Introduction
Receptor tyrosine kinases (RTKs) serve pivotal roles
in tumor initiation and malignant progression in various types of
human cancers. Epidermal growth factor receptor (EGFR) is one of
the most characterized RTK and is overexpressed in approximately
50% of non-small cell lung cancers (NSCLCs) (1–4).
Activating EGFR mutations, typically the exon 19 deletion or L858R
point mutation, are also frequently observed. EGFR tyrosine kinase
inhibitors (TKIs) contribute to the treatment of lung cancer
patients harboring EGFR mutations in the clinical setting (5–7).
EGFR becomes activated by the asymmetric
homo-dimerization of intracellular TK domains and subsequent
tyrosine autophosphorylation (8,9). In
contrast to canonical activation, it has become evident that the
TK-independent non-canonical serine/threonine phosphorylation of
EGFR plays a key role in the regulation of EGFR activity.
Non-canonical EGFR regulation is triggered by various conditions,
including inflammatory cytokines, ultraviolet radiation, and
DNA-damaging agents (10–14). The treatment of cells with these
stimuli induces the clathrin-mediated endocytosis (CME) of EGFR,
which is triggered by the activation of p38, in a TK-independent
manner. It has recently been reported that the TK-independent
functions of EGFR contribute to the initiation of autophagy and
prevention of TNF-α-induced apoptosis (10,15,16).
Therefore, a more complete understanding of the TK-independent
functions of EGFR under cellular stress conditions is needed in the
field of EGFR biology and EGFR-targeting therapeutics.
Treatments with cytotoxic platinum-containing
agents, including cisplatin and carboplatin, are currently standard
chemotherapy for NSCLC patients (12,17). In
the present study, we characterized the cisplatin-induced
non-canonical phosphorylation and endocytosis of EGFR by focusing
on two major p38 target regions, Ser-1015 and Ser-1047, in lung
cancer cells.
Materials and methods
Antibodies and reagents
Phospho-specific antibodies against p38
(Thr-180/Tyr-182) and EGFR (Tyr-1068) were purchased from Cell
Signaling Technology. Monoclonal antibodies against phospho-EGFR
(Ser-1047) (clone 1H9) and EGFR (clone LA1) were obtained from
Abcam (Cambridge, UK) and EMD Millipore (Billerica, MA, USA),
respectively. Antibodies against EGFR (1005) and β-actin (C-11)
were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). A phospho-EGFR (Ser-1015) rabbit monoclonal antibody was
generated using the rabbit-immunospot array assay on a chip (ISAAC)
system (18). Recombinant human EGF
and TNF-α were obtained from R&D Systems, Inc. (Minneapolis,
MN, USA). Cisplatin (CDDP), gefitinib, and a Phos-tag ligand were
obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
SB203580, trametinib, and PD153035 were purchased from Merck KGaA
(Darmstadt, Germany). All chemical inhibitors were dissolved in
dimethyl sulfoxide (DMSO), and the final concentration of DMSO was
less than 0.1%.
Cell culture
PC-9 and RPC-9 cells were kind gifts from Dr Kiura
(Okayama University, Okayama, Japan). A549, PC-9, and RPC-9 cells
were maintained in RPMI-1640 medium supplemented with 10% fetal
calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 µg/ml
streptomycin at 37°C in 5% CO2. HeLa and 293 cells were
maintained in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal calf serum, 4 mM glutamine, 100 U/ml
penicillin, and 100 µg/ml streptomycin at 37°C in 5%
CO2.
Immunoblotting
Whole cell lysates were prepared as described
previously (19). Cell lysates were
resolved by SDS-PAGE and transferred to an Immobilon-P nylon
membrane (EMD Millipore). The membrane was treated with Block Ace
(Dainippon Pharmaceutical Co., Ltd., Suita, Japan) and proved with
the primary antibodies described above. Antibodies were detected
using horseradish peroxidase-conjugated anti-rabbit, mouse, or goat
immunoglobulin G (Dako; Agilent Technologies, Inc., Santa Clara,
CA, USA) and visualized with an enhanced chemiluminescence system
(GE Healthcare, Chicago, IL, USA). Some antibody reactions were
performed in Can Get Signal solution (Toyobo Life Science, Osaka,
Japan).
Zn2+-Phos-tag SDS-PAGE
Cell lysates were prepared with RIPA buffer [50 mM
Tris-HCl (pH7.4), 0.15 M NaCl, 0.25% sodium deoxycholate, 1% NP-40,
1 mM EDTA, 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1
mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, 10 µg/ml
aprotinin, and 10 µg/ml leupeptin]. Each sample was mixed with a
half volume of SDS-PAGE sample buffer (195 mM Tris-HCl (pH 6.8), 3%
SDS, 15% 2-mercaptoethanol, 30% glycerol, and 0.1% bromophenol
blue), and heated at 95°C for 5 min. The procedures for
Zn2+-Phos-tag SDS-PAGE were described previously
(20). In brief, the acrylamide
pendant phos-tag ligand and two equivalents of ZnCl2
were added to the separating gel before polymerization. The running
buffer consisted of 100 mM Tris and 100 mM MOPS containing 0.1% SDS
and 5 mM sodium bisulfite. After electrophoresis, the gel was
washed twice with a solution containing 25 mM Tris, 192 mM glycine,
10% methanol, and 1 mM EDTA for 20 min and then once with a
solution containing 25 mM Tris, 192 mM glycine, and 10% methanol
for 20 min. Gel transfer, blocking, the antibody reaction, and
detection were conducted according to the normal immunoblotting
protocol described above.
Transfection of plasmid DNAs
HeLa and 293 cells were transfected using
Lipofectamine 2000 and 3000, respectively (Thermo Fisher
Scientific, Inc., Waltham, MA, USA), in accordance with the
manufacturer's instructions. The pEGFP-N expression vector for the
dimer-deficient EGFR mutant (dd-EGFR) with DCR1/I682Q/V924R
mutations and its S1015A/T1017A/S1018A (R1m) and S1046A/S1047A
(R2m) mutants were generated by PCR with KOD FX Neo polymerase or
KOD-Plus-Neo polymerase (Toyobo Life Science).
RNA interference
A small interfering RNA (siRNA) against p38α (target
sequence; 5′-GCAUUACAACCAGACAGUUGAUAUU-3′) was synthesized by
Hokkaido System Science Co., Ltd. (Sapporo, Japan). Negative
control siRNA was purchased from Thermo Fisher Scientific, Inc.
A549 cells were transfected with siRNAs at a final concentration of
50 nM using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.).
Cells were used for experiments 72 h post-transfection.
Immunofluorescence
Cells were seeded on coverslip glass (Thermo Fisher
Scientific, Inc.). Two days after seeding, cells were incubated
with inhibitors and ligands or transfected with plasmid DNAs. Cells
were rinsed in cold PBS and fixed in 4% paraformaldehyde (PFA) at
room temperature for 15 min or in methanol at −20°C for 10 min.
After PFA fixation, cells were permeabilized in PBS containing 0.5%
Triton X-100 and washed by PBS. Cells were proved for 40 min with
primary antibodies and then washed and incubated with
isotype-specific secondary antibodies conjugated to Alexa Fluor
(nvitrogen; Thermo Fisher Scientific, Inc.) for 30 min. These
antibodies were diluted in PBS containing 0.5% BSA. Microscopy was
performed using a LSM 700 confocal microscope (Zeiss, Oberkochen,
Germany).
Results
Cisplatin induces the non-canonical
phosphorylation of EGFR
We previously reported that cisplatin induces the
non-canonical phosphorylation of EGFR at Thr-669 in the
juxtamembrane domain and Ser-1046/1047 in the C-terminal region via
the p38 and ERK pathways, respectively (11). We and others have also suggested the
importance of another p38 target, Ser-1015 in the endocytosis of
EGFR (13); however, this has not
been fully characterized under physiological conditions. Therefore,
we generated a phospho-specific monoclonal antibody against
Ser-1015. We demonstrated that cisplatin induced Ser-1015
phosphorylation with similar dynamics to previously reported
Ser-1047 phosphorylation in A549 (wild type EGFR), PC-9 (exon 19
deletion), RPC-9 (erlotinib-resistant PC-9 cells harboring the
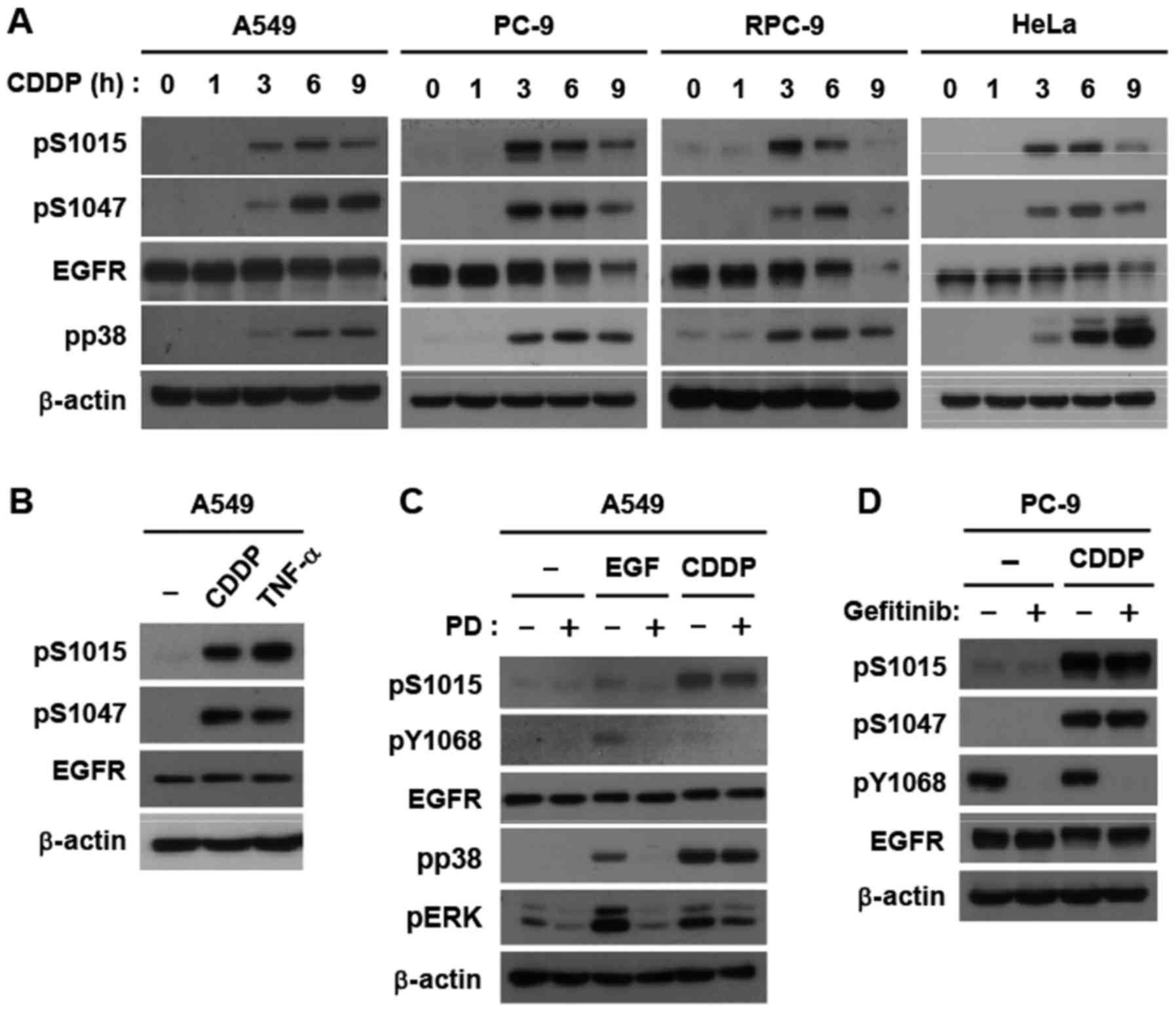
T790M secondary mutation), and HeLa cells (Fig. 1A). TNF-α also triggered Ser-1015
phosphorylation (Fig. 1B). In
addition, a stimulation with EGF weakly induced Ser-1015
phosphorylation in a TK-dependent manner; however, potent
phosphorylation by cisplatin was independent of TK activity in A549
cells (Fig. 1C). This correlated with
p38 activation (Fig. 1C). Similarly,
gefitinib, an EGFR-TKI, did not inhibit the cisplatin-induced
phosphorylation of Ser-1015 and Ser-1047, but completely suppressed
the constitutive phosphorylation of Tyr-1068, a major
autophosphorylation site, in PC-9 cells (Fig. 1D). Collectively, these results
demonstrated that Ser-1015 is a target site for the
cisplatin-induced signaling pathway in lung cancer cells.
p38 triggers the phosphorylation of
EGFR in cells treated with cisplatin
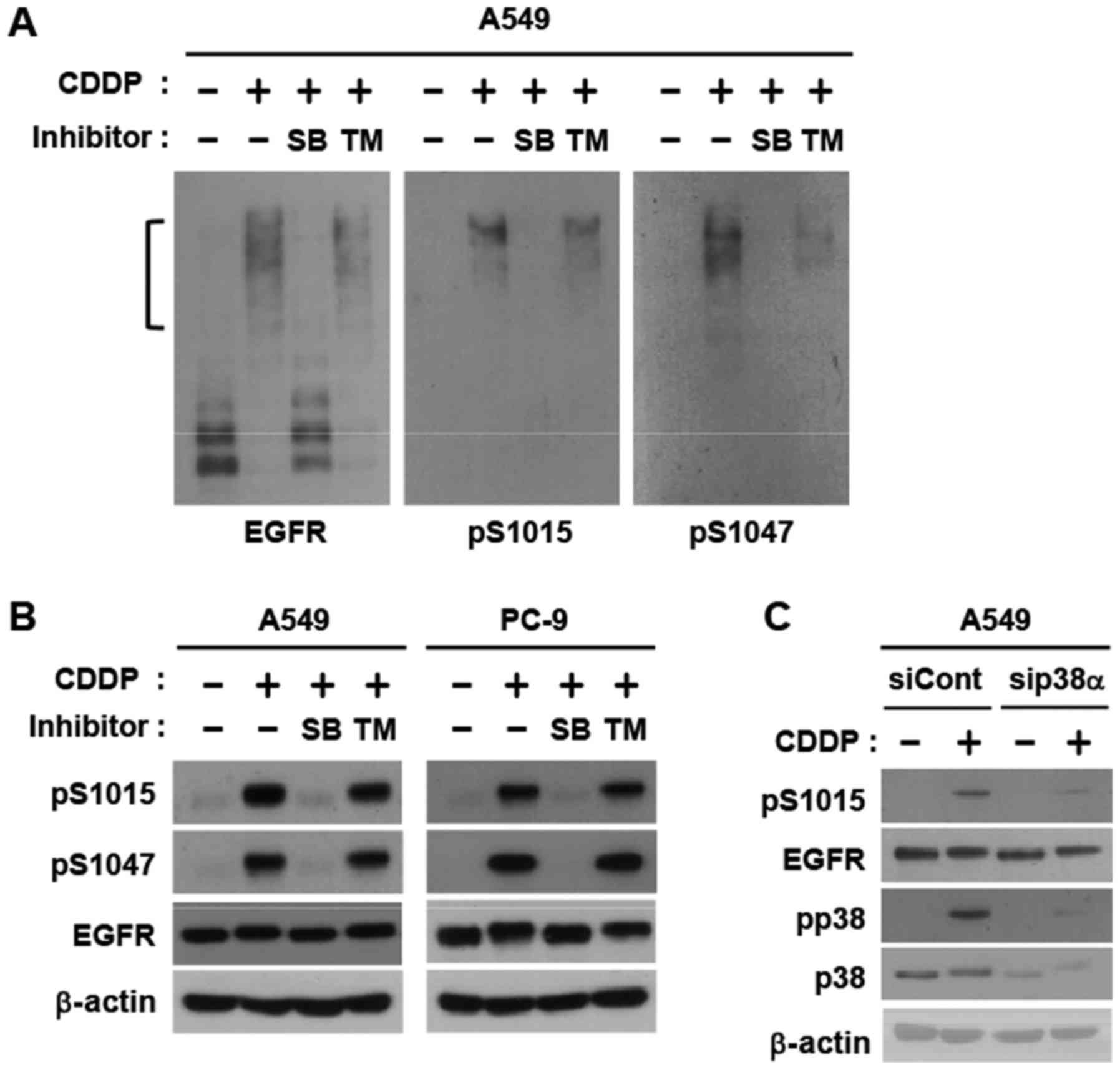
In order to elucidate the ratio of phosphorylated
EGFR upon the cisplatin stimulation, we employed immunoblotting
using a Phos-tag gel, which detects phosphorylated proteins as
shifted bands (21,22). Cisplatin caused band shifts in all
EGFR proteins expressed, indicating high phosphorylating activity
(Fig. 2A). Phospho-specific
antibodies against Ser-1015 and Ser-1047 detected the only shifted
EGFR band. SB203580, a p38 inhibitor, strongly abolished the band
shift, which correlated with the disappearance of phospho-specific
bands (Fig. 2A). A western blot
analysis using normal SDS-PAGE clearly demonstrated that the
phosphorylation of these two serines was completely inhibited by
SB203580, but not by trametinib, a MEK inhibitor (Fig. 2B). RNAi-mediated knockdown of p38α, a
major isoform, abrogated phosphorylation of Ser-1015 (Fig. 2C). These results indicate that severe
DNA-damaging conditions with cisplatin control most EGFR proteins
via the phosphorylation of two p38 target regions.
Cisplatin-induced EGFR endocytosis via
Ser-1015 phosphorylation
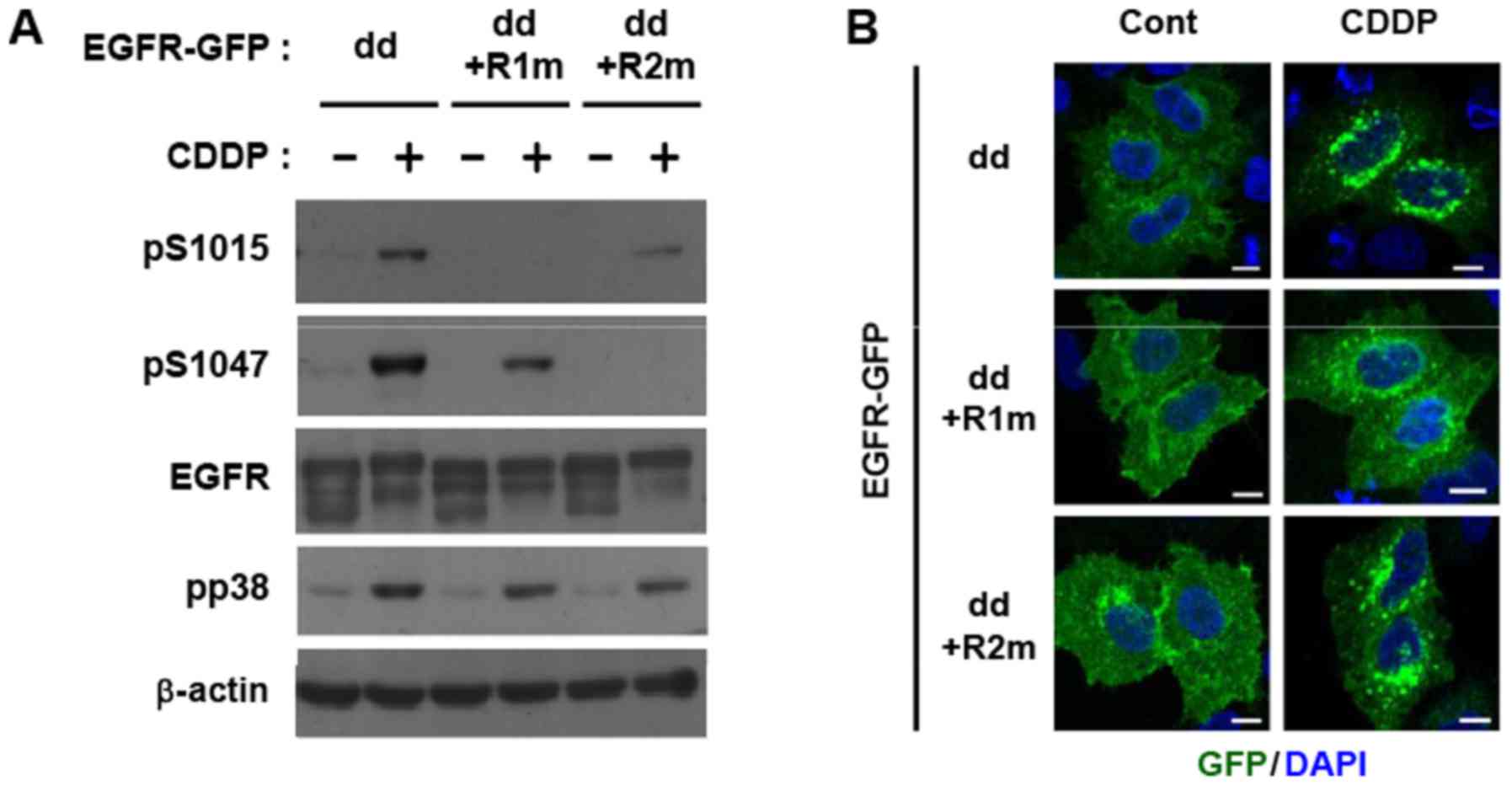
We recently demonstrated that TNF-α-induced
endocytosis occurred on inactive monomeric EGFR in a TK-independent
manner using a dimer-deficient EGFR mutant (dd-EGFR) lacking extra-
and intracellular dimerization sequences (9,18,23,24). We
detected the cisplatin-induced phosphorylation of dd-EGFR at
Ser-1015 and Ser-1047 (Fig. 3A). An
analysis using alanine substitution mutants of dd-EGFR at
Ser-1015/Thr-1017/Ser-1018 (region 1 mutant; dd-R1m) or
Ser-1046/Ser-1047 (dd-R2m) clarified the selectivity of antibodies
to the corresponding phosphorylation sites (Fig. 3A). Moreover, an immunofluorescence
analysis of GFP-tagged dd-EGFR demonstrated that cisplatin
augmented the endocytosis of EGFR monomers, shown as green doted
particles in the cytoplasm (Fig. 3B).
The cisplatin-induced endocytosis of dd-R1m, but not dd-R2m
(Fig. 3B) was reduced, indicating
that the p38 phosphorylation of region 1 containing Ser-1015
triggered non-canonical EGFR endocytosis under DNA-damaging
conditions.
Cisplatin-induced EGFR endocytosis in
lung cancer cells
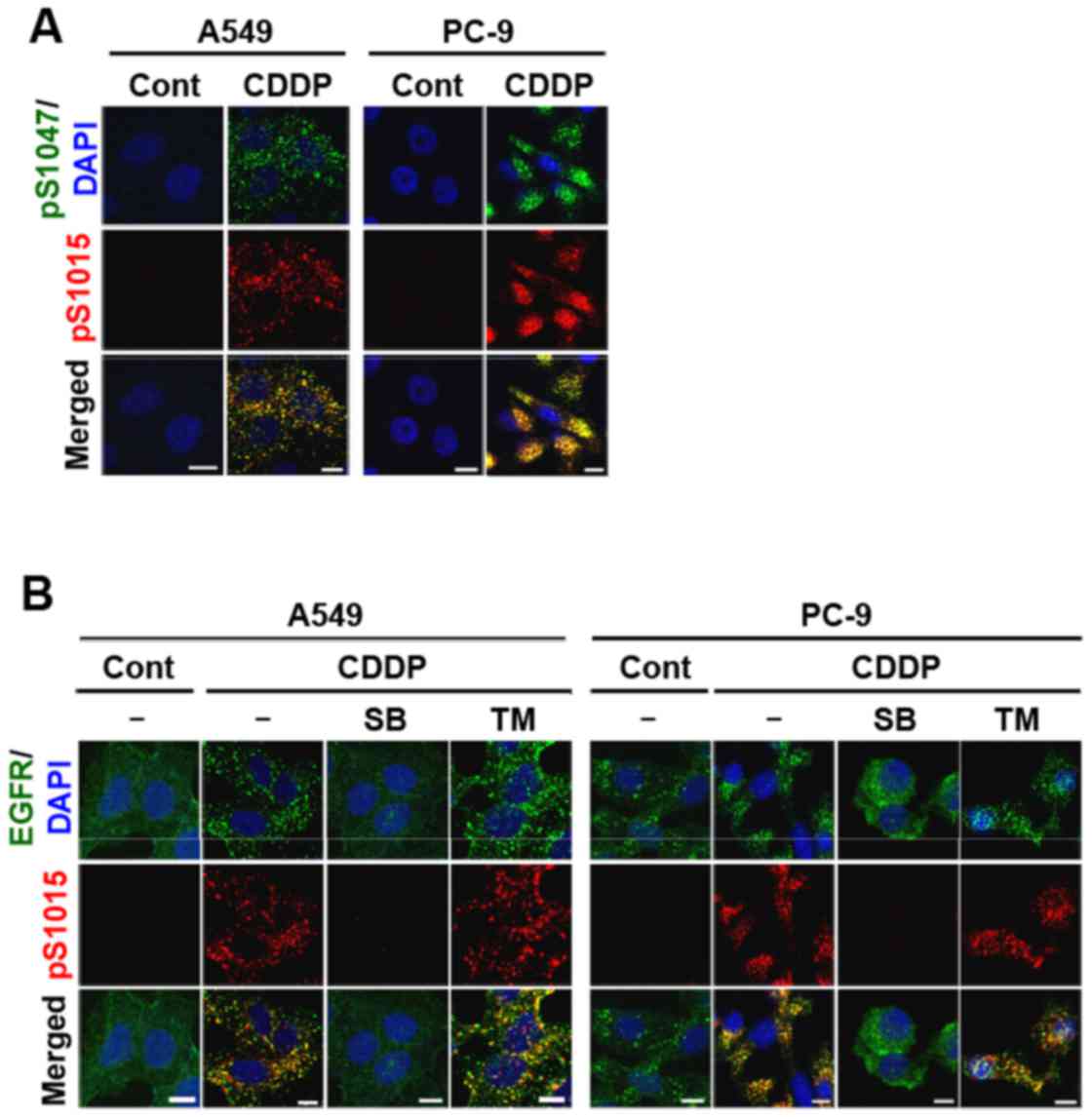
Internalized endogenous EGFR was stained with
pS1015, pS1047, and total EGFR antibodies in NSCLC cell lines. As
expected, pS1015-EGFR co-localized with pS1047-EGFR in A549 and
PC-9 cells (Fig. 4A). The staining
patterns of pS1015-EGFR and total EGFR also overlapped (Fig. 4B). Moreover, p38 inhibition, but not
MEK inhibition abolished cisplatin-induced EGFR endocytosis and
Ser-1015 phosphorylation (Fig. 4B).
Similar immunofluorescence staining was observed in
cisplatin-treated RPC-9 cells (our unpublished data). These results
indicate that although only Ser-1015 is involved in the
internalization process, Ser-1015 and Ser-1047 were both
simultaneously phosphorylated on internalized EGFR proteins via p38
activation.
Discussion
A phosphoproteomic analysis identified more than 30
Ser/Thr phosphorylation sites in the intracellular domain of EGFR
(25). Thr-669 in the juxtamembrane
domain is the most characterized site, is phosphorylated by ERK,
and is involved in the negative feedback regulation of tyrosine
kinase activity (26). Ser-1046 and
Ser-1047, target sites of the p38 pathway, are considered to
regulate receptor desensitization via the internalization of EGFR
(12,14,27).
Commercially available phospho-specific antibodies to these Ser/Thr
sites have been widely used in analyses of these sites. Another
important p38 target Ser-1015 is suggested to be involved in
receptor endocytosis (13,18); however, its physiological
characterization has not yet been conducted due to the lack of a
high-quality phospho-specific antibody. In the present study, we
demonstrated that Ser-1015 was phosphorylated with similar dynamics
to Ser-1047 phosphorylation using a newly generated antibody.
Nevertheless, only Ser-1015 was essential for the non-canonical
endocytosis of wild type EGFR as well as EGFR harboring primary and
secondary mutations. Taken together with previous findings on
Ser-1046/1047, cooperative regulation with Ser-1015 needs to be
reconsidered and the original unknown functions of Ser-1046/1047
elucidated.
Evidence for the physiological functions of
TK-inactive EGFR has been increasing. For example,
lysosomal-associated protein transmembrane 4B (LAPTM4B) binds to
inactive EGFR on endosomes, which participates in the initiation of
autophagy (16,28). Furthermore, cellular stress conditions
with p38 activation have been shown to affect autophagy (15). Distinct from activation with ligands,
EGFR phosphorylated by p38 is sorted to lipid lysobisphosphatidic
acid (LBPA)-rich perinuclear MVBs upon a UVC or cisplatin
stimulation, demonstrating that canonical and non-canonical EGFR
endocytic trafficking is involved in different cellular responses
(29). It is essential to investigate
the role of the MAPK-dependent non-canonical regulation of EGFR in
the initiation of autophagy in cisplatin-treated cancer cells.
p38-induced EGFR endocytosis is known to be
dependent on clathrin (12,30). In NSCLCs, clathrin light chain isoform
b (CLCb) is up-regulated, and this is associated with a poor
patient prognosis, particularly in the advanced stages of tumors.
This type of CME entirely depends on dynamin-1 (Dyn1) and increases
the activation of EGFR downstream pathways, particularly Akt
(31,32). However, the role of CLC in EGFR
trafficking has yet to be characterized; therefore, it is of
interest to investigate whether the CLCb/Dyn1 machinery is involved
in the p38-mediated endocytosis of Ser/Thr-phosphorylated EGFR in
NSCLC cells, and the findings obtained will contribute to our
understanding of new functions of non-canonical EGFR in tumor
malignant alterations.
Collectively, our results showing the precise
phosphorylation patterns of EGFR using a new phospho-specific
antibody will drive new research directions that will provide a
clearer understanding of the TK-independent functions of EGFR in
cisplatin-treated NSCLC cells.
Acknowledgements
We thank Dr Katsuyuki Kiura (Department of Allergy
and Respiratory Medicine, Okayama University Hospital, Okayama,
Japan) for providing PC-9 and RPC-9 cells.
Funding
This work was supported in part by JSPS KAKENHI
(grant no. JP16H04694), JSPS Core-to-Core Program, B. Asia-Africa
Science Platforms, and the Platform Project for Supporting Drug
Discovery and Life Science Research from the Japan Agency for
Medical Research and Development.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
TT contributed to the experimental design,
conduction of experiments, data analysis and interpretation and
manuscript writing. TO and AM contributed to the antibody
preparation. EO performed the cell culture experiments. HS
contributed to the experimental design, data analysis and
interpretation and manuscript writing. All authors approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yarden Y and Pines G: The ERBB network: At
last, cancer therapy meets systems biology. Nat Rev Cancer.
12:553–563. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Casaletto JB and McClatchey AI: Spatial
regulation of receptor tyrosine kinases in development and cancer.
Nat Rev Cancer. 12:387–400. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Health Quality Ontario: Epidermal growth
factor receptor mutation (EGFR) testing for prediction of response
to EGFR-targeting tyrosine kinase inhibitor (TKI) drugs in patients
with advanced non-small-cell lung cancer: an evidence-based
analysis. Ont Health Technol Assess Ser. 10:1–48. 2010.
|
|
5
|
Chung C: Tyrosine kinase inhibitors for
epidermal growth factor receptor gene mutation-positive non-small
cell lung cancers: An update for recent advances in therapeutics. J
Oncol Pharm Pract. 22:461–476. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bennasroune A, Gardin A, Aunis D, Cremel G
and Hubert P: Tyrosine kinase receptors as attractive targets of
cancer therapy. Crit Rev Oncol Hematol. 50:23–38. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mendelsohn J and Baselga J: Epidermal
growth factor receptor targeting in cancer. Semin Oncol.
33:369–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jura N, Endres NF, Engel K, Deindl S, Das
R, Lamers MH, Wemmer DE, Zhang X and Kuriyan J: Mechanism for
activation of the EGF receptor catalytic domain by the
juxtamembrane segment. Cell. 137:1293–1307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nishimura M, Shin MS, Singhirunnusorn P,
Suzuki S, Kawanishi M, Koizumi K, Saiki I and Sakurai H:
TAK1-mediated serine/threonine phosphorylation of epidermal growth
factor receptor via p38/extracellular signal-regulated kinase:
NF-{kappa}B-independent survival pathways in tumor necrosis factor
alpha signaling. Mol Cell Biol. 29:5529–5539. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Refaat A, Aminullah, Zhou Y, Kawanishi M,
Tomaru R, Abdelhamed S, Shin MS, Koizumi K, Yokoyama S, Saiki I and
Sakurai H: Role of tyrosine kinase-independent phosphorylation of
EGFR with activating mutation in cisplatin-treated lung cancer
cells. Biochem Biophys Res Commun. 458:856–861. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zwang Y and Yarden Y: p38 MAP kinase
mediates stress-induced internalization of EGFR: Implications for
cancer chemotherapy. EMBO J. 25:4195–4206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tong J, Taylor P and Moran MF: Proteomic
analysis of the EGFR interactome and post-translational
modifications associated with receptor endocytosis in response to
EGF and stress. Mol Cell Proteomics. 13:1644–1658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Adachi S, Natsume H, Yamauchi J,
Matsushima-Nishiwaki R, Joe AK, Moriwaki H and Kozawa O: p38 MAP
kinase controls EGF receptor downregulation via phosphorylation at
Ser1046/1047. Cancer Lett. 277:108–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan X, Lambert PF, Rapraeger AC and
Anderson RA: Stress-induced EGFR trafficking: Mechanisms, functions
and therapeutic implications. Trends Cell Biol. 26:352–366. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka T, Zhou Y, Ozawa T, Okizono R,
Banba A, Yamamura T, Oga E, Muraguchi A and Sakurai H:
Ligand-activated epidermal growth factor receptor (EGFR) signaling
governs endocytic trafficking of unliganded receptor monomers by
non-canonical phosphorylation. J Biol Chem. 293:2288–2301. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Refaat A, Zhou Y, Suzuki S, Takasaki I,
Koizumi K, Yamaoka S, Tabuchi Y, Saiki I and Sakurai H: Distinct
roles of transforming growth factor-beta-activated kinase 1
(TAK1)-c-Rel and interferon regulatory factor 4 (IRF4) pathways in
human T cell lymphotropic virus 1-transformed T helper 17 cells
producing interleukin-9. J Biol Chem. 286:21092–21099. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou Y, Yamada N, Tanaka T, Hori T,
Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I and
Sakurai H: Crucial roles of RSK in cell motility by catalysing
serine phosphorylation of EphA2. Nat Commun. 6:76792015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou Y, Tanaka T, Sugiyama N, Yokoyama S,
Kawasaki Y, Sakuma T, Ishihama Y, Saiki I and Sakurai H:
p38-Mediated phosphorylation of Eps15 endocytic adaptor protein.
FEBS Lett. 588:131–137. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kinoshita E and Kinoshita-Kikuta E:
Improved Phos-tag SDS-PAGE under neutral pH conditions for advanced
protein phosphorylation profiling. Proteomics. 11:319–323. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Q, Villeneuve G and Wang Z: Control
of epidermal growth factor receptor endocytosis by receptor
dimerization, rather than receptor kinase activation. EMBO Rep.
6:942–948. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garrett TP, McKern NM, Lou M, Elleman TC,
Adams TE, Lovrecz GO, Zhu HJ, Walker F, Frenkel MJ, Hoyne PA, et
al: Crystal structure of a truncated epidermal growth factor
receptor extracellular domain bound to transforming growth factor
alpha. Cell. 110:763–773. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hornbeck PV, Kornhauser JM, Tkachev S,
Zhang B, Skrzypek E, Murray B, Latham V and Sullivan M:
PhosphoSitePlus: A comprehensive resource for investigating the
structure and function of experimentally determined
post-translational modifications in man and mouse. Nucleic Acids
Res. 40:D261–D270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sato K, Shin MS, Sakimura A, Zhou Y,
Tanaka T, Kawanishi M, Kawasaki Y, Yokoyama S, Koizumi K, Saiki I
and Sakurai H: Inverse correlation between Thr-669 and constitutive
tyrosine phosphorylation in the asymmetric epidermal growth factor
receptor dimer conformation. Cancer Sci. 104:1315–1322. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Countaway JL, Nairn AC and Davis RJ:
Mechanism of desensitization of the epidermal growth factor
receptor protein-tyrosine kinase. J Biol Chem. 267:1129–1140.
1992.PubMed/NCBI
|
|
28
|
Tan X, Sun Y, Thapa N, Liao Y, Hedman AC
and Anderson RA: LAPTM4B is a PtdIns(4,5)P2 effector that regulates
EGFR signaling, lysosomal sorting and degradation. EMBO J.
34:475–490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tomas A, Vaughan SO, Burgoyne T, Sorkin A,
Hartley JA, Hochhauser D and Futter CE: WASH and
Tsg101/ALIX-dependent diversion of stress-internalized EGFR from
the canonical endocytic pathway. Nat Commun. 6:73242015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tomas A, Futter CE and Eden ER: EGF
receptor trafficking: Consequences for signaling and cancer. Trends
Cell Biol. 24:26–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen PH, Bendris N, Hsiao YJ, Reis CR,
Mettlen M, Chen HY, Yu SL and Schmid SL: Crosstalk between
CLCb/Dyn1-mediated adaptive clathrin-mediated endocytosis and
epidermal growth factor receptor signaling increases metastasis.
Dev Cell. 40:278–288.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Schmid SL: Reciprocal regulation of
signaling and endocytosis: Implications for the evolving cancer
cell. J Cell Biol. 216:2623–2632. 2017.PubMed/NCBI
|