Introduction
Cervical cancer is one of most common types of
cancer; there are ~470,000 new cases and 233,000 associated
mortalities per year worldwide (1,2). The
reported five-year survival rate is 68% in the United States
(3) and patient outcomes depend on
how early the cancer is detected (4).
The occurrence rate is high, at ~70% in developing countries
(5). In developed countries, the rate
has been markedly reduced with the application of cervical
screening programs (5). The greatest
risk factor for cervical cancer is type 16 and 18 human
papillomavirus (HPV) infection, which accounts for 75% of cervical
cancer cases (6). Smoking is the next
most significantly associated risk factor for cervical cancer; the
incidence of invasive cervical cancer is 2–3 times higher in
current or former smokers among HPV-infected women (7).
HPV infection is one of the most important causative
molecular mechanisms for cervical cancer (8). The high-risk HPV types encode two
oncoproteins, E6 and E7, which can inactivate tumor suppressor
proteins and abrogate apoptosis (8).
Using advanced microarray and next-generation sequencing
technologies, researchers have explored the gene expression profile
and molecular mechanisms of cervical cancer. In 2006, Wong et
al (9) used oligonucleotide
microarray analysis and reverse transcription-polymerase chain
reaction to demonstrate that secreted phosphoprotein 1, cyclin
dependent kinase inhibitor 2A (CDKN2A), ribosomal protein
L39 and C1orf10 were differentially regulated in
cervical cancer compared with normal cervix tissue. Connective
tissue growth factor and regulator of G protein signaling 1 were
identified as upregulated in late stage cervical cancer (9). The pelvic lymph node metastasis ability
of early-stage cervical cancer has been associated with barrier to
autointegration factor 1, La ribonucleoprotein domain family member
7, Secretory carrier membrane protein, CUE domain containing 1 and
phosphatidylethanolamine binding protein 1 by comparing gene
expression profiles of tumor samples from patients with and without
metastasis (10).
Several critical microRNAs (miRs/miRNAs) have also
been identified in cervical cancer. Hu et al (11) indicated that miR-200a and miR-9 can
predict patient survival, and that miR-200 likely affects the
metastatic process of cervical cancer cells by suppressing the
genes controlling cell motility. The expression of miR-424 has been
demonstrated as significantly downregulated in 147 cervical cancer
tissues vs. 74 normal tissues (12).
miR-424 has also been identified as a crucial tumor suppressor
through the mechanism of upregulating the expression and
phosphorylation of CHEK1 (12).
Although various studies have explored the
expression profiles of cervical cancer, a combination of mRNA and
miRNA expression profiles has rarely been studied in cervical
cancer. A large number of microarray expression datasets are
publicly available in the Gene Expression Omnibus (GEO) database,
and data-mining the deposited datasets using bioinformatics methods
may advance the understanding of cervical cancer (13). In the present study, the significantly
differentially expressed genes (DEGs) in cervical cancer were first
identified based on two expression datasets from independent labs.
DEGs were then subjected to functional annotation based on the Gene
Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
databases. The regulation mechanisms between the identified DEGs
and reported miRNAs in cervical cancer were then explored. The
prognostic performance of the identified DEGs were virtually
validated using the SurvExpress online database.
Materials and methods
mRNA and miRNA expression
profiles
Datasets for cervical cancer were obtained from the
publicly available GEO database (http://www.ncbi.nlm.nih.gov/geo/). Two mRNA expression
profile datasets that used the same platform and contained normal
tissue controls were selected. GSE63678 was submitted by Pappa
et al in 2014 (14), and
contains 5 normal tissues and 5 cancer tissues, whereas GSE63514
was submitted by den Boon et al in 2014 (15) and includes 24 normal tissue and 28
cancer tissue expression profiles. Both of these datasets had been
produced using the Affymetrix Human Genome U133 Plus 2.0 array.
Further information regarding the original samples and experiments
are documented in the referenced manuscripts.
Identification of differentially
expressed genes
An R script and database produced in-house were used
for the data analysis and annotation. In brief, mRNA expression
profiles underwent background correction, normalization and
log2 transformation with the GeneChip Robust Multi-array
Analysis algorithm (16). Control
probe sets were filtered out, and the mean expression was
calculated for genes with multiple probes. Finally, the Linear
Models for Microarray Analysis algorithm in Bioconductor was
applied for DEG screening (17). The
common DEGs between the two data sets were also identified.
Criteria to indicate a statistically significant difference were
set to P≤0.05 and absolute log2 (fold-change) ≥2.
GO and KEGG pathway annotation
The identified DEGs were subjected to GO and KEGG
pathway enrichment analysis using the Database for Annotation,
Visualization and Integrated Discovery (DAVID) online (18). GO terms were identified in the
Biological Process (BP), Cellular Component (CC) and Molecular
Function (MF) categories. P<0.05 was set as the significance
threshold.
miRNAs may be critical in carcinogenesis and
metastasis through their regulation of mRNA expression; by
searching PubMed, 6 critical miRNAs involved in the development of
cervical cancer were identified: miR-200a-5p, miR-9-5p, miR-424-5p,
miR-133b, miR-224-3p and miR-506-5p (11,12,19–21).
A regulation network between the common DEGs and 4 miRNAs was
constructed. miRNA targets were predicted based on the microcosm
(22), mirTarbase (23) and TargetScan (24) databases, and the associations between
the common DEGs and the target genes were identified. Finally, the
regulation network was plotted using CyTargetLinker plugin
(25) in Cytoscape v3.5.1 (26).
Co-expression and interaction network
analysis
The concept of co-expression can be used for the
identification of novel mechanisms that contribute to tumorigenesis
and progression (27). To identify
patterns of co-expression, the odds ratio (OR) between each pair of
query genes was calculated, and significant pairs were selected
based on the cervical carcinoma data (28) from The Cancer Genome Atlas (TCGA)
(28) using cBioPortal (29). The interactions between the identified
DEGs and potential cancer drug targets were predicted based on
various databases, including Reactome (30), DrugBank (31), CancerRxGene (32) and PANTHER (33).
In-silico validation
To validate the clinical significance of the
identified DEGs, they were first validated in large cohort cervical
cancer samples from TCGA (28) using
cBioPortal (30). The prognostic
performance of the 16 selected DEGs was then virtually evaluated
using the SurvExpress database, which includes gene expression
datasets with clinical outcome data (34). One cervical cancer dataset (28), including clinical information, was
selected from TCGA for virtual validation. Detailed information
regarding the dataset can be found in the original study (28). Parameters were carefully selected
according to the developer's guide that provides optimized
parameters (34). A heatmap for all
samples, based on the miRNA target genes, was plotted using the
heatmap module in Bioconductor (35).
Results
DEGs in cervical cancer
Subsequent to background correction and
normalization, the median gene expression values for the two
datasets were similar (data not shown). A total of 975 and 402 DEGs
were screened in GSE63514 and GSE63678, respectively. In GSE63514,
this included 523 upregulated (53.6%) and 452 downregulated genes
(46.4%), and in GSE63678, 160 upregulated (39.8%) and 242
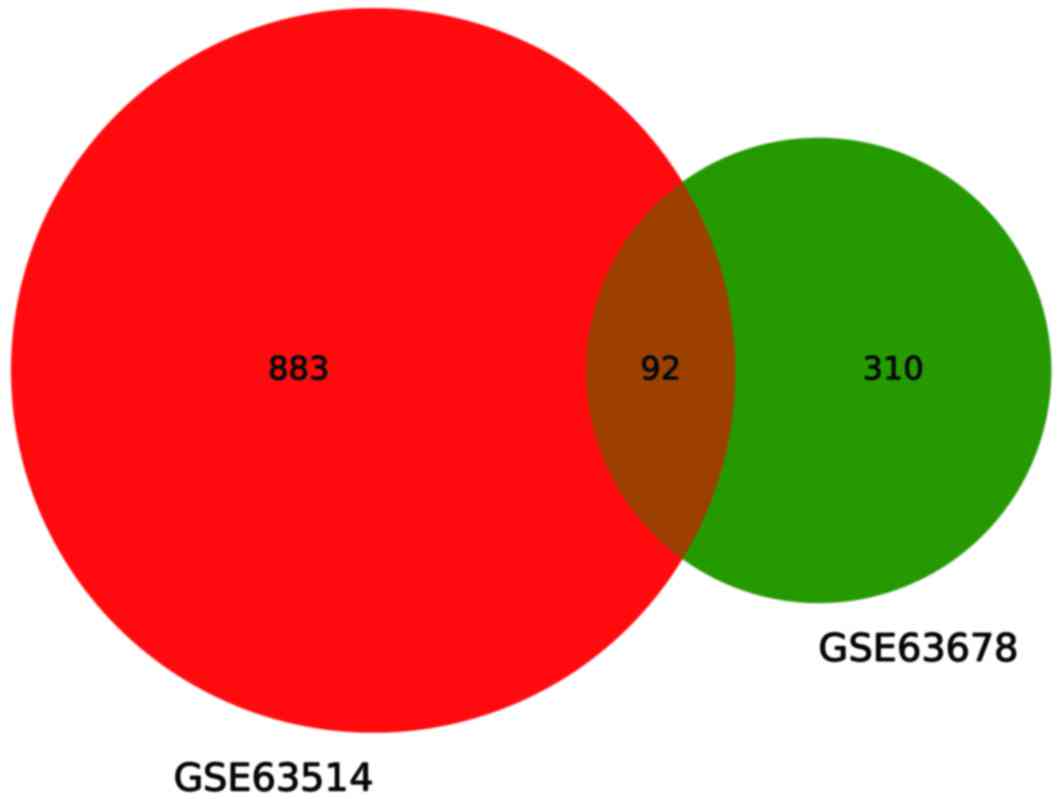
downregulated genes (60.2%). Fig. 1
illustrates that 92 common genes were differentially expressed
between GSE63514 and GSE63678, and fold-changes for the top 20
up/downregulated common DEGs are listed in Table I.
 | Table I.Top 20 differentially expressed genes
in the GSE63514 and GSE63678 datasets. |
Table I.
Top 20 differentially expressed genes
in the GSE63514 and GSE63678 datasets.
|
| Fold-change |
|---|
|
|
|
|---|
| Gene | GSE63514 | GSE63678 |
|---|
|
C1orf116 | −2.57 | 2.17 |
| CEACAM6 | −2.05 | 3.32 |
| BIRC5 | 2.10 | 2.14 |
| CCNB1 | 2.11 | 2.45 |
| CENPF | 2.27 | 2.18 |
| AURKA | 2.31 | 2.43 |
| BUB1B | 2.37 | 2.06 |
| CDKN3 | 2.41 | 2.20 |
| CHEK1 | 2.44 | 2.12 |
| CEP55 | 2.50 | 2.51 |
| ASPM | 2.55 | 2.77 |
|
APOBEC3B | 2.58 | 2.06 |
| CENPN | 2.63 | 2.02 |
| CDK1 | 2.68 | 2.35 |
| CCNE2 | 2.75 | 2.56 |
| CENPE | 2.98 | 2.06 |
| CDC7 | 3.00 | 2.54 |
| ATAD2 | 3.59 | 2.36 |
| CA9 | 4.19 | 2.39 |
| CDKN2A | 6.41 | 2.46 |
GO and KEGG pathway annotation
To identify the biological functions of the common
DEGs, GO and KEGG pathway enrichment analysis were performed using
the DAVID online tool. The results indicate that the common DEGs
are significantly enriched in three KEGG pathways (Table II). A total of 10 genes were
associated with ‘cell cycle’ P=9×10−9), 6 with ‘p53
signaling’ (P=2.2×10−5) and 5 genes with ‘oocyte
meiosis’ (P=0.002). In GO terms, DEGs were most commonly associated
with molecular functions of ‘microtubule binding’
(P=9.4×10−5), ‘ATP binding’ (P=1.7×10−4) and
‘protein binding’ (P=5.2×10−4; Table III). The top 5 most significantly
enriched terms in biological process were ‘S transition of mitotic
cell cycle’ (P=8.3×10−11), ‘cell division’
(P=1.6×10−9), ‘mitotic nuclear division’
(P=4.5×10−8), ‘G2/M transition of mitotic cell cycle’
(P=4.3×10−7) and ‘sister chromatid cohesion’
(P=7.6×10−7; Table III).
The top 5 significantly enriched terms in cellular component were
‘midbody’ (P=2.4×10−11), ‘nucleoplasm’
(P=3.2×10−11), ‘nucleus’ (P=9.5×10−10),
‘chromosome centromeric region’ (P=8.9×10−9) and
‘spindle microtubule’ (P=5.7×10−8; Table III).
| Table II.Kyoto Encyclopedia of Genes and
Genomes pathway enrichment results for the common differentially
expressed genes. |
Table II.
Kyoto Encyclopedia of Genes and
Genomes pathway enrichment results for the common differentially
expressed genes.
| Term | P-value | Genes |
|---|
| hsa04110: cell
cycle |
9.02×10−9 | CDKN2A, CDC7,
MCM2, CDK1, CCNE2, CHEK1, BUB1B, CCNB1, MAD2L1, MCM4 |
| hsa04115: p53
signaling pathway | 2.20×10-5 | CDKN2A, CDK1,
CCNE2, RRM2, CHEK1, CCNB1 |
| hsa04114: oocyte
meiosis | 0.00234 | CDK1, AURKA,
CCNE2, MAD2L1, PGR |
| Table III.Top 5 GO terms for the common
differentially expressed genes. |
Table III.
Top 5 GO terms for the common
differentially expressed genes.
| A, Molecular
function |
|---|
|
|---|
| ID | GO Term | P-value | Genes |
|---|
| GO:0008017 | microtubule
binding |
9.49×10−5 | KIF4A, PRC1,
CENPE, KIF11, KIF20A, BIRC5, CRYAB, NUSAP1 |
| GO:0005524 | ATP binding |
1.72×10−4 | ATAD2, KIF4A,
CDC7, OAS2, FGFR2 |
| GO:0005515 | protein
binding |
5.28×10−4 | CDKN2A, DTL,
KIF4A, UBD, PGR |
| GO:0019901 | protein kinase
binding | 7.13×10-4 | CDKN2A, PRC1,
AURKA, FOXM1, CCNE2, KIF11, KIF20A, CCNB1, KRT17 |
| GO:0003777 | microtubule motor
activity |
7.33×10−4 | KIF4A, CENPE,
KIF2C, KIF11, KIF20A |
|
| B, Biological
process |
|
| ID | GO Term | P-value | Genes |
|
| GO:0000082 | G1/S transition of
mitotic cell cycle | 8.32×10-11 | CDKN2A, CDC7,
MCM2, CDKN3, ID4 |
| GO:0051301 | cell division |
1.66×10−9 | CDC7, SMC4,
CDK1, CCNB1, MAD2L1 |
| GO:0007067 | mitotic nuclear
division | 4.57×10-8 | CDK1, AURKA,
NEK2, CEP55, BIRC5 |
| GO:0000086 | G2/M transition of
mitotic cell cycle |
4.38×10−7 | MELK, CDK1,
HMMR, AURKA, NEK2, FOXM1, CHEK1, BIRC5, CCNB1 |
| GO:0007062 | sister chromatid
cohesion | 7.67×10-7 | CENPE, CENPN,
CENPF, SLC35F6, KIF2C, BUB1B, BIRC5, MAD2L1 |
|
| C, Cellular
component |
|
| ID | GO Term | P-value | Genes |
|
| GO:0030496 | midbody |
2.49×10−11 | KIF4A, ECT2,
PRC1, CEP55, BIRC5 |
| GO:0005654 | nucleoplasm | 3.29×10-11 | ATAD2, CDKN2A,
DTL, SOX17, PGR |
| GO:0005634 | nucleus |
9.58×10−10 | ATAD2, CDKN2A,
DTL, SOX17, PGR |
| GO:0000775 | chromosome,
centromeric region | 8.95×10-9 | CENPE, CENPN,
MKI67, CENPF, SLC35F6, KIF2C, OIP5, BIRC5 |
| GO:0005876 | spindle
microtubule |
5.70×10−8 | KIF4A, PRC1,
CDK1, AURKA, KIF11, BIRC5, NUSAP1 |
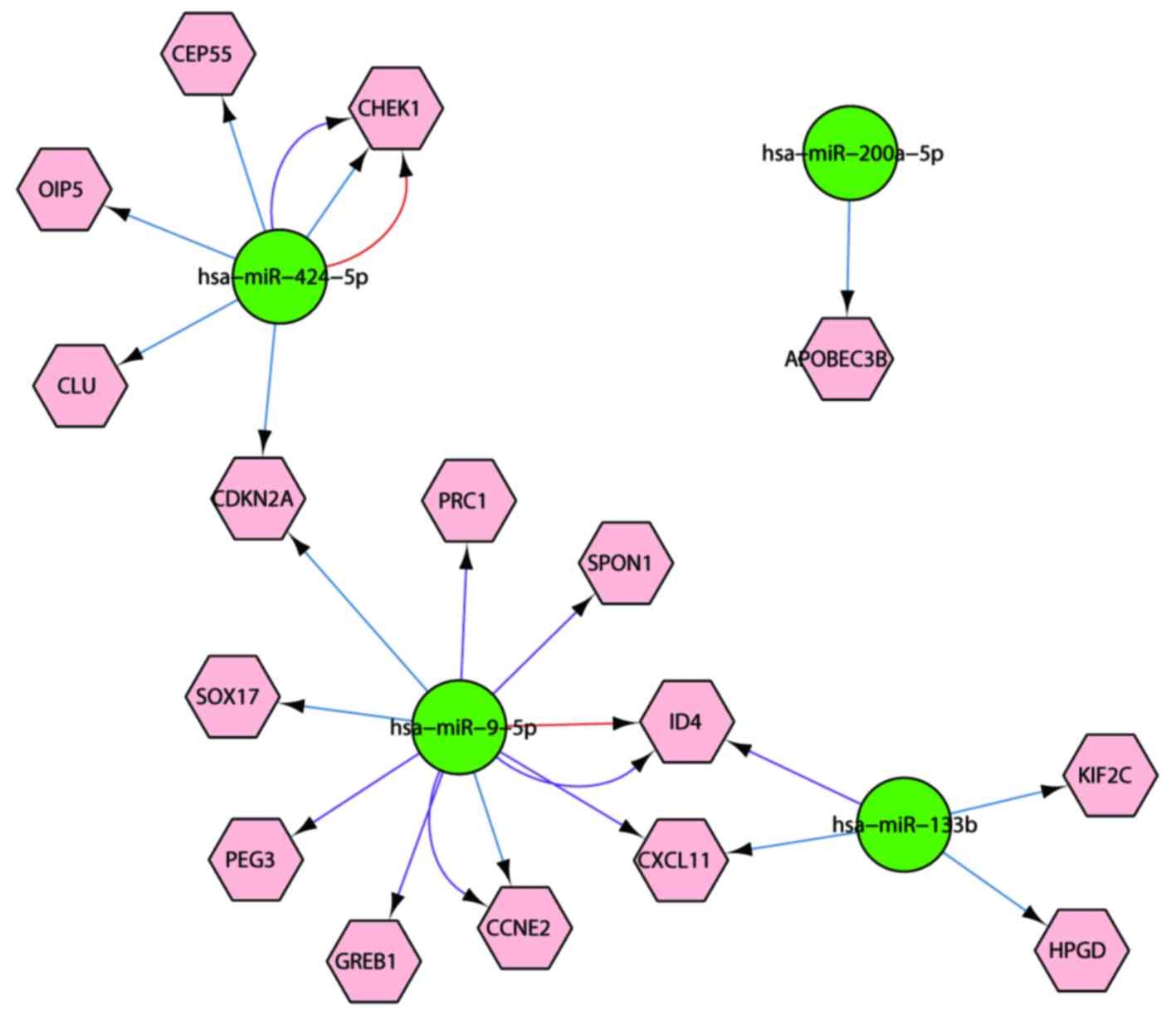
mRNA-miRNA network construction
The 6 identified miRNAs could target 231, 2,792 and
3,143 genes in the miRTarBase, MicroCosm and TargetScan databases,
respectively. Among the target genes, 16 genes were common DEGs.
Based on the interaction network between the 16 genes and 4 miRNAs
(Fig. 2), it was identified that
miR-424-5p could regulate 5 genes, miR-9-5p could regulate 9 genes,
miR-133b could regulate 4 genes and miR-200a-5p could regulate one
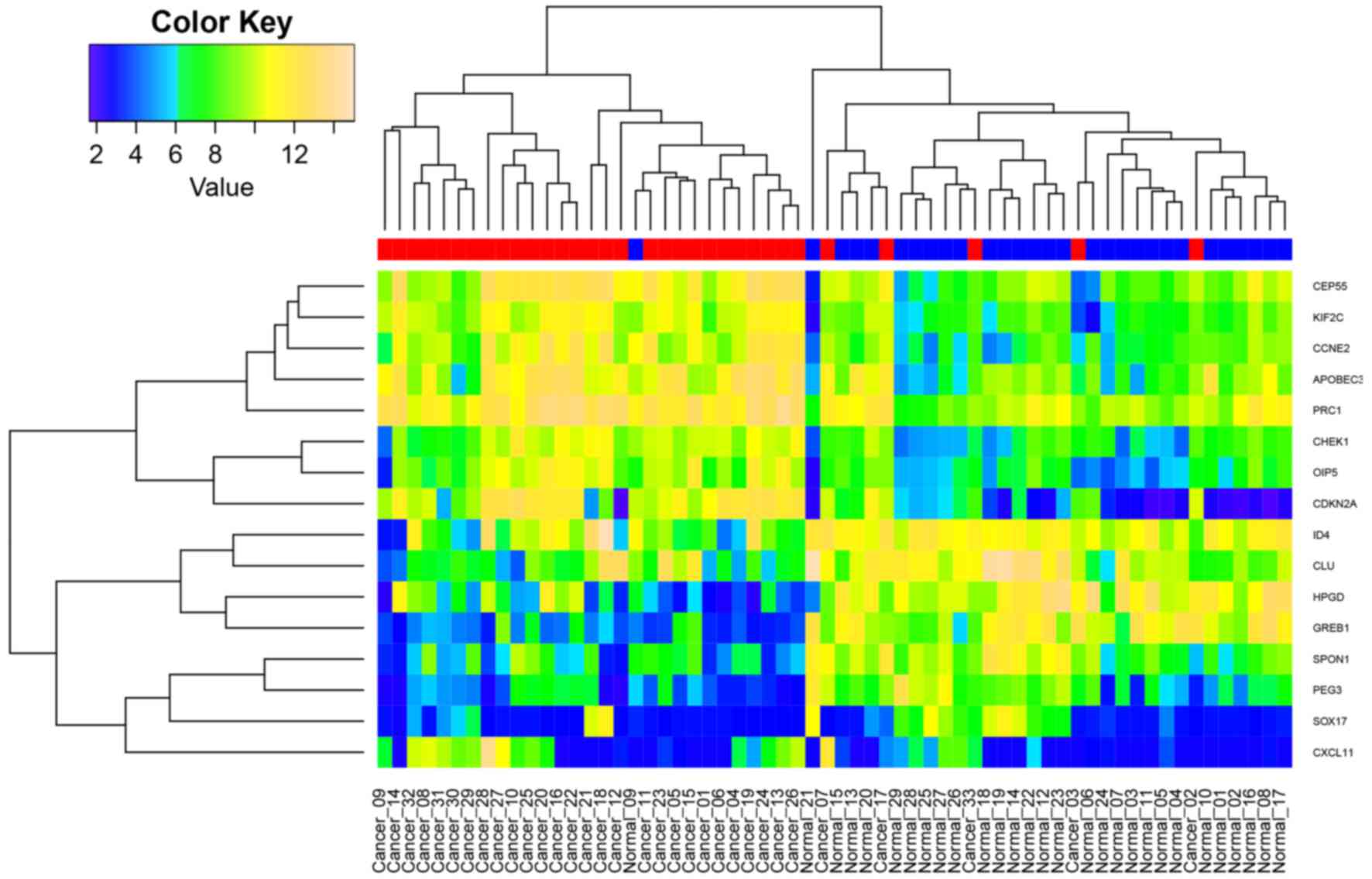
gene. All samples from the two datasets were subjected to
hierarchical clustering using the expression of the 16 DEGs.
Fig. 3 illustrates that tumor (red)
and normal (blue) samples can almost be classified into two groups.
Data for a number of tumor and normal samples were confounded; this
was most likely due to tumor heterogeneity or expression value
variation.
Co-expression and interaction network
analysis
To reveal gene co-expression patterns, the
significant gene pairings were selected based on odds ratio. This
analysis identified 12 gene pairings among the 16 DEGs. SRY-box 17
(SOX17) tended to be co-expressed with paternally expressed
3 (PEG3), growth regulation by estrogen in breast cancer 1
(GREB1), inhibitor of DNA binding 4 (ID4) and Spondin
1 (SPON1), respectively (P<0.01; Table IV). PEG3 was often
co-expressed with ID4 and GREB1 (P<0.05; Table IV). In addition, the interaction
network indicated that Checkpoint kinase 1 (CHEK1), Cyclin
E2 (CCNE2), CDKN2A and Wings apart-like protein
homolog interact via Transcription factor Dp-2, E2F transcription
factor 1, Topoisomerase (DNA) II binding protein 1, ATR and
karyopherin subunit α1 (Fig. 4).
Cancer drugs Roscovitine, AZD7762 and 681640 can target
CDKN2A and CHEK1 respectively (Fig. 4).
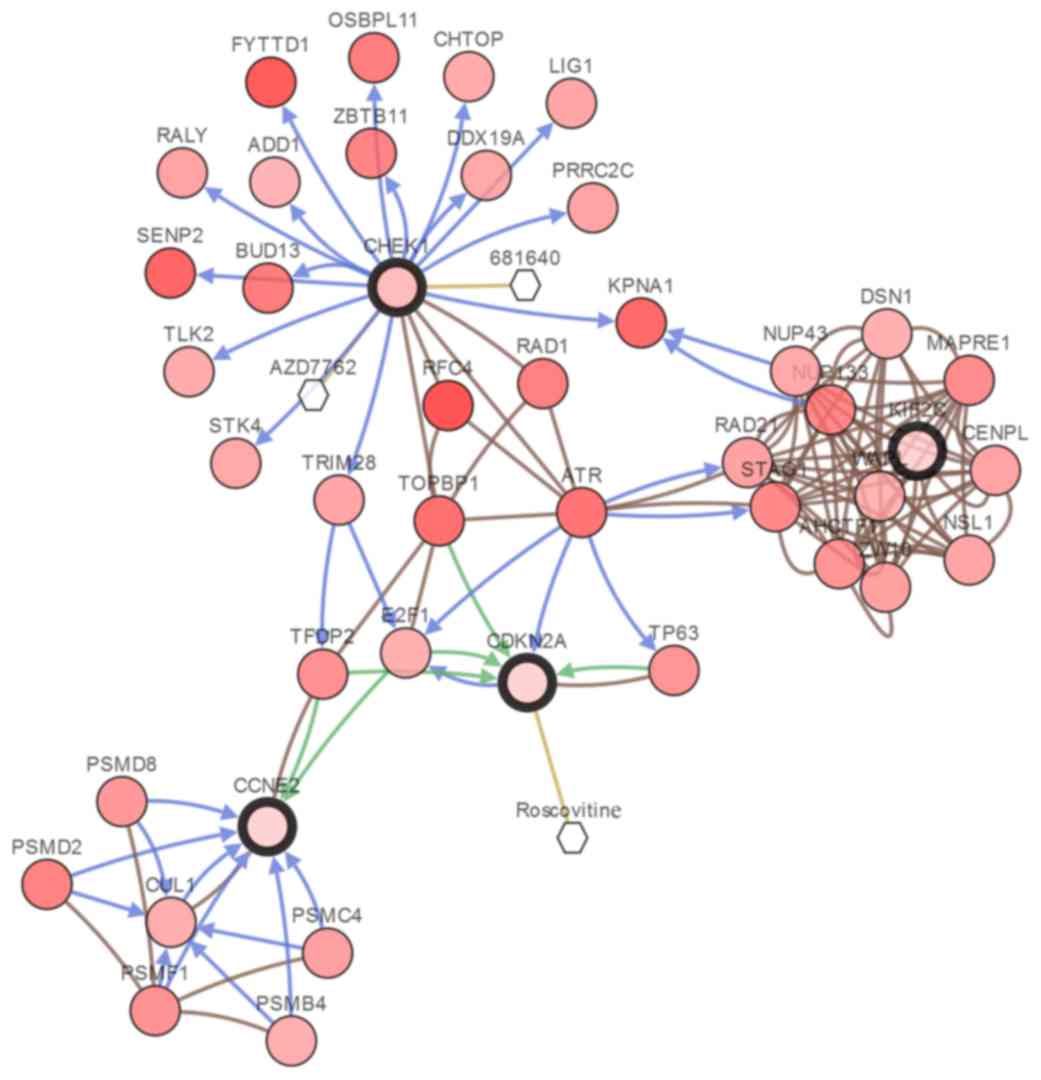 | Figure 4.Interaction network between the
identified differentially expressed genes and potential drugs. Each
node is color coded along a white to red color gradient, indicating
the total frequency of alteration across the selected case set, the
deeper the red, the higher the frequency of alteration). The
hexagons represent a drug and the circles represent a gene. Node
border: Thin, linker nodes or targets of DEGs; thick, selected
DEGs. Edge colors: Blue, controls state change of; brown, controls
transport of; green, controls expression of; yellow, controls
phosphorylation of. DEGs, differentially expressed genes. |
| Table IV.The significant gene co-expression
pairs among the differentially expressed genes. |
Table IV.
The significant gene co-expression
pairs among the differentially expressed genes.
| Gene A | Gene B | P-value | Log odds ratio |
|---|
| SOX17 | PEG3 | <0.001 | >3 |
| SOX17 | GREB1 | <0.001 | >3 |
| SOX17 | ID4 | <0.001 | >3 |
| SOX17 | SPON1 | <0.001 | >3 |
| PEG3 | GREB1 | <0.001 | >3 |
| PEG3 | ID4 | 0.023 | 2.759 |
| GREB1 | ID4 | 0.007 | 2.521 |
| CDKN2A | OIP5 | 0.011 | 1.858 |
| KIF2C | CCNE2 | 0.004 | 1.709 |
| CDKN2A |
APOBEC3B | 0.030 | 1.475 |
| CCNE2 | PRC1 | 0.035 | 1.243 |
| CHEK1 |
APOBEC3B | 0.041 | 1.208 |
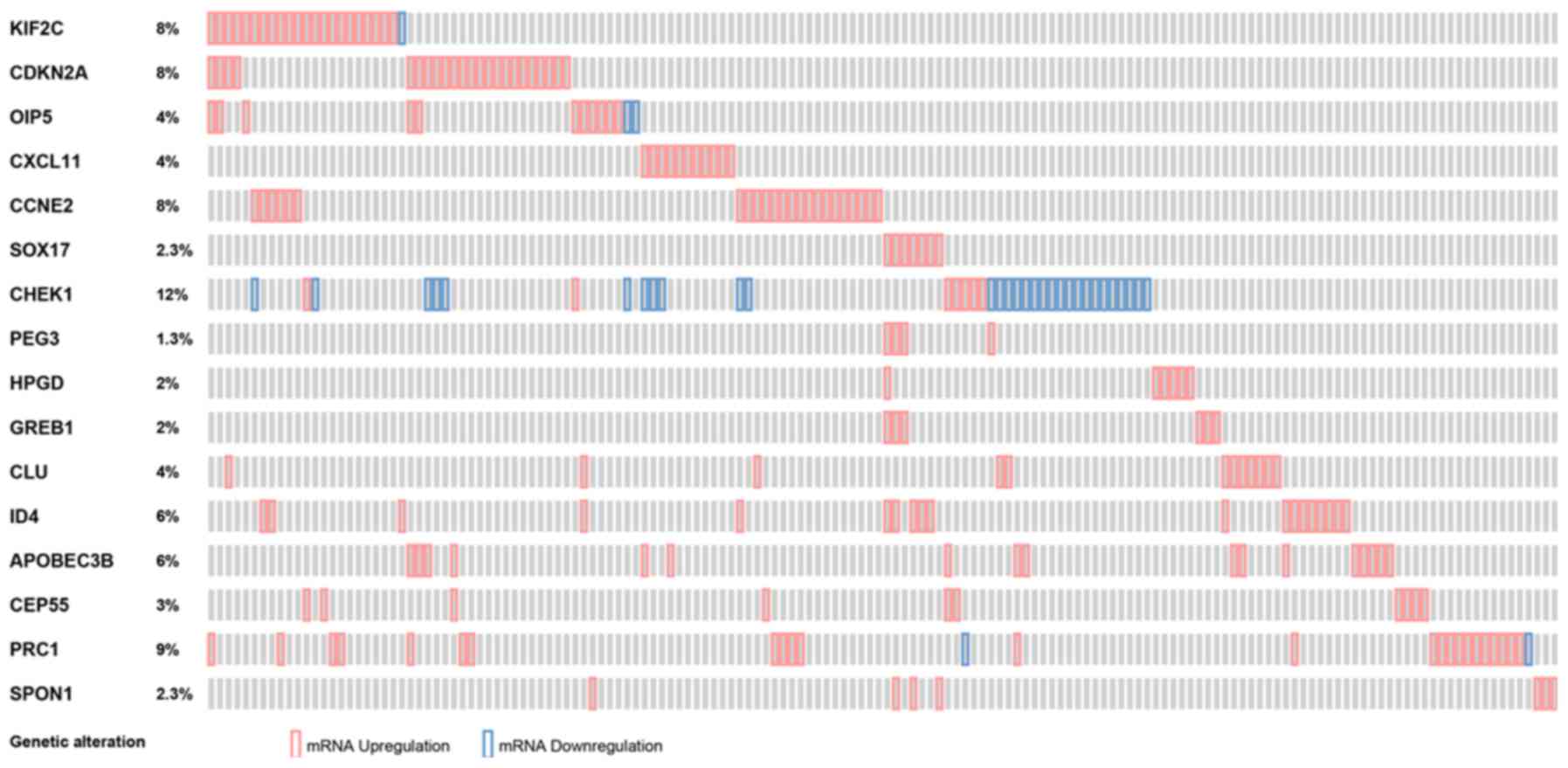
In-silico validation
In a large cohort of cervical cancer samples, the
identified common DEGs were all differentially expressed (z-score,
<-2 or >2). The percentage of sample ranged from 1.3 to 12%
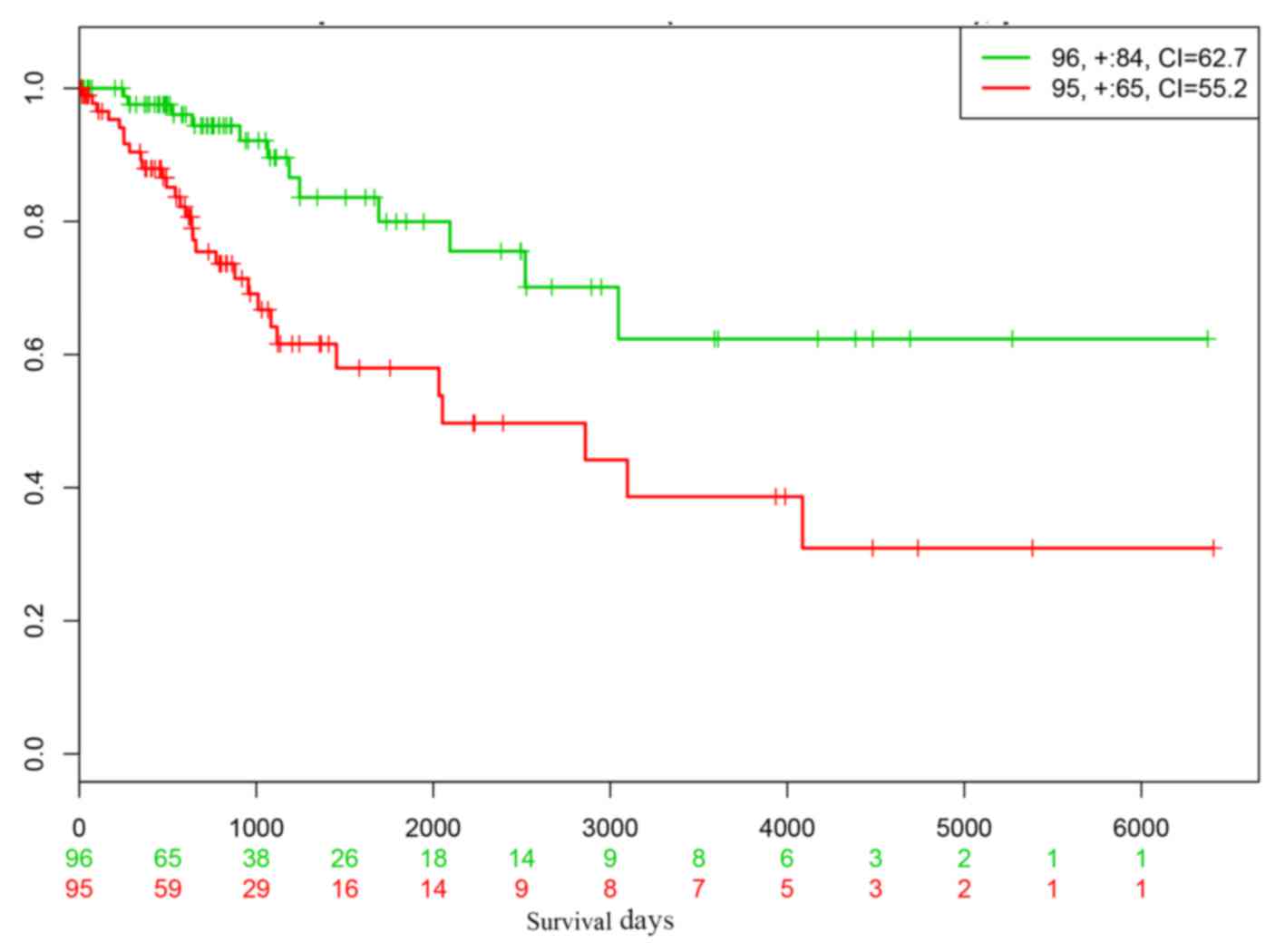
(Fig. 5). The prognostic performance
of the 16 genes was virtually validated using the SurvExpress
online tool's cervical cancer dataset. This analysis indicated that
low- and high-risk cervical cancer groups could be significantly
differentiated based on the 16 genes (Fig. 6). The low P-value (P=0.001) and high
concordance index suggested that an accurate prediction of
prognosis could be achieved based on the DEGs.
Discussion
With the rapid development of microarray and NGS
technologies, more and more cervical cancer molecular biomarkers
have been identified. However, there has been limited integration
of multi-omic data for the exploration of the molecular mechanisms
of cervical cancer. In this study, mRNA expression profiles and
previously identified miRNA-mRNA interactions were integrated to
contribute to the characterization of the complex mechanism of
cervical cancer. Systematic analysis revealed that 92 genes were
simultaneously differentially expressed in 33 tumor tissues and
that 16 DEGs may have been regulated by 4 critical miRNAs. The
tumor and normal tissues were clearly classified into two groups
based on the expression of the 16 DEGs.
Pathway enrichment analysis indicated that 6 DEGs
were associated with ‘p53 signaling’. The study by Xiao et
al (36) indicated that
fra-1 was significantly downregulated in cervical cancer
compared with adjacent normal tissue. fra-1 can dysregulate
p53 signaling via regulating the expression of p53 and
MDM2 in vivo.
Based on an mRNA-miRNA interaction network, the
mechanism of cervical cancer was further characterized, indicating
that miR-424-5p can regulate CHEK1. The protein encoded by
this gene is required for checkpoint-mediated cell cycle arrest in
response to DNA damage (37).
Immunohistochemical study has indicated the ubiquitous expression
of CHEK1 protein in cervical cancer, ovarian carcinoma and other
types of cancer (38). In 2011,
Mazumder et al (39)
demonstrated that the deletion and methylation of CHEK1 were
associated with the progression of cervical carcinoma, and that the
inactivation of the ATM-CHEK1 DNA damage response pathway
participated in cervical cancer. ATM is activated by
phosphorylation in response to DNA damage, which in turn activates
its downstream target CHEK1, suggesting that inactivation of
the ATM-CHEK1-associated DNA damage response pathway may
have an important role in the development of cervical carcinoma
(39). In addition, Xu et al
(12) identified a critical tumor
suppressive role for miR-424 in the progression of cervical cancer,
at least partly via upregulating the expression of CHEK1 and
p-CHEK1. Overexpression of miR-424 inhibited the expression of
CHEK1 and p-CHEK1 at residue Ser345, and decreased the activity of
the luciferase-reporter containing the 3′-untranslated region of
CHEK1. Hsieh et al (40)
revealed that Euphorbia antiquorum extracts could
downregulate topoisomerase and activate ATM kinase, inducing the
CHEK1/2 and mitogen activated protein kinase
signaling pathways, and promoting the degradation of cell division
cycle 25A to induce S-phase arrest in HeLa cells.
An additional critical gene is CDKN2A, which
may be regulated by miR-424-5p or miR-9-5p. CDKN2A encodes 2
main proteins, including p16INK4, which is a cyclin-dependent
kinase inhibitor, and p14ARF, which can bind the p53-stabilizing
protein MDM2 (41). These proteins
participate in the regulation of two critical cell cycle regulatory
pathways, the p53 pathway and the RB1 pathway. p16 can decelerate
cell cycle progression from G1 phase to S phase. Wijetunga et
al (42) reported a novel
potential link between early cervical cancer disease progression
and CpG-DNA methylation within the area 700 bp downstream of the
transcriptional start site of CDKN2A, which may lead to
increased p16INK4A/p14ARF expression prior to the development of
malignant disease. In advanced cervical cancers, the majority of
cells exhibited methylated CDKN2A, a lack of p16INK4A
protein and no expression of HPV E7, suggesting p16INK4A
inactivation may be a mechanism of blocking the cyclin D-RB1
pathway in invasive cervical cancer (43).
SOX17 encodes a member of the SOX family
involved in the regulation of embryonic development and the
determination of cell fate, and can be regulated by miR-9-5p
(44). SOX17 methylation
frequency has been reported to be significantly higher in squamous
cell carcinoma than in cervical adenocarcinoma, indicating that
SOX17 silencing may contribute to the aberrant activation of
Wnt signaling in cervical cancer (45). Furthermore, SOX17 exhibited
general hypermethylation in CpG sites analyzed in cervical cancer
samples and may interact directly with CTNNB1 (46). Genes previously unreported, to the
best of our knowledge, including CEP55 and ID4, were
also identified in this study.
In summary, the development and progression of
cervical cancer is likely to be induced by various processes. The
present study identified DEGs that may be potential targets for
regulation in cervical cancer.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81373075,
8167140, 81371748 and 81571395).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bosch FX and de Sanjosé S: Chapter 1:
Human papillomavirus and cervical cancer-burden and assessment of
causality. J Natl Cancer Inst Monogr. 3–13. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Thanagumtorn K: Survival rate of recurrent
cervical carcinoma. J Med Assoc Thai. 95 Suppl 3:S125–S130.
2012.PubMed/NCBI
|
|
4
|
zur Hausen H: Papillomaviruses and cancer:
From basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Canavan TP and Doshi NR: Cervical cancer.
Am Fam Physician. 61:1369–1376. 2000.PubMed/NCBI
|
|
6
|
Bosch FX, Manos MM, Muñoz N, Sherman M,
Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R and Shah KV:
Prevalence of human papillomavirus in cervical cancer: A worldwide
perspective. International biological study on cervical cancer
(IBSCC) Study Group. J Natl Cancer Inst. 87:796–802. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Winkelstein W Jr: Smoking and cervical
cancer-current status: A review. Am J Epidemiol. 131:945–960. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ledwaba T, Dlamini Z, Naicker S and Bhoola
K: Molecular genetics of human cervical cancer: Role of
papillomavirus and the apoptotic cascade. Biol Chem. 385:671–682.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong YF, Cheung TH, Tsao GS, Lo KW, Yim
SF, Wang VW, Heung MM, Chan SC, Chan LK, Ho TW, et al: Genome-wide
gene expression profiling of cervical cancer in Hong Kong women by
oligonucleotide microarray. Int J Cancer. 118:2461–2469. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Biewenga P, Buist MR, Moerland PD, Ver
Loren van Themaat E, van Kampen AH, ten Kate FJ and Baas F: Gene
expression in early stage cervical cancer. Gynecol Oncol.
108:520–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hu X, Schwarz JK, Lewis JS Jr, Huettner
PC, Rader JS, Deasy JO, Grigsby PW and Wang X: A microRNA
expression signature for cervical cancer prognosis. Cancer Res.
70:1441–1448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F,
Xie X, Zhou C and Lu W: Suppressed miR-424 expression via
upregulation of target gene Chk1 contributes to the progression of
cervical cancer. Oncogene. 32:976–987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berger B, Peng J and Singh M:
Computational solutions for omics data. Nat Rev Genet. 14:333–346.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pappa KI, Polyzos A, Jacob-Hirsch J,
Amariglio N, Vlachos GD, Loutradis D and Anagnou NP: Profiling of
discrete gynecological cancers reveals novel transcriptional
modules and common features shared by other cancer types and
embryonic stem cells. PLoS One. 10:e01422292015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
den Boon JA, Pyeon D, Wang SS, Horswill M,
Schiffman M, Sherman M, Zuna RE, Wang Z, Hewitt SM, Pearson R, et
al: Molecular transitions from papillomavirus infection to cervical
precancer and cancer: Role of stromal estrogen receptor signaling.
Proc Natl Acad Sci USA. 112:E3255–E3264. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gentleman RC, Carey VJ, Bates DM, Bolstad
B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al:
Bioconductor: Open software development for computational biology
and bioinformatics. Genome Biol. 5:R802004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kerr MK: Linear models for microarray data
analysis: Hidden similarities and differences. J Comput Biol.
10:891–901. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qin W, Dong P, Ma C, Mitchelson K, Deng T,
Zhang L, Sun Y, Feng X, Ding Y, Lu X, et al: MicroRNA-133b is a key
promoter of cervical carcinoma development through the activation
of the ERK and AKT1 pathways. Oncogene. 31:4067–4075. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fang W, Shu S, Yongmei L, Endong Z, Lirong
Y and Bei S: miR-224-3p inhibits autophagy in cervical cancer cells
by targeting FIP200. Sci Rep. 6:332292016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wen SY, Lin Y, Yu YQ, Cao SJ, Zhang R,
Yang XM, Li J, Zhang YL, Wang YH, Ma MZ, et al: miR-506 acts as a
tumor suppressor by directly targeting the hedgehog pathway
transcription factor Gli3 in human cervical cancer. Oncogene.
34:717–725. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Griffiths-Jones S, Saini HK, van Dongen S
and Enright AJ: miRBase: Tools for microRNA genomics. Nucleic Acids
Res. 36:(Database Issue). D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin
YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, et al: miRTarBase
2016: Updates to the experimentally validated miRNA-target
interactions database. Nucleic Acids Res. 44:D239–D247. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lewis BP, Shih IH, Jones-Rhoades MW,
Bartel DP and Burge CB: Prediction of mammalian microRNA targets.
Cell. 115:787–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kutmon M, Kelder T, Mandaviya P, Evelo CT
and Coort SL: CyTargetLinker: A cytoscape app to integrate
regulatory interactions in network analysis. PLoS One.
8:e821602013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Smoot ME, Ono K, Ruscheinski J, Wang PL
and Ideker T: Cytoscape 2.8: New features for data integration and
network visualization. Bioinformatics. 27:431–432. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ciriello G, Cerami E, Sander C and Schultz
N: Mutual exclusivity analysis identifies oncogenic network
modules. Genome Res. 22:398–406. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cancer Genome Atlas Research Network;
Albert Einstein College of Medicine; Analytical Biological
Services; Barretos Cancer Hospital; Baylor College of Medicine;
Beckman Research Institute of City of Hope; Buck Institute for
Research on Aging; Canada's Michael Smith Genome Sciences Centre;
Harvard Medical School, . Helen F. Graham Cancer Center &
Research Institute at Christiana Care Health Services, et
al: Integrated genomic and molecular characterization of
cervical cancer. Nature. 543:378–384. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fabregat A, Sidiropoulos K, Garapati P,
Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F,
McKay S, et al: The Reactome pathway Knowledgebase. Nucleic Acids
Res. 44:D481–D487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Law V, Knox C, Djoumbou Y, Jewison T, Guo
AC, Liu Y, Maciejewski A, Arndt D, Wilson M, Neveu V, et al:
DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids
Res. 42:(Database Issue). D1091–D1097. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang W, Soares J, Greninger P, Edelman EJ,
Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et
al: Genomics of drug sensitivity in cancer (GDSC): A resource for
therapeutic biomarker discovery in cancer cells. Nucleic Acids Res.
41:(Database Issue). D955–D961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mi H, Huang X, Muruganujan A, Tang H,
Mills C, Kang D and Thomas PD: PANTHER version 11: Expanded
annotation data from Gene Ontology and Reactome pathways, and data
analysis tool enhancements. Nucleic Acids Res. 45:D183–D189. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aguirre-Gamboa R, Gomez-Rueda H,
Martínez-Ledesma E, Martínez-Torteya A, Chacolla-Huaringa R,
Rodriguez-Barrientos A, Tamez-Peña JG and Treviño V: SurvExpress:
An online biomarker validation tool and database for cancer gene
expression data using survival analysis. PLoS One. 8:e742502013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hadley W: Ggplot2: Elegrant graphics for
data analysis. Springer; Switzerland: 2016
|
|
36
|
Xiao S, Zhou Y, Yi W, Luo G, Jiang B, Tian
Q, Li Y and Xue M: Fra-1 is downregulated in cervical cancer
tissues and promotes cervical cancer cell apoptosis by p53
signaling pathway in vitro. Int J Oncol. 46:1677–1684. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han X, Tang J, Wang J, Ren F, Zheng J,
Gragg M, Kiser P, Park PS, Palczewski K, Yao X and Zhang Y:
Conformational change of human checkpoint kinase 1 (Chk1) induced
by DNA damage. J Biol Chem. 291:12951–12959. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gao Q, Huang X, Tang D, Cao Y, Chen G, Lu
Y, Zhuang L, Wang S, Xu G, Zhou J and Ma D: Influence of chk1 and
plk1 silencing on radiation- or cisplatin-induced cytotoxicity in
human malignant cells. Apoptosis. 11:1789–1800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mazumder Indra D, Mitra S, Roy A, Mondal
RK, Basu PS, Roychoudhury S, Chakravarty R and Panda CK:
Alterations of ATM and CADM1 in chromosomal 11q22.3–23.2 region are
associated with the development of invasive cervical carcinoma. Hum
Genet. 130:735–748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hsieh WT, Lin HY, Chen JH, Lin WC, Kuo YH,
Wood WG, Lu HF and Chung JG: Latex of Euphorbia antiquorum-induced
S-phase arrest via active ATM kinase and MAPK pathways in human
cervical cancer HeLa cells. Environ Toxicol. 30:1205–1215. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robertson KD and Jones PA: Tissue-specific
alternative splicing in the human INK4a/ARF cell cycle regulatory
locus. Oncogene. 18:3810–3820. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wijetunga NA, Belbin TJ, Burk RD, Whitney
K, Abadi M, Greally JM, Einstein MH and Schlecht NF: Novel
epigenetic changes in CDKN2A are associated with progression of
cervical intraepithelial neoplasia. Gynecol Oncol. 142:566–573.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB
and Herman JG: In situ detection of the
hypermethylation-induced inactivation of the p16 gene as an early
event in oncogenesis. Proc Natl Acad Sci USA. 96:12754–12759. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Irie N, Weinberger L, Tang WW, Kobayashi
T, Viukov S, Manor YS, Dietmann S, Hanna JH and Surani MA: SOX17 is
a critical specifier of human primordial germ cell fate. Cell.
160:253–268. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
van der Meide WF, Snellenberg S, Meijer
CJ, Baalbergen A, Helmerhorst TJ, van der Sluis WB, Snijders PJ and
Steenbergen RD: Promoter methylation analysis of WNT/β-catenin
signaling pathway regulators to detect adenocarcinoma or its
precursor lesion of the cervix. Gynecol Oncol. 123:116–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen YC, Huang RL, Huang YK, Liao YP, Su
PH, Wang HC, Chang CC, Lin YW, Yu MH, Chu TY and Lai HC:
Methylomics analysis identifies epigenetically silenced genes and
implies an activation of β-catenin signaling in cervical cancer.
Int J Cancer. 135:117–127. 2014. View Article : Google Scholar : PubMed/NCBI
|