiR-22 regulates 5-FU sensitivity by inhibiting
autophagy and promoting apoptosis in colorectal cancer cells.
Cancer Lett. 356:781–790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nachmias B, Ashhab Y and Ben-Yehuda D: The
inhibitor of apoptosis protein family (IAPs): An emerging
therapeutic target in cancer. Semin Cancer Biol. 14:231–243. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Endo T, Abe S, Seidlar HB, Nagaoka S,
Takemura T, Utsuyama M, Kitagawa M and Hirokawa K: Expression of
IAP family proteins in colon cancers from patients with different
age groups. Cancer Immunol Immunother. 53:770–776. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu B, Han M, Wen JK and Wang L:
Livin/ML-IAP as a new target for cancer treatment. Cancer Lett.
250:168–176. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xi RC, Biao WS and Gang ZZ: Significant
elevation of survivin and livin expression in human colorectal
cancer: Inverse correlation between expression and overall
survival. Onkologie. 34:428–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Myung DS, Park YL, Chung CY, Park HC, Kim
JS, Cho SB, Lee WS, Lee KH, Lee JH and Joo YE: Expression of Livin
in colorectal cancer and its relationship to tumor cell behavior
and prognosis. PLoS One. 8:e732622013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhuang L, Shen LD, Li K, Yang RX, Zhang
QY, Chen Y, Gao CL, Dong C, Bi Q, Tao JN, et al: Inhibition of
livin expression suppresses cell proliferation and enhances
chemosensitivity to cisplatin in human lung adenocarcinoma cells.
Mol Med Rep. 12:547–552. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ding ZY, Zhang H, Adell G, Olsson B and
Sun XF: Livin expression is an independent factor in rectal cancer
patients with or without preoperative radiotherapy. Radiat Oncol.
8:2812013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Grem JL: 5-Fluorouracil: Forty-plus and
still ticking. A review of its preclinical and clinical
development. Invest New Drugs. 18:299–313. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X, Xu J, Ju S, Ni H, Zhu J and Wang
H: Livin gene plays a role in drug resistance of colon cancer
cells. Clin Biochem. 43:655–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang SR, Hu GR, Fang LJ, Huang SJ, Li JS,
Zhao MY and Meng MJ: CpG oligodeoxynucleotides enhance
chemosensitivity of 5-fluorouracil in HepG2 human hepatoma cells
via downregulation of the antiapoptotic factors survivin and livin.
Cancer Cell Int. 13:1062013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Green DR: Apoptotic pathways: Ten minutes
to dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sui X, Jin L, Huang X, Geng S, He C and Hu
X: p53 signaling and autophagy in cancer: A revolutionary strategy
could be developed for cancer treatment. Autophagy. 7:565–571.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y,
Chen W, Wang K, Li D, Jin W, et al: JNK confers 5-fluorouracil
resistance in p53-deficient and mutant p53-expressing colon cancer
cells by inducing survival autophagy. Sci Rep. 4:46942014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lum JJ, DeBerardinis RJ and Thompson CB:
Autophagy in metazoans: Cell survival in the land of plenty. Nat
Rev Mol Cell Biol. 6:439–448. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang J and Yao S: JNK-Bcl-2/Bcl-xL-Bax/Bak
pathway mediates the crosstalk between matrine-induced autophagy
and apoptosis via interplay with Beclin 1. Int J Mol Sci.
16:25744–25758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cheng Y, Ren X, Zhang Y, Patel R, Sharma
A, Wu H, Robertson GP, Yan L, Rubin E and Yang JM: eEF-2 kinase
dictates cross-talk between autophagy and apoptosis induced by Akt
inhibition, thereby modulating cytotoxicity of novel Akt inhibitor
MK-2206. Cancer Res. 71:2654–2663. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Elbashir SM, Harborth J, Weber K and
Tuschl T: Analysis of gene function in somatic mammalian cells
using small interfering RNAs. Methods. 26:199–213. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McManus MT and Sharp PA: Gene silencing in
mammals by small interfering RNAs. Nat Rev Genet. 3:737–747. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Overbergh L, Kyama CM, Valckx D, Debrock
S, Mwenda JM, Mathieu C and D'Hooghe T: Validation of real-time
RT-PCR assays for mRNA quantification in baboons. Cytokine.
31:454–458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hirano S: Western blot analysis. Methods
Mol Biol. 926:87–97. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Janes KA: An analysis of critical factors
for quantitative immunoblotting. Sci Signal. 8:rs22015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang J, Guo H, Zhang H, Wang H, Qian G,
Fan X, Hoffman AR, Hu JF and Ge S: Putative tumor suppressor
miR-145 inhibits colon cancer cell growth by targeting oncogene
friend leukemia virus integration 1 gene. Cancer. 117:86–95. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu J, Li X, Tashiro S, Onodera S and
Ikejima T: Bcl-2 family proteins were involved in pseudolaric acid
B-induced autophagy in murine fibrosarcoma L929 cells. J Pharmacol
Sci. 107:295–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
An WW, Wang MW, Tashiro S, Onodera S and
Ikejima T: Norcantharidin induces human melanoma A375-S2 cell
apoptosis through mitochondrial and caspase pathways. J Korean Med
Sci. 19:560–566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kasibhatla S, Amarante-Mendes GP, Finucane
D, Brunner T, Bossy-Wetzel E and Green DR: Acridine orange/ethidium
bromide (AO/EB) staining to detect apoptosis. CSH Protoc.
2006:pdb.prot44932006.PubMed/NCBI
|
|
37
|
Wang R, Lin F, Wang X, Gao P, Dong K, Zou
AM, Cheng SY, Wei SH and Zhang HZ: Silencing Livin gene expression
to inhibit proliferation and enhance chemosensitivity in tumor
cells. Cancer Gene Ther. 15:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Miura K, Fujibuchi W, Ishida K, Naitoh T,
Ogawa H, Ando T, Yazaki N, Watanabe K, Haneda S, Shibata C and
Sasaki I: Inhibitor of apoptosis protein family as diagnostic
markers and therapeutic targets of colorectal cancer. Surg Today.
41:175–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ding ZY, Liu GH, Olsson B and Sun XF:
Upregulation of the antiapoptotic factor Livin contributes to
cisplatin resistance in colon cancer cells. Tumor Biol. 34:683–693.
2013. View Article : Google Scholar
|
|
40
|
Hannun YA: Apoptosis and the dilemma of
cancer chemotherapy. Blood. 89:1845–1853. 1997.PubMed/NCBI
|
|
41
|
Lopes RB, Gangeswaran R, McNeish IA, Wang
Y and Lemoine NR: Expression of the IAP protein family is
dysregulated in pancreatic cancer cells and is important for
resistance to chemotherapy. Int J Cancer. 120:2344–2352. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12 Suppl 2:S1509–S1518. 2005. View Article : Google Scholar
|
|
43
|
Ogata M, Hino S, Saito A, Morikawa K,
Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K,
et al: Autophagy is activated for cell survival after endoplasmic
reticulum stress. Mol Cell Biol. 26:9220–9231. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pietrocola F, Pol J, Vacchelli E, Baracco
EE, Levesque S, Castoldi F, Maiuri MC, Madeo F and Kroemer G:
Autophagy induction for the treatment of cancer. Autophagy.
12:1962–1964. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gozuacik D and Kimchi A: Autophagy as a
cell death and tumor suppressor mechanism. Oncogene. 23:2891–2906.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bialik S and Kimchi A: Lethal weapons:
DAP-kinase, autophagy and cell death. Curr Opin Cell Biol.
22:199–205. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nakamura Y, Yogosawa S, Izutani Y,
Watanabe H, Otsuji E and Sakai T: A combination of
indole-3-carbinol and genistein synergistically induces apoptosis
in human colon cancer HT-29 cells by inhibiting Akt phosphorylation
and progression of autophagy. Mol Cancer. 8:1002009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li S, Chen JW, Xie X, Tian J, Deng C, Wang
J, Gan HN and Li F: Autophagy inhibitor regulates apoptosis and
proliferation of synovial fibroblasts through the inhibition of
PI3K/AKT pathway in collagen-induced arthritis rat model. Am J
Transl Res. 9:2065–2076. 2017.PubMed/NCBI
|