Introduction
Solid pseudopapillary neoplasia of the pancreas
(SPN) is a rare pancreatic neoplasm accounting for <2% of
pancreatic exocrine tumors. SPN usually occurs in young female
patients during the second to third decade of life (1). In the majority of cases, the patient
presents with a mixed cystic-solid mass in any region of the
pancreas, which may compress adjacent organs and cause symptoms
(2). SPN is generally regarded as a
low-grade malignant tumor that is confined to the pancreas, for
which en-bloc resection is the primary therapeutic method (3). The prognosis is generally favorable,
except for patients with invasion and metastasis at the time of
diagnosis, which is reported in ~5% of cases (4,5).
Histopathologically, SPN displays solid
pseudopapillary areas intermixed with cystic regions, and consist
of uniform polygonal-shaped cells with oval nuclei and abundant
cytoplasm, with occasional pleomorphism and mitosis (6). The majority of SPNs are marked with
neuron-specific enolase, vimentin and α1-antitrypsin and are
immunohistochemically negative for cytokeratin AE1/AE3 (3). In addition, Ki-67 immunoreactivity is
associated with tumor aggressiveness and poorer patient prognosis.
A recent study has revealed that the nuclear accumulation of the
β-catenin protein is one of the pathological hallmarks of SPN
(7). Further investigations reported
somatic mutations to catenin β1 (CTNNB1) in the tumor
tissue, with the majority being point mutations at serine/threonine
residues that are phosphorylation sites of glycogen synthase
kinase-3β (GSK-3β), including at codons 33, 37 and 41, and the
flanking residues of codon 33 (codons 32 and 34) (8). The most common mutation was reported at
codon 32 (8).
β-catenin, encoded by CTNNB1, is centrally
involved in the Wnt signaling pathway, which serves physiological
roles in embryonic organ development (9). In differentiated cells, the cytosolic
level of β-catenin is kept low by a ubiquitination process driven
by phosphorylation of β-catenin at its clustered serine/threonine
residues in exon 3 by casein kinase 1 (CK1), and glycogen synthase
kinase 3 (GSK3), which are the scaffolding proteins that serve a
role in β-catenin ubiquitination and proteasomal degradation
(9). Mutations involving these
phosphorylation sites are involved in the molecular pathogenesis of
various pediatric embryonal tumors, including hepatoblastoma, Wilms
tumor, medulloblastoma and pancreatoblastoma (10–12). Codon
32, encoding for aspartic acid, is not the phosphorylation target
itself. However, point mutations at Asp32 have been reported in
various types of human cancer, including SPN (8). The juxtaposition of Asp32 mutation with
Ser33 may result in conformational changes that prevent effective
phosphorylation, hence the cytosolic retention of β-catenin and its
translocation into the nucleus (13).
Functional genetic studies focusing on codon 32
revealed a reduction in ubiquitination activity in D32G, D32N and
D32Y mutants when compared to the wild-type sequence (13,14).
However, details regarding structural changes caused by codon 32
mutations have not been purposed. The present study investigated
mutations to CTNNB1 detected in samples from three patients
SPN and used molecular dynamics (MD) simulation to attempt to
predict alterations to protein conformations caused by the
mutations.
Materials and methods
Samples and polymerase chain
reaction
The present study was approved by the Research
Ethics Committee of the Faculty of Medicine, Prince of Songkla
University (Hat Yai, Thailand) and patients provided written
informed consent agreeing to their inclusion. Snap-frozen tumor
specimens from three patients with SPN that underwent surgical
resection in Songklanagarind Hospital were retrieved for DNA
extraction. The cases included 1 male and 2 females, aged 12, 13
and 61 years, respectively. DNA extraction was done using GeneJET
genomic DNA purification kit (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), following manufacturer's protocol. A mutation
study covering the exon 2–4 of CTNNB1 was performed by
polymerase chain reaction and direct nucleotide sequencing using 2
primer sets designed by Koch et al (15) and the PCR conditions reported by the
study of Udatsu et al (16).
PCR polymerase was performed by using TopTaq Master Mix kit
(Qiagen, Hilden, Germany) with the condition as follows: 5 min at
95°C, 30 cycles (30 sec at 95°C, 30 sec at 58°C, 45 sec at 72°C)
and 10 min at 72°C. All amplicon was then purified by GeneJET PCR
Purification kit (Thermo scientific, Massachusetts, USA).
Nucleotide sequencing was performed by the Scientific Equipment
Center, Prince of Songkla University. Mutations to CTNNB1 in each
case involved codon 32, consisting of two incidences of D32A and
one of D32Y (Table I).
 | Table I.Characteristics of the solid
pseudopapillary neoplasias that were used in the present study. |
Table I.
Characteristics of the solid
pseudopapillary neoplasias that were used in the present study.
| Patient | CTNNB1
mutation | Tumor details | Case data |
|---|
| 1 | D32A | A 6-cm cyst at the
pancreatic head with pancreatic duct dilatation | 12-year-old male,
alive 16 years following surgery |
| 2 | D32Y | A 3-cm cyst at the
pancreatic body with bilateral pleural effusion | 61-year-old female,
recurrence at 8 years following surgery |
| 3 | D32A | A 5-cm
well-encapsulated cyst at the head of the pancreas | 13-year-old female,
alive 4 years following surgery |
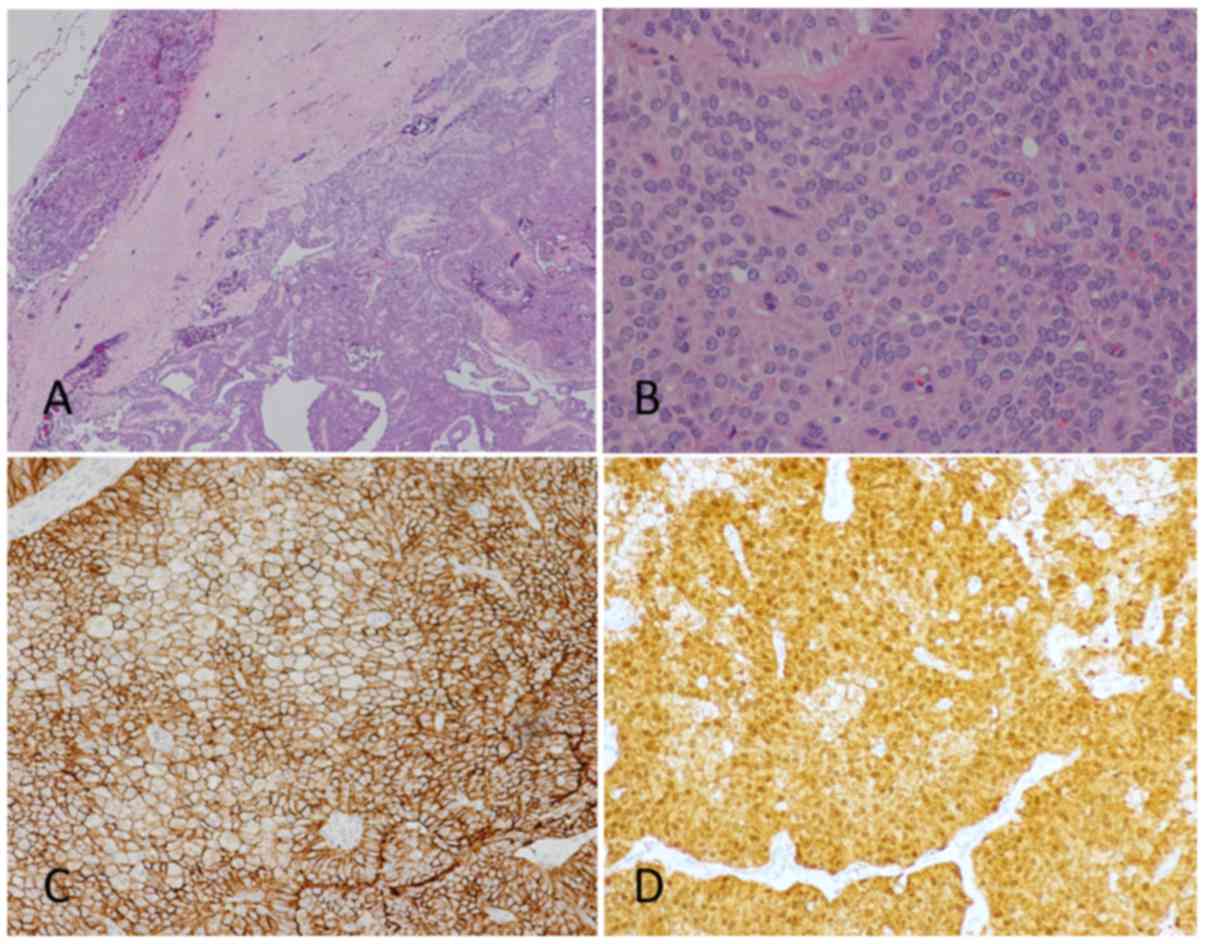
Immunohistochemistry
Immunohistochemistry of the tumor tissue used the
protocol presented in our previous work (17). Sections of 3-µm thickness were stained
using β-catenin monoclonal antibody (cat. no. 6B3; BD Biosciences,
Franklin Lakes, NJ, USA) at a 1:1,000 dilution. Detection was then
performed by using Bond Polymer Refine Detection system (Leica,
Ltd., Milton Keynes, UK). This included all reagents: Peroxide
blocking reagent (3–4%), polymer anti-mouse poly-HRP-IgG (<25
µg/ml) containing 10% (v/v) animal serum in tris-buffered
saline/0.09% ProClin™ 950, DAB Part 1 (66 mM 3,3′-diaminobenzidine
tetrahydrochloride hydrate, in a stabilizer solution), DAB Part B
(≤0.1% (v/v) hydrogen peroxide in a stabilizer solution), and
hematoxylin. The protocol used in this study was performed as
followed: After endogenous peroxidase blocking, slides were
incubated at room temperature with primary antibody for 120 min,
followed by a 30 min incubation at room temperature with peroxidase
labeled polymer conjugated to rabbit anti-mouse immunoglobulins.
Color was then developed by the liquid 3,3′-diaminobenzidine
tetrahydrochloride hydrate (DAB) chromogen. Counterstaining was
performed with hematoxylin and imaged using a light microscope
(magnification, ×20 and ×40). All procedures were performed
according to the Bond Polymer Refine Detection system manufacturer
protocol. Nuclear accumulation of β-catenin was demonstrated in the
tissue of all cases (Fig. 1).
MD simulation of wild-type and mutated
forms of β-catenin
To elucidate the role of the aforementioned
mutations on β-catenin function, a three-dimensional (3D) structure
was required. The wild-type β-catenin template was adopted from the
first structure of an experimental nuclear magnetic
resonance-derived structure from a rabbit (pdb code, 2G57), in
which the sequence identity in the region of interest is identical
to its human counterpart. As mutant structures of human β-catenin
are not available, bioinformatics tools were introduced in the
present study. In addition, MD simulation was performed to
investigate the effects of point mutations on the protein
conformation in detail.
The structure prediction was commenced using
residues 11–50 of the chosen β-catenin sequence. The residue
numbers refer to those of the human β-catenin protein sequence
(http://www.uniprot.org/uniprot/P35222). For each
β-catenin (wild-type, D32A or D32Y), a secondary structure was
independently predicted using two bioinformatics tools, PSIPRED
Protein Sequence Analysis Workbench (18,19) and
NetSurfP version 1.1 (20). A
prediction of the 3D structure of the wild type was performed using
PEP-fold server (21–23), using the structure of rabbit β-catenin
as a molecular template. The wild-type structure was finally chosen
from 50 possible structures corresponding to the aforementioned
predicted secondary structure predictions. For the mutant, the 3D
structures of D32A and D32Y were constructed using the point
mutation module of the Rosetta Backrub server (23), and the aforementioned wild-type
structure was exploited as a template. The selection of the
predicted structure was performed using the best result from 50
possible structures based on the highest prediction score.
Each constructed β-catenin structure was solvated in
the TIP3P water rectangular box, ~4,400 water molecules, with a
distance of 12 Å from the protein surface using the command from
tleap module in AMBER16 package. Sodium chloride was then added
into the system to make it equivalent to 0.15 M NaCl solution.
Protonation states in all ionizable amino acid side chains were set
at pH 7. The protein-solution system was modeled using AMBER10
force field, which was included in an AMBER 16 package (24). Prior to MD simulation, the system was
energy-minimized to remove unusual inter-atomic contacts, using the
steepest descent method for 2,000 steps. The system was then
equilibrated in a constant number (N), volume (V), and temperature
(T) (NVT) ensemble for 600 psec, where the protein was
position-restrained using force constants of 250, 150, 100, 50, 20,
and 10 kcal/mol/Å2 for each 100 psec simulation. A time
step of 1 sec was applied in each NVT run and a temperature of 310
K (37°C) was controlled using Langevin dynamics (25). The simulation was subsequently
switched to an isobar/isothermal (constant number (N), pressure
(P), and temperature (T); NPT) ensemble, with a temperature of 310
K and a pressure of 1 atm, regulated by Berendsen algorithms
(26). Short- and long-range
interactions were computed using a 12 Å cutoff and electrostatic
forces were computed using Lennard-Jones 6–12 potential and
Particle Mesh Ewald (PME) method (27), in which both are implemented in the
AMBER16 simulation program. The NPT simulation was performed for
200 nsec, with a time step of 2 fsec. The energy minimization and
MD simulations were performed using SANDER and PMEMD modules,
respectively, using the AMBER 16 package (28). The first 100-nsec NPT simulation was
omitted as an equilibrated phase and 100 equidistant snapshots from
the last 100 nsec were taken for analysis. The simulations of
wild-type and mutant β-catenins followed an identical protocol. All
structure visualization was performed using Visual Molecular
Dynamics package version 1.9.1 (29)
which is freely available online from Theoretical and Computational
Biophysics Group of University of Illinois at Urbana Champaign.
Results
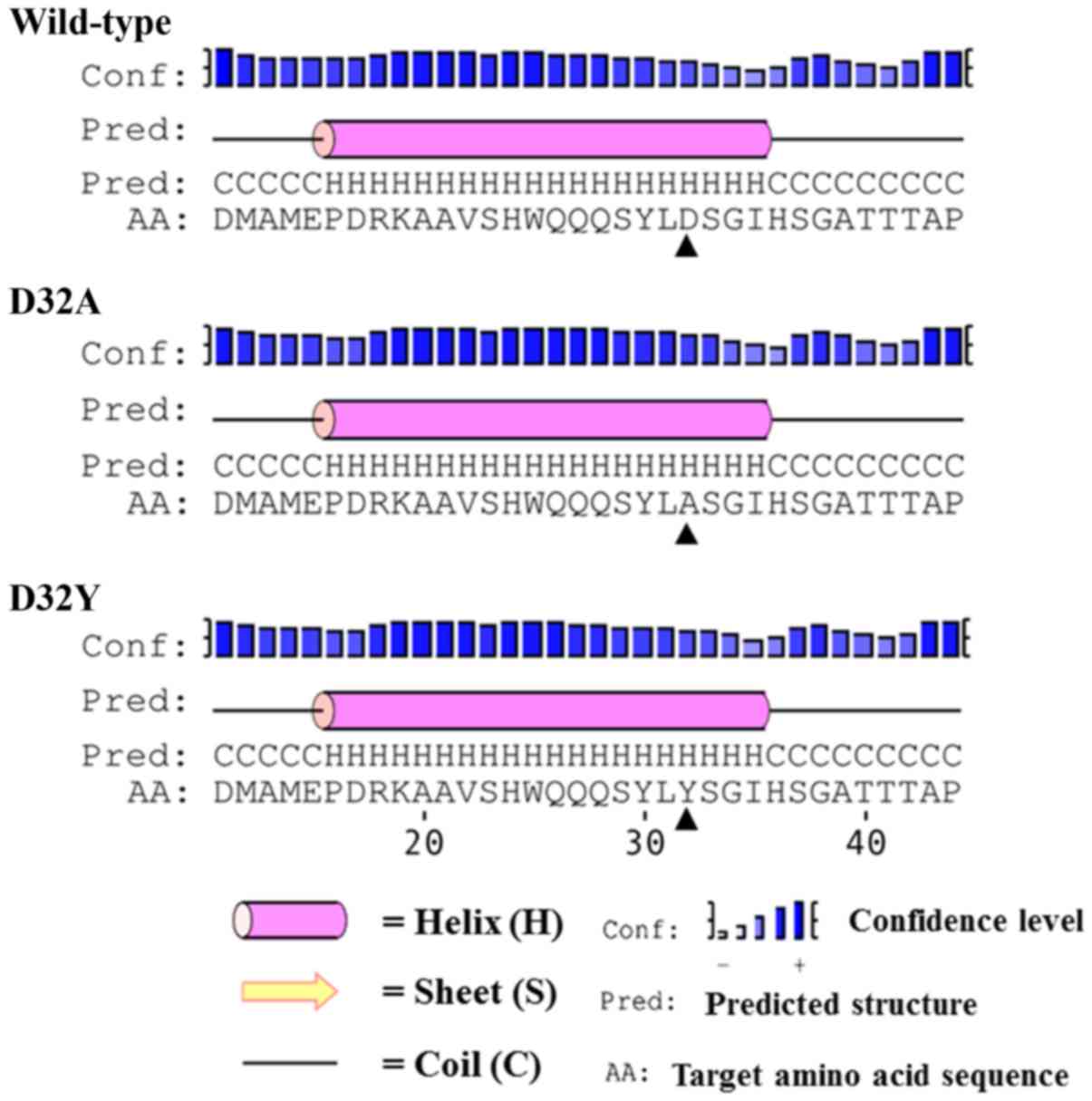
Each modeled tertiary structure of the β-catenin
fragment (M11-G50) was in good agreement with the predicted
secondary structure of its counterpart (Fig. 2). Since the protein fragment (M11-G50)
also consists of S33, an important phosphorylation site for
β-catenin functions (30–34), the simulation was performed with the
hypothesis that the domain requires a specific conformation for
phosphorylation to occur, and a point mutation could affect the
phosphorylation by inducing a conformational change. Therefore,
three MD simulations (wild-type, D32A, and D32Y) were performed to
visualize the β-catenin conformations under in vivo,
aqueous-phase conditions at 37°C. Conformational abnormalities in
mutant proteins may interfere with the phosphorylation of S33 and
eventually contribute to β-catenin dysfunction. Additionally, the
secondary structure of each residue in β-catenin residue was
plotted against the simulation time to investigate the
conformational changes of the 3D structure at the S33 site.
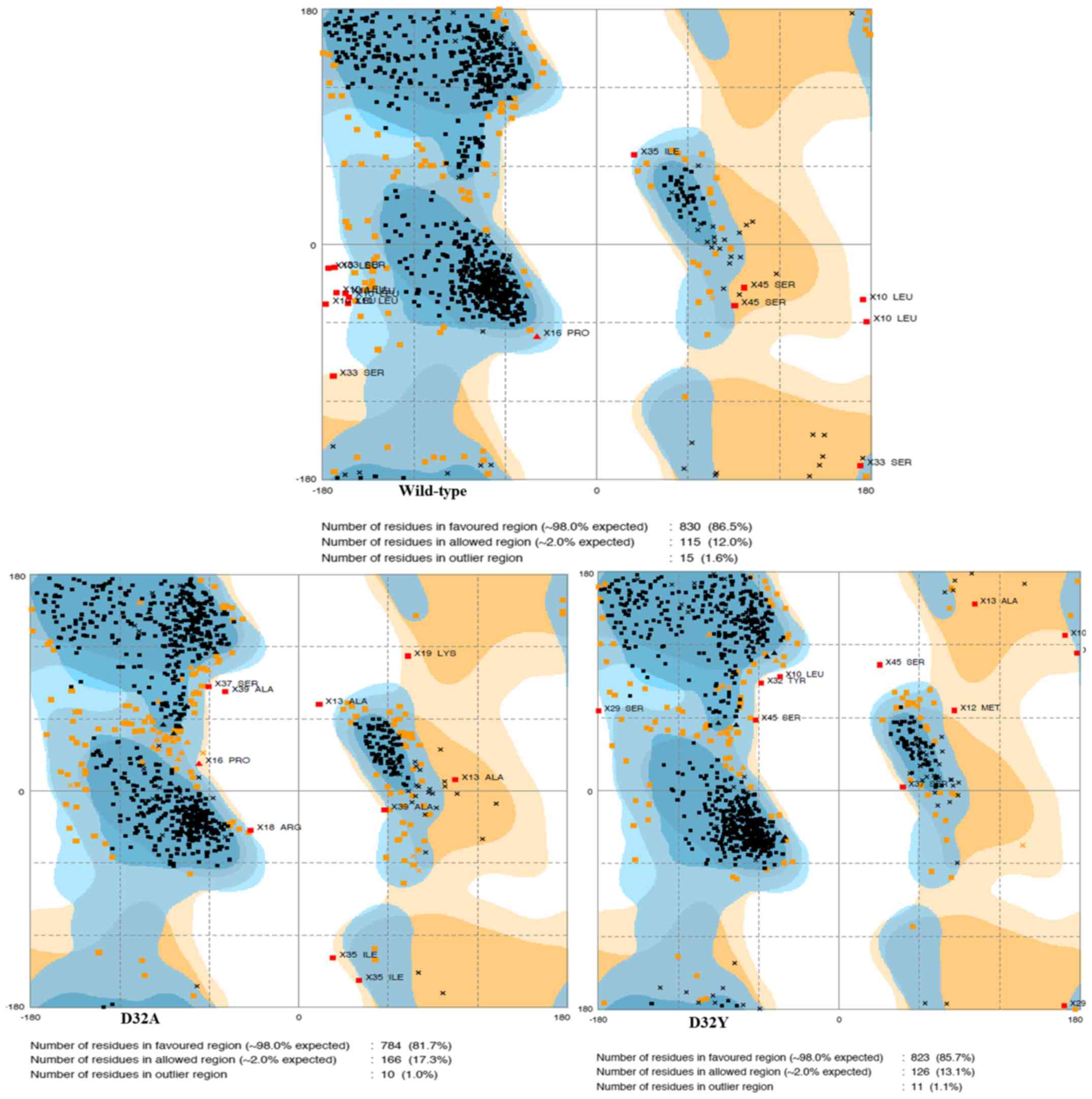
The stability of structural dynamics was observed
via the Ramachandran plot from average structure, rather than the
root-mean-square-displacement, owing to the high flexible coil in
the fragment structure. The plot illustrated that <1% of the
overall amino acids in the dynamics trajectories fell into the
outlier (disallowed) region (Fig. 3).
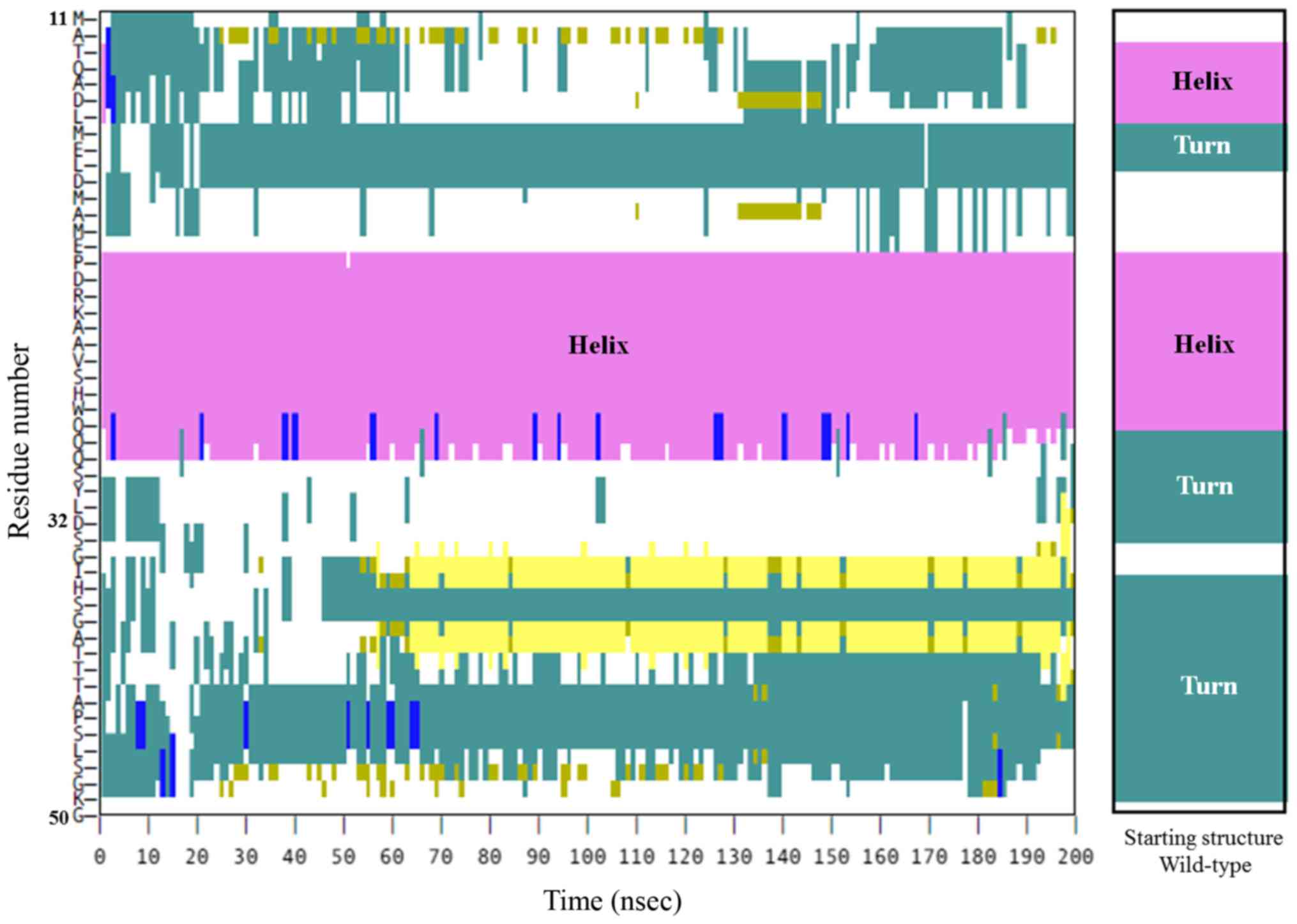
Initially, the P16-H28 fragment was predicted to be a helical
structure in the first place (Fig.
2). In the wild-type protein, after 100 nsec, the secondary
structure of P16-H28 remained helical, similar to the starting
structure (Fig. 4). These results
indicated that the helical structure of this protein fragment
(P16-H28) is a prerequisite to S33 phosphorylation. A similar
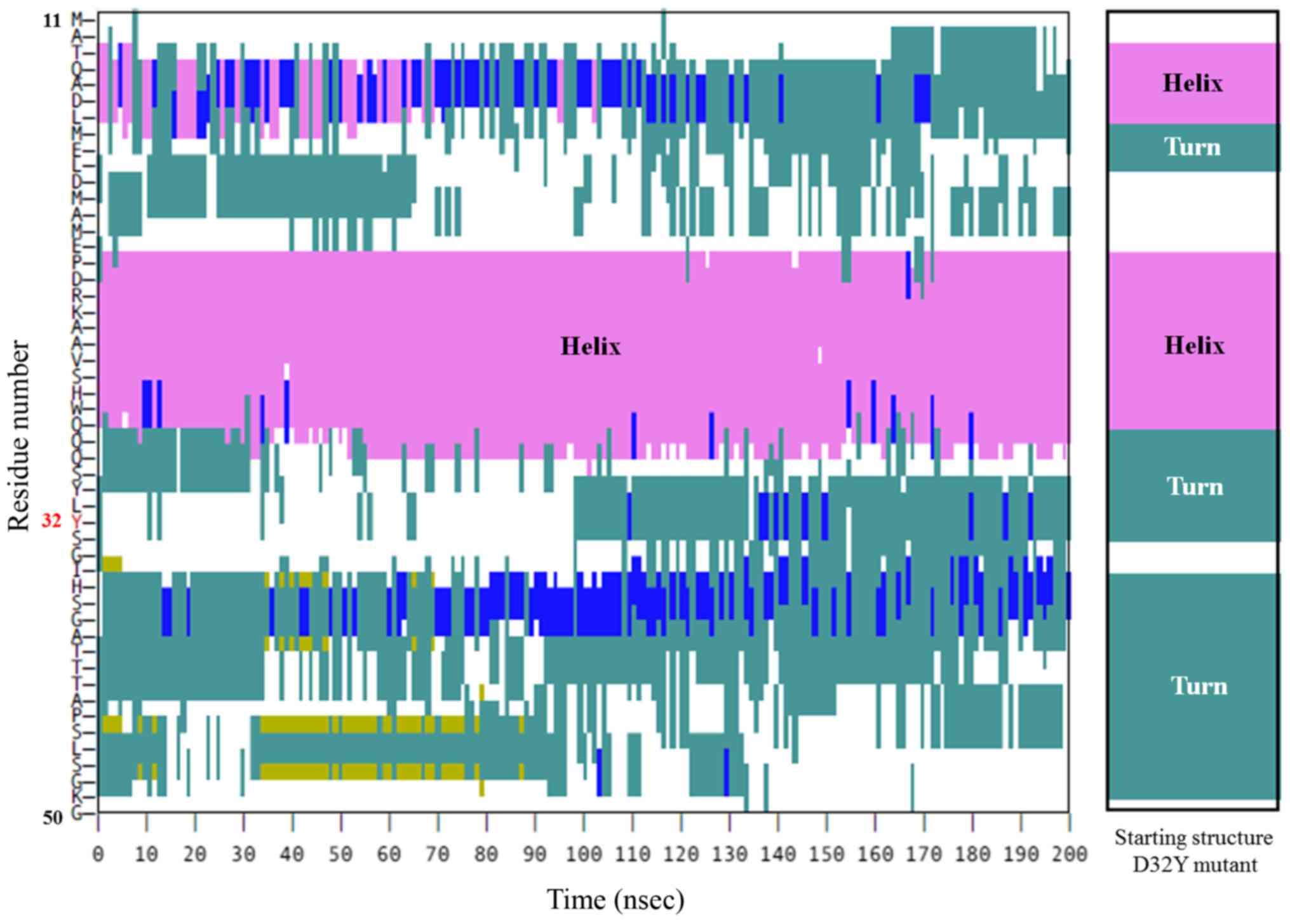
pattern of secondary structure was observed in the D32Y fragment
(Fig. 5). This indicated that D32Y
mutation, at least, may not affect phosphorylation activity via
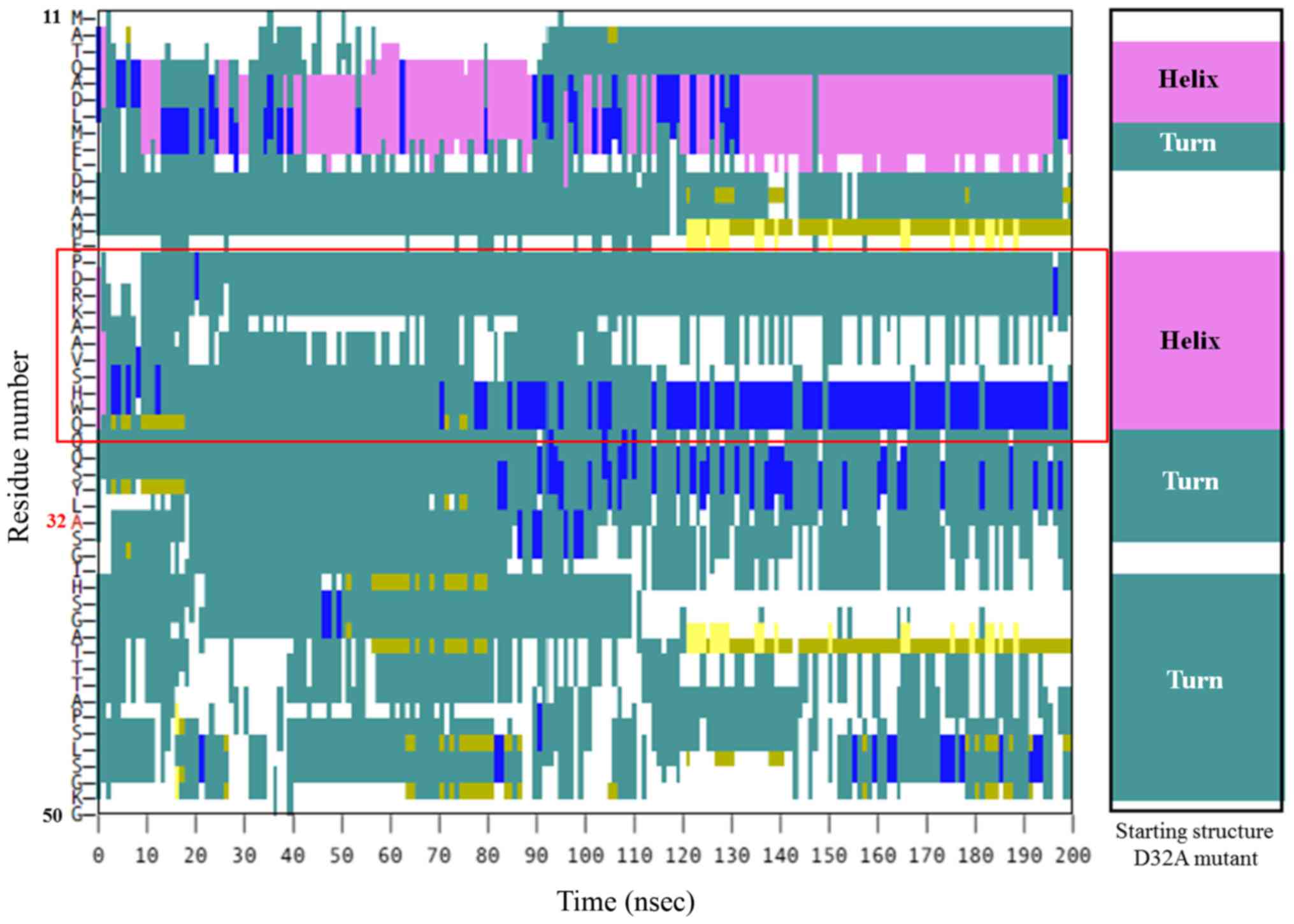
conformation distortion in this area. However, in the D32A mutant,
a loss in the helical secondary structure of P16-H28 was observed,
moving from a helical shape into a turn (Fig. 6). The D32A mutant therefore clearly
possesses structural differences to the wild type β-catenin, and
the mutation could enhance protein unfolding via alternation of the
helical structure to interfere with the functional S33
position.
Discussion
SPN belongs to a group of human cancer types that
occur in developing organs with somatic mutations to CTNNB1.
Patterns of CTNNB1 mutation differ, and are specific to
tumor types. In nephroblastoma, CTNNB1 mutations usually
occur to codon 45, whereas the majority of mutations in
hepatoblastoma are large deletions involving exon 3 (10,35).
Defective phosphorylation caused by β-catenin sequence alterations
involves the priming phosphorylation sites for casein kinase I
proteins, underlying the molecular mechanism of tumorigenesis of
those neoplasms. Tumors containing mutations on the main
phosphorylation sites are relatively fast-growing, invasive and
respond well to chemotherapy. The mutation spots in
medulloblastomas and pancreatoblastomas are confined to residues 33
and 37, which are sequential phosphorylation sites for GSK-3β
(10). Tumors harboring lesions on
those secondary phosphorylation sites are usually found in older
children and are relatively non-invasive (10).
Alterations to CTNNB1 codon 32 have been
reported in rare tumor types, including SPN, pilomatrixomas and
medulloblastomas (36–38). These tumors are relatively low-grade
and rarely undergo distant metastasis. The study of Ellison et
al (36) demonstrated that codon
32 was the most commonly mutated in childhood medulloblastoma. The
current study detected mutations to this codon in each of the three
cases studied. Together, this evidence supports the relevance of
the molecular pathology in these rare tumors.
Three-dimensional molecular simulation is a useful
computational tool for the prediction of the molecular structure of
biomolecules, particularly proteins. The present study demonstrated
that amino acid alterations to codon 32 tend to interfere with a
helical structure within β-catenin. The MD simulations indicated
that the D32A mutation was responsible for hindrance of
phosphorylation at S33 in β-catenin by contributing to a loss of
secondary structure, although D32Y may not act in the same way.
Data from the structural prediction were consistent with a previous
functional genetic study by Al-Fageeh et al (13), which demonstrated increased T-cell
factor transactivation in a 293 cell culture model.
In conclusion, the present study used a
computer-generated molecular structure model to successfully
predicted conformational changes to β-catenin caused by point
mutations at codon 32. These data indicate at the mechanism of
tumorigenesis in patients with SPN that possess D32 β-catenin
mutations.
Acknowledgements
Not applicable.
Funding
The study was partially supported by the Faculty of
Medicine, Prince of Songkla University (Kho Hong, Thailand; grant
no. 59-221-10-1).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author, on reasonable
request.
Authors' contributions
VT and NCP performed in silico molecular
modeling of the β-catenin protein. KK interpreted the pathological
and immunohistochemistry results. JS performed the mutation study
and wrote the manuscript. SS collected the clinical data.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committee of the Faculty of Medicine, Prince of Songkla
University (Hat Yai, Thailand) and all patients provided written
informed consent.
Consent for publication
Patients provided written informed consent for the
publication of their data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Guo N, Zhou QB, Chen RF, Zou SQ, Li ZH,
Lin Q, Wang J and Chen JS: Diagnosis and surgical treatment of
solid pseudopapillary neoplasm of the pancreas: Analysis of 24
cases. Can J Surg. 54:368–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Papavramidis T and Papavramidis S: Solid
pseudopapillary tumors of the pancreas: Review of 718 patients
reported in English literature. J Am Coll Surg. 200:965–972. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yagcı A, Yakan S, Coskun A, Erkan N,
Yıldırım M, Yalcın E and Postacı H: Diagnosis and treatment of
solid pseudopapillary tumor of the pancreas: Experience of one
single institution from Turkey. World J Surg Oncol. 11:3082013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee JS, Han HJ, Choi SB, Jung CW, Song TJ
and Choi SY: Surgical outcomes of solid pseudopapillary neoplasm of
the pancreas: A single institution's experience for the last ten
years. Am Surg. 78:216–219. 2012.PubMed/NCBI
|
|
5
|
Ansari D, Elebro J, Tingstedt B, Ygland E,
Fabricius M, Andersson B and Andersson R: Single-institution
experience with solid pseudopapillary neoplasm of the pancreas.
Scand J Gastroenterol. 46:1492–1497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adams AL, Siegal GP and Jhala NC: Solid
pseudopapillary tumor of the pancreas: A review of salient clinical
and pathologic features. Adv Anat Pathol. 15:39–45. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang SC, Ng KF, Yeh TS, Chang HC, Su CY
and Chen TC: Clinicopathological analysis of β-catenin and Axin-1
in solid pseudopapillary neoplasms of the pancreas. Ann Surg Oncol.
19 Suppl 3:S438–S446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kobayashi T, Ozasa M, Miyashita K, Saga A,
Miwa K, Saito M, Morioka M, Takeuchi M, Takenouchi N, Yabiku T, et
al: Large solid-pseudopapillary neoplasm of the pancreas with
aberrant protein expression and mutation of β-catenin: A case
report and literature review of the distribution of β-catenin
mutation. Intern Med. 52:2051–2056. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
MacDonald BT, Tamai K and He X:
Wnt/beta-catenin signaling: Components, mechanisms, and diseases.
Dev Cell. 17:9–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Koesters R and von Knebel Doeberitz M: The
Wnt signaling pathway in solid childhood tumors. Cancer Lett.
198:123–138. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sangkhathat S, Kusafuka T, Miao J, Yoneda
A, Nara K, Yamamoto S, Kaneda Y and Fukuzawa M: In vitro RNA
interference against β-catenin inhibits the proliferation of
pediatric hepatic tumors. Int J Oncol. 28:715–722. 2006.PubMed/NCBI
|
|
12
|
Sangkhathat S, Kanngurn S, Chaiyapan W,
Gridist P and Maneechay W: Wilms' tumor 1 gene (WT1) is
overexpressed and provides an oncogenic function in pediatric
nephroblastomas harboring the wild-type WT1. Oncol Lett. 1:615–619.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Al-Fageeh M, Li Q, Dashwood WM, Myzak MC
and Dashwood RH: Phosphorylation and ubiquitination of oncogenic
mutants of beta-catenin containing substitutions at Asp32.
Oncogene. 23:4839–4846. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Provost E, McCabe A, Stern J, Lizardi I,
D'Aquila TG and Rimm DL: Functional correlates of mutation of the
Asp32 and Gly34 residues of beta-catenin. Oncogene. 24:2667–2676.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koch A, Denkhaus D, Albrecht S, Leuschner
I, von Schweinitz D and Pietsch T: Childhood hepatoblastomas
frequently carry a mutated degradation targeting box of the
beta-catenin gene. Cancer Res. 59:269–273. 1999.PubMed/NCBI
|
|
16
|
Udatsu Y, Kusafuka T, Kuroda S, Miao J and
Okada A: High frequency of beta-catenin mutations in
hepatoblastoma. Pediatr Surg Int. 17:508–512. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wanitsuwan W, Kanngurn S,
Boonpipattanapong T, Sangthong R and Sangkhathat S: Overall
expression of beta-catenin outperforms its nuclear accumulation in
predicting outcomes of colorectal cancers. World J Gastroenterol.
14:6052–6059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Buchan DW, Minneci F, Nugent TC, Bryson K
and Jones DT: Scalable web services for the PSIPRED protein
analysis workbench. Nucleic Acids Res. 41:(Web Server issue).
W349–W357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jones DT: Protein secondary structure
prediction based on position-specific scoring matrices. J Mol Biol.
292:195–202. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Petersen B, Petersen TN, Andersen P,
Nielsen M and Lundegaard C: A generic method for assignment of
reliability scores applied to solvent accessibility predictions.
BMC Struct Biol. 9:512009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maupetit J, Derreumaux P and Tuffery P:
PEP-FOLD: An online resource for de novo peptide structure
prediction. Nucleic Acids Res. 37:(Web Server Issue). W498–W503.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maupetit J, Derreumaux P and Tufféry P: A
fast method for large-scale de novo peptide and miniprotein
structure prediction. J Comput Chem. 31:726–738. 2010.PubMed/NCBI
|
|
23
|
Thévenet P, Shen Y, Maupetit J, Guyon F,
Derreumaux P and Tufféry P: PEP-FOLD: An updated de novo structure
prediction server for both linear and disulfide bonded cyclic
peptides. Nucleic Acids Res. 40:(Web Server Issue). W288–W293.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Case DA, Cerutti DS, Cheatham TE III,
Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Greene D, Homeyer
N, et al: AMBER 2017. University of California, SF; 2017
|
|
25
|
Pastor RW, Brooks BR and Szabo A: An
analysis of the accuracy of Langevin and molecular dynamics
algorithms. Mol Phys. 65:1409–1419. 1988. View Article : Google Scholar
|
|
26
|
Berendsen HJC, Postma JPM, Gunsteren WF,
van DiNola A and Haak JR: Molecular dynamics with coupling to an
external bath. J Chem Phys. 81:3684–3690. 1984. View Article : Google Scholar
|
|
27
|
Darden TA and Pedersen LG: Molecular
modeling: An experimental tool. Environ Health Perspect.
101:410–412. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Case DA, Darden TA, Cheatham TE III,
Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM,
et al: AMBER 12. University of California, SF; 2012
|
|
29
|
Humphrey W, Dalke A and Schulten K: VMD:
Visual molecular dynamics. J Mol Graph. 14(33–38): 27–28. 1996.
|
|
30
|
Morin PJ, Sparks AB, Korinek V, Barker N,
Clevers H, Vogelstein B and Kinzler KW: Activation of
beta-catenin-Tcf signaling in colon cancer by mutations in
beta-catenin or APC. Science. 275:1787–1790. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chan EF, Gat U, McNiff JM and Fuchs E: A
common human skin tumour is caused by activating mutations in
beta-catenin. Nat Genet. 21:410–413. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Legoix P, Bluteau O, Bayer J, Perret C,
Balabaud C, Belghiti J, Franco D, Thomas G, Laurent-Puig P and
Zucman-Rossi J: Beta-catenin mutations in hepatocellular carcinoma
correlate with a low rate of loss of heterozygosity. Oncogene.
18:4044–4046. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Huang H, Mahler-Araujo BM, Sankila A,
Chimelli L, Yonekawa Y, Kleihues P and Ohgaki H: APC mutations in
sporadic medulloblastomas. Am J Pathol. 156:433–437. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
van Noort M, van de Wetering M and Clevers
H: Identification of two novel regulated serines in the N terminus
of beta-catenin. Exp Cell Res. 276:264–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Koesters R, Ridder R, Kopp-Schneider A,
Betts D, Adams V, Niggli F, Briner J and von Knebel Doeberitz M:
Mutational activation of the beta-catenin proto-oncogene is a
common event in the development of Wilms' tumors. Cancer Res.
59:3880–3882. 1999.PubMed/NCBI
|
|
36
|
Ellison DW, Onilude OE, Lindsey JC, Lusher
ME, Weston CL, Taylor RE, Pearson AD and Clifford SC: United
Kingdom Children's Cancer Study Group Brain Tumour Committee:
beta-Catenin status predicts a favorable outcome in childhood
medulloblastoma: The United Kingdom Children's Cancer Study Group
Brain Tumour Committee. J Clin Oncol. 23:7951–7957. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chmara M, Wernstedt A, Wasag B, Peeters H,
Renard M, Beert E, Brems H, Giner T, Bieber I, Hamm H, et al:
Multiple pilomatricomas with somatic CTNNB1 mutations in children
with constitutive mismatch repair deficiency. Genes Chromosomes
Cancer. 52:656–664. 2013.PubMed/NCBI
|
|
38
|
Silva RD, Marie SK, Uno M, Matushita H,
Wakamatsu A, Rosemberg S and Oba-Shinjo SM: CTNNB1, AXIN1 and APC
expression analysis of different medulloblastoma variants. Clinics
(Sao Paulo). 68:167–172. 2013. View Article : Google Scholar : PubMed/NCBI
|