Introduction
Worldwide, cancer is the second-leading cause of
mortality following cardiovascular disease. A certain proportion of
several cancer types, including colorectal cancer, can only be
diagnosed at an advanced stage (1).
However, certain cancer types, such as hepatocellular carcinoma
(HCC), can easily recur following a short duration despite
effective treatment (2). In addition,
certain other cancer types are accompanied by severe complications,
including failure of the vital organs, despite diagnosis at an
early stage and surgery is the contraindication. Established
chemotherapies are not suitable for certain patients (1). Hence, the development of a novel
therapeutic approach to enhance the overall prognosis of patients
with cancer is essential.
Vitamin K (VK) is an essential lipid-soluble vitamin
that is comprised of three types, VK1, VK2, and VK3. VK can
activate coagulation factors (factor II, VII, IX, and X), protein C
and protein S by facilitating γ-glutamyl carboxylase to catalyze
the carboxylation of glutamic acid residues (3). In addition, VK-dependent γ-carboxylation
has an essential role in maintaining bone homeostasis (4). A lack of VK can lead to severe neonatal
bleeding and osteoporosis, which can be treated by the clinical
application of VK2 (5).
Previous reports have demonstrated that VK1, VK2 and
VK3 can inhibit several neoplastic cell lines at different levels,
primarily by inducing apoptosis and cell cycle arrest of cancer
cells (6), including HCC, leukemia,
colorectal cancer, ovarian cancer, pancreatic cancer and lung
cancer. Although the inhibition caused by VK3 is highly potent, VK3
is also highly toxic. By contrast, VK2 is milder, but causes no
side effects, whereas VK1 has the least strong function (7). Hence, VK2 is a potential
chemotherapeutic candidate for the treatment of cancer. The present
review summarizes the results of VK2 against cancer in clinical,
animal and in vitro experiments and aims to elucidate the
mechanisms of anticancer effects of VK2.
Administration of VK2 in patients with
cancer
To date, several case reports have highlighted the
utility of VK2 as a potential antitumor agent. A prior study
reported that daily administration of VK2 alleviated pancytopenia
in an 80-year-old woman with myelodysplastic syndrome (MDS) and
rendered red-cell transfusions redundant after 14 months (8). Similarly, a 72-year-old woman with
relapsing acute promyelocytic leukemia was reported to attain
complete remission following the combination treatment of VK2 and
all-trans retinoic acid (9).
Treatment with a combination of VK2 and an angiotensin-converting
enzyme inhibitor was shown to shrink a hepatic dysplastic nodule in
a 66-year-old Japanese woman with liver cirrhosis (10). Furthermore, the combination of VK2 and
vitamin E suppressed the growth of the primary tumor and
obliterated the intraperitoneal dissemination in a 65-year-old man
with ruptured HCC (11).
These encouraging case reports led to several
clinical studies on the anticancer functions of VK2. A multicenter
pilot study on VK2 treatment of MDS and post-MDS acute myeloid
leukemia (AML) revealed that VK2 could significantly reduce blastic
cell numbers in the bone marrow and/or peripheral blood and enhance
hematopoiesis, particularly in patients with post-MDS AML (12). Sada et al (13) demonstrated an association between
improvements in hematopoiesis and the anti-apoptotic effect of VK2
on normal erythroid progenitors. In addition, the results of
several studies (14–18) indicate that VK2 could potentially
suppress the development and recurrence of HCC in patients. A study
aiming to investigate the VK2-mediated prevention of osteoporosis
in female cirrhotic patients deduced that VK2 may decrease the risk
of HCC in female cirrhotic patients (14). Another study investigating the
function of VK2 in patients with type C cirrhosis concluded that
VK2 exerted inhibitory effects on HCC development in patients with
type C cirrhosis (15). Mizuta et
al (16) reported that VK2 could
reduce the recurrence rates of HCC and enhance the survival rates.
Kakizaki et al (17)
investigated the effects of VK2 on recurrence in patients with HCC
derived from HCV infection, with the results corroborating those
obtained by Mizuta et al (16)
Ishizuka et al (18) suggested
that VK2 moderately inhibited HCC recurrence following curative
hepatectomy. Although the results of certain studies (16–18) did
not find statistically significant results, the majority of studies
at present, except that conducted by Yoshida et al,
considered VK2 as a valuable agent for clinical therapy in patients
with cancer. Yoshida et al demonstrated that the
VK2-dependent inhibition of HCC recurrence was not proven in a
double-blind, randomized, placebo-controlled study (2). However, this study may have had problems
with its design. First, Yoshida et al (2) enrolled patients with an increased
recurrence and who had recurred after the first treatment. Second,
the quality of VK2, which is susceptible to decrease following
exposure to light, may also have affected the results. In addition,
Zhong et al (19) conducted a
meta-analysis based on six recent randomized control trials and one
cohort study; the authenticity assessment of this meta-analysis was
high. The results indicated that VK2 treatment could significantly
decrease the 2- and 3-year tumor recurrence rate, but could not
significantly decrease the 1-year recurrence rate, and could also
increase the 1-, 2-, and 3-year survival rate (19). Overall, we hypothesize that VK2 can
exert positive effects on the therapy of patients with cancer.
Anticancer effect of VK2 in animal
research
Consistent with the results of clinical studies,
data from animal studies indicated that VK2 treatment significantly
inhibited tumor growth, without any evident side effects. For
instance, exposing male BALB/c-nu/nu mice implanted with PLC/PRF/5
HCC cells to VK2 exhibited evident suppression of the growth of
subcutaneous HCC tumors. In addition, decreases in cyclin D1 and
cyclin-dependent kinase 4 (CDK4) levels indicated that VK2 may
suppress tumor cells in vivo by inducing G1
arrest (7). Notably, mice bearing
established colorectal cancer cells in the VK2 group exhibited no
apparent changes compared with the control group, where the fur and
weight of mice changed substantially. Following examination of
apoptotic cells in vivo, researchers deduced that VK2 could
potentially inhibit colorectal cancer cells by accelerating
apoptosis (1). Hence, the induction
of the cell-cycle arrest and apoptosis has a crucial role in the
antitumor mechanism of VK2. Besides, it is established that VK2
could protect affected cells from forming precancerous lesions to
reduce hepatocarcinogenesis in animals (20).
The combination of VK2 with other anticancer agents
can be synergistic in tumor-bearing animals. For instance,
pretreatment with VK2 prior to sorafenib treatment is proven to
exert more effective HCC growth inhibition in animals than
treatment with either alone (21).
Similarly, VK2 and phosphatidylcholine together can exert a
stronger inhibition on tumorigenesis, which can be applied to
prevent hepatocarcinogenesis in patients at a high risk of HCC,
particularly those with chronic hepatitis, while preserving the
hepatic function (22). To summarize,
animal studies have demonstrated that VK2 could repress cancer
growth, which is likely to be associated with the induction of
cell-cycle arrest and apoptosis.
Inhibitory effect of VK2 on cancer
cells
Several in vitro experiments (1,23–28) have certified the anticancer effect of
VK2 against several neoplastic cell lines, including HCC, leukemia,
cholangiocarcinoma, ovarian cancer, pancreatic cancer, and
colorectal cancer. These studies also investigated the mechanism of
VK2 inhibition of cancer cells. Although several details concerning
this mechanism require clarification, the results of these studies
mainly focus on the growth inhibition of cancer cells caused by the
induction of cell-cycle arrest, cell differentiation, apoptosis and
autophagy, and the suppression of cancer cell invasion.
Inhibition of proliferation of cancer
cells by VK2
VK2 can inhibit the proliferation of cancer cells by
inducing the cell-cycle arrest of cancer cells, in which inhibition
of nuclear factor-κB (NF-κB) activity has a crucial role. NF-κB is
a regulatory factor that can be simulated by cytokines to
participate in the immune and inflammatory reaction (29). In addition, as a nuclear transcription
factor, NF-κB is associated with cell growth by regulating the
cyclin D1 gene (23). Cyclin D1 can
contribute to the G1-S transformation during the cell
cycle by binding to CDK4 or CDK6 (30). Reportedly, NF-κB and cyclin D1 are
involved in carcinogenesis. In cancer cells, VK2 can downregulate
the expression of cyclin D1 by inhibiting the binding of NF-κB to
the cyclin D1 promoter, which is followed by cell-cycle arrest in
the G1 phase (23).
Regarding the suppression of the aberrant activity of NF-κB by VK2,
certain studies reported results using HCC cells as follows. First,
VK2 can inhibit the IκB kinase (IKK)/IκB/NF-κB pathway (23). Usually, NF-κB exists in the cytosol
and is inactivated by the inhibitor of NF-κB (IκB). IκB can be
phosphorylated by IKK in response to certain stimulatory factors
prior to degrading IκB, which leads to the nuclear translocation of
NF-κB and the activation of associated genes regulated by NF-κB
(31). VK2 inhibited the function of
IKK, which suppressed the phosphorylation of IkB and activity of
NF-κB (23). Second, VK2 can inhibit
the protein kinase Cα (PKCα)/NF-κB and PKCε/protein kinase D1
(PKD1)/NF-κB pathways (24). Protein
kinase C (PKC) is a phospholipid-dependent kinase family, which is
reportedly involved in the activation of NF-kB and cell growth.
PKD1 is the effector of PKC and has been confirmed to be
phosphorylated entirely by PKCε (32). Xia et al (24) suggested that VK2 inhibited NF-κB
activation by inhibiting the catalytic action of activated PKCα,
not by phosphorylation of PKCα, and that VK2 could also inhibit
NF-κB activation by hindering the phosphorylation of PKCε and thus
further suppressing PKD1 phosphorylation. Besides, PKD1 was found
to promote IκB phosphorylation, from which the PKC/PKD1 pathway may
be one of the upstream pathways through which VK2 restrains IKK
activity.
In addition to PKC, protein kinase A (PKA), which
can lead to cell-cycle arrest at the G1 and
G2-M phase, is another type of kinase involved in the
mechanism of VK2 against tumor cells. VK2 has been identified to
stimulate the phosphorylation of PKA and activate activating
protein 2 (AP-2), upstream transcription factor-1 (USF-1), and
cAMP-response element binding protein (CREB) transcriptional
factors to inhibit the proliferation of HCC cells (33). At present, the activities of these
transcriptional factors are possibly the downstream pathways of VK2
activating PKA, because PKA is the only known regulator of AP-2,
USF-1, and CREB. However, the details about the mechanism by which
VK2 activates PKA continue to require clarification.
The promotion of VK2 on the transcription of p21 and
p27 is another way to effectively promote cell-cycle arrest of
cancer cells. Cell-cycle regulatory proteins p27 and p21 are the
two members of the Cip/Kip family, working as CDK inhibitors, which
have a negative role in the cell-cycle progression of the
G1-S transition. In HCC cells, VK2 induces G1
arrest by activating the promoter of the p21 gene and increasing
the expression of p21; however, the activity of p27 is not
interfered with by VK2. It has been identified that the primary
interaction sites of VK2 and the promoter region of the p21 gene
are the sequences between −2,130 and −102 bp of p21 (34). By contrast, VK2 upregulates the
expression of p27 to result in the G0-G1
arrest in leukemic cells (28). The
difference between VK2 regulating p21 and p27 in HCC cells and
leukemic cells is likely attributed to cancer cell types, and the
role p21 and p27 have in other tumor cell lines inhibited by VK2
warrants further investigation.
Furthermore, suppression of the c-MYC expression is
possibly associated with the cell-cycle arrest of cancer cells
induced by VK2. c-MYC is highly expressed in a variety of human
tumors, and the induction of cell-cycle arrest and differentiation
of AML cells by all-trans retinoic acid is primarily
attributed to c-MYC downregulation (35). Maniwa et al (35) identified VK2 suppression on c-MYC
expression and deduced that 5 µΜ VK2 exposure inhibited c-MYC
expression in HL-60 leukemia cells to ~80% that of control
cells.
Finally, in HCC cells, it has been identified that
VK2 could evidently downregulate the expression of hepatoma-derived
growth factor (HDGF) by interfering in the initiation of HDGF gene
transcription and subsequently inhibiting the proliferation of
cancer cells. The region −1 to −150 bp in the promoter of the HDGF
gene was detected to be the binding site of VK2 (36). HDGF is highly expressed in various
cancer cells and can transmit a proliferative signal to cells in
the early G1 phase and then stimulate the increase of
cyclin D (36). Hence, the inhibitory
activity of VK2 on HDGF may lead to G1 arrest and then
suppress the cell growth. There are two problems that remain to be
solved: i) Which pathways exert functions in the regulation of VK2
to HDGF; and ii) whether VK2 could suppress cancer cell growth by
downregulating other growth factors (Fig.
1).
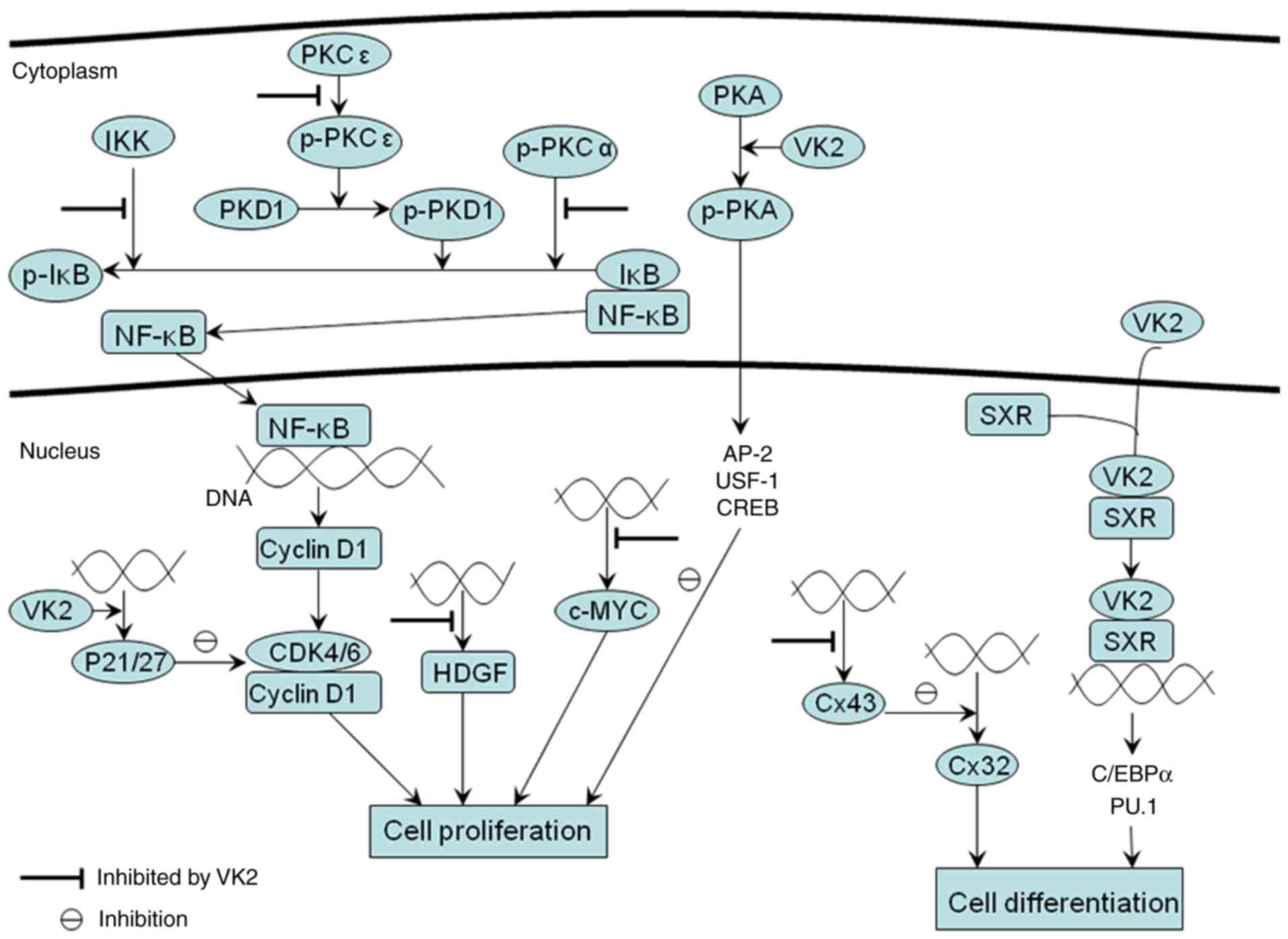 | Figure 1.Inhibition of the proliferation of
cancer cells by VK2 and induction of cell differentiation in cancer
cells by VK2. VK2 inhibits the proliferation of cancer cells by
inducing cell-cycle arrest. In cancer cells, IκB is phosphorylated
by IKK, PKD1 or p-PKCα, followed by the nuclear translocation of
NF-kB and the activation of cyclin D1 genes regulated by NF-κB,
which enhances the expression of cyclin D1 and its binding to
CDK4/6, promoting the proliferation of cancer cells. PKD1 is
phosphorylated entirely by p-PKCε. Results of studies using HCC
cells revealed that VK2 could inhibit the function of IKK, the
phosphorylation of PKCε and the catalytic action of activated PKCα,
but not the phosphorylation of PKCα. This suppresses the aberrant
activity of NF-κB and induces cell-cycle arrest in cancer cells.
The cell-cycle regulatory proteins p27 and p21 are CDK inhibitors,
acting to hinder cell-cycle progression. In HCC cells, VK2
increases the expression of P21 and then leads to cell cycle
arrest. However, in leukemic cells, VK2 upregulates the expression
p27 not p21 to induce cell cycle arrest. Besides, VK2 suppresses
the high expression of HDGF in HCC cells and c-MYC in HL-60
leukemia cells at the transcriptional level, and then induces
cell-cycle arrest. VK2 has been identified to stimulate the
phosphorylation of PKA and activate AP-2, USF-1 and CREB
transcriptional factors to inhibit the proliferation of HCC cells.
At present, the activities of these transcriptional factors are
potential downstream pathways of VK2 activation of PKA. In HCC
cells, Cx43 expression is highly upregulated, which inhibits Cx32
expression. The VK2-dependent suppression of Cx43 expression at the
transcriptional level enhances Cx32 activity, altering cancer cells
differentiation. In addition, VK2 promotes differentiation of
myeloid progenitors partly due to its binding to SXR and
upregulation of transcriptional factors C/EBPα and PU.1 crucial for
myeloid development. VK2 binding to SXR may subsequently improve
the expression of C/EBPα and PU.1, which may elucidate the reason
behind the therapeutic effect of VK2 on patients with MDS. NF-κB,
nuclear factor-κB; IκB, inhibitor of NF-κB; Cdk, cyclin-dependent
kinase; CREB, cAMP-response element binding protein; Cx, connexin;
HCC, hepatocellular carcinoma; HDGF, hepatoma-derived growth
factor; IKK, IκB kinase; MDS, myelodysplastic syndrome; p-PKA,
phosphorylated protein kinase A; p-PKCα, phosphorylated PKCα;
p-PKCε, phosphorylated PKCε; PKD1, protein kinase D1; p-PKD1,
phosphorylated protein kinase D1; SXR, steroid and xenobiotic
receptor; VK2, vitamin K2; AP-2, activating protein2; USF-1,
upstream transcription factor; C/EBPα, CCAAT/enhancer-binding
protein-α. |
Induction of cell differentiation in
cancer cells by VK2
It has been certified in various cell lines that
cell-cycle arrest is closely associated with cell differentiation.
VK2 can exhibit differentiation-inducing results by the induction
of the G0-G1 arrest in cancer cells. For
instance, Miyazawa et al (28)
reported that VK2 treatment induced monocytic differentiation in
HL-60-Bcl-2 leukemia cells, with an evident increase in the
proportion of cells in the G0-G1 phase.
Maniwa et al (35) determined
that VK2 reduced c-MYC expression, which subsequently contributed
to cell growth arrest and cell differentiation. Hence, the
molecular activities regulated by VK2, including the induction of
cell-cycle arrest in cancer cells, are likely to be crucial to the
mechanism of the VK2-dependent stimulation of cancer cell
differentiation. Notably, cell-cycle arrest is only one of the
processes that can lead to the differentiation of cancer cells.
VK2 suppresses connexin 43 (Cx43) expression and
enhances Cx32 activity, which is another mechanism of cancer cell
differentiation. Cx proteins are comprised of gap junction channel
structures that mediate intercellular communication to maintain
tissue homeostasis. Studies indicate that Cx genes have
tumor-suppressing effects and the distribution of Cx is tissue
specific (37). Cx32 is mainly
expressed in hepatocytes. The risk of tumor development can be
raised when knocking out the Cx32 gene in mice. However, Cx43 is
not found in regular hepatocytes, and its expression is highly
upregulated in HCC cells (38).
Regarding cell differentiation induced by VK2, Kaneda et al
(37) reported that VK2 exposure
could drive Huh7 HCC cells to adopt a normal liver cell phenotype
by upregulating Cx32 indirectly via downregulation of Cx43 at the
transcriptional level, and the increase of gap junction
intercellular communication enhanced by VK2 is potentially due to
Cx32 upregulation.
VK2-induced differentiation is partly associated
with steroid and xenobiotic receptor (SXR). VK2 can work as the
ligand of SXR, a nuclear receptor, and their binding largely exerts
osteoprotective function. Sada et al (13) investigated the effects of VK2 on
normal hematopoietic progenitor cells and revealed that VK2
promotes the differentiation of myeloid progenitors partly owing to
its binding to SXR and the upregulation of the transcriptional
factors CCAAT/enhancer-binding protein α (C/EBPα) and PU.1, which
are crucial for myeloid development. In addition, VK2 binding to
SXR may subsequently improve the expression of C/EBPα and PU.1,
which may aid elucidation of the therapeutic effect of VK2 on
patients with MDS (Fig. 1) (13).
Induction of apoptosis of cancer cells
by VK2
The mitochondrial pathway is a crucial process
through which VK2 exerts its pro-apoptotic function. Following
treatment with VK2, the mitochondrial membrane potential is
depolarized and cytochrome c is released from the mitochondria into
the cytosol to form the apoptosome, driving activation of
caspase-9, which ultimately leads to the activation of caspase-3
and initiation of cell apoptosis (39–41). It is
anticipated that the selective binding of VK2 and VK2-2,3 epoxide
(VK2-O) to the mitochondrial protein Bcl-2 antagonist killer 1
(Bak) is an essential molecular mechanism of VK2 dissipating the
mitochondrial membrane potential of leukemia cells. In addition,
the direct binding of VK2 to Bak specifically resulted in a
post-translational modification of Bak (39). It has been revealed that VK2 and VK2-O
metabolized from VK2 could non-covalently bind to Bak through the
Arg-169 and Trp-170 residues, and that VK2-O further covalently
attached to the Cys-166 residue of Bak in HL-60 leukemia cells
(39). In addition, VK2 treatment
increased the intracellular level of the reactive oxygen species
(ROS) (39,40). Evidence indicates that VK2-induced ROS
generation occurred prior to the induction of apoptosis. Assumedly,
ROS contributes by converting VK2 to VK2-O (39). Besides Bak, other Bal-2 family members
also have crucial roles in the mitochondrial apoptosis pathway
induced by VK2. VK2 decreases Bcl-2 expression and increases the
expression of Bcl-2-associated X protein (Bax) in a newly
established MDS cell line, which was associated with apoptosis
(42). Furthermore, Tsujioka et
al (41) determined that the
ratio of B-cell lymphoma (Bcl)-extra large and Bcl-extra small
expression decreased when exposing myeloma cells to 10 µΜ VK2;
however, the expression of Bcl-2 and Bax remained unaffected. VK2
can thus alter the expression of the Bcl-2 family, tending to a
pro-apoptotic balance and activating the mitochondrial apoptosis
pathway of cancer cells. It is hypothesized that the release of
cytochrome c from the mitochondria partly results from the acidic
phospholipid cardiolipin (CL), abundant in the outer membrane of
the mitochondria, being peroxidated by ROS (40), which remains to be confirmed.
Furthermore, the mitogen-activated protein kinase
(MAPK) pathway is essential for the VK2-mediated mitochondrial
apoptosis pathway (25,26,41,43). In
the MAPK superfamily, p38 MAPK and c-Jun N-terminal kinase (JNK)
pathways respond to stress and contribute to inflammation or even
apoptosis, whereas the extracellular signal-related kinase (ERK)
pathway reacts to growth factors or other external mitogenic
signals by stimulating cell proliferation and resisting apoptotic
signals. VK2 activates p38 MAPK to its phosphorylated form and
subsequently results in apoptosis of the neoplastic cells (41,44).
Tsujioka et al (41)
identified this in myeloma cells, following which they detected
that the mitochondrial membrane potential was depolarized and
caspase-9 was activated, indicating that the phosphorylation of p38
MAPK stimulated by VK2 possibly induces apoptosis by initiating the
mitochondrial pathway. In addition, these authors found that ROS
generation stimulated by VK2 is associated with this apoptosis
(41). Sibayama-Imazu et al
(25) investigated the apoptosis
induction of PA-1 ovarian cancer cells by VK2 and concluded that
the increase of synthesis and accumulation in the mitochondria of
TR3, also known as Nur77 and neuron growth factor inducible factor
I-B, highly expressed in various tumor cell lines, is possibly
associated with the mitochondrial apoptosis pathway induced by VK2.
In addition, Sibayama-Imazu et al (25) deduced that VK2 may activate JNK to
phosphorylate TR3 and increase TR3 levels in the mitochondria. VK2
is reported to inhibit ERK phosphorylation by suppressing Ras
activation and subsequently suppressing the activation of MAPK
kinase (MEK), which causes the apoptosis of HCC cells (43). Conversely, research investigating the
inhibitory role of VK2 on pancreatic cancer demonstrated that
apoptosis of cancer cells treated with VK2 was primarily associated
with an increase in phosphorylated ERK (26). This contradiction may be due to
differences in the type of cancer cells, and requires further
investigation. Another unsolved problem is that not all pancreatic
cancer cell lines are sensitive to VK2 treatment. Hence, the
effects of VK2 on pancreatic cancer cells warrant further
investigation.
In addition to the mitochondrial pathway, described
as an intrinsic pathway, the extrinsic apoptosis pathway also
participates in the mechanism of VK2-dependent induction of cell
death. Evidence indicates that VK2 can induce apoptosis in cancer
cells by activating p53 and initiating the extrinsic apoptosis
pathway (45). Notably, p53 is a
multi-faceted tumor-repressor gene capable of inducing cell-cycle
arrest, cell differentiation or apoptosis in reaction to oncogenic
stress (46). The extrinsic apoptosis
pathway is depedent on the death receptors binding to ligands to
form the death-inducing signaling complex, which then contributes
to caspase-8 activation and further activates caspase-3 (47). A study investigating the antitumor
effects of VK2 in Smmc-7721 HCC cells established that VK2
stimulated the extrinsic apoptosis pathway by increasing p53
phosphorylation and then activating caspase-8 (Fig. 2) (45).
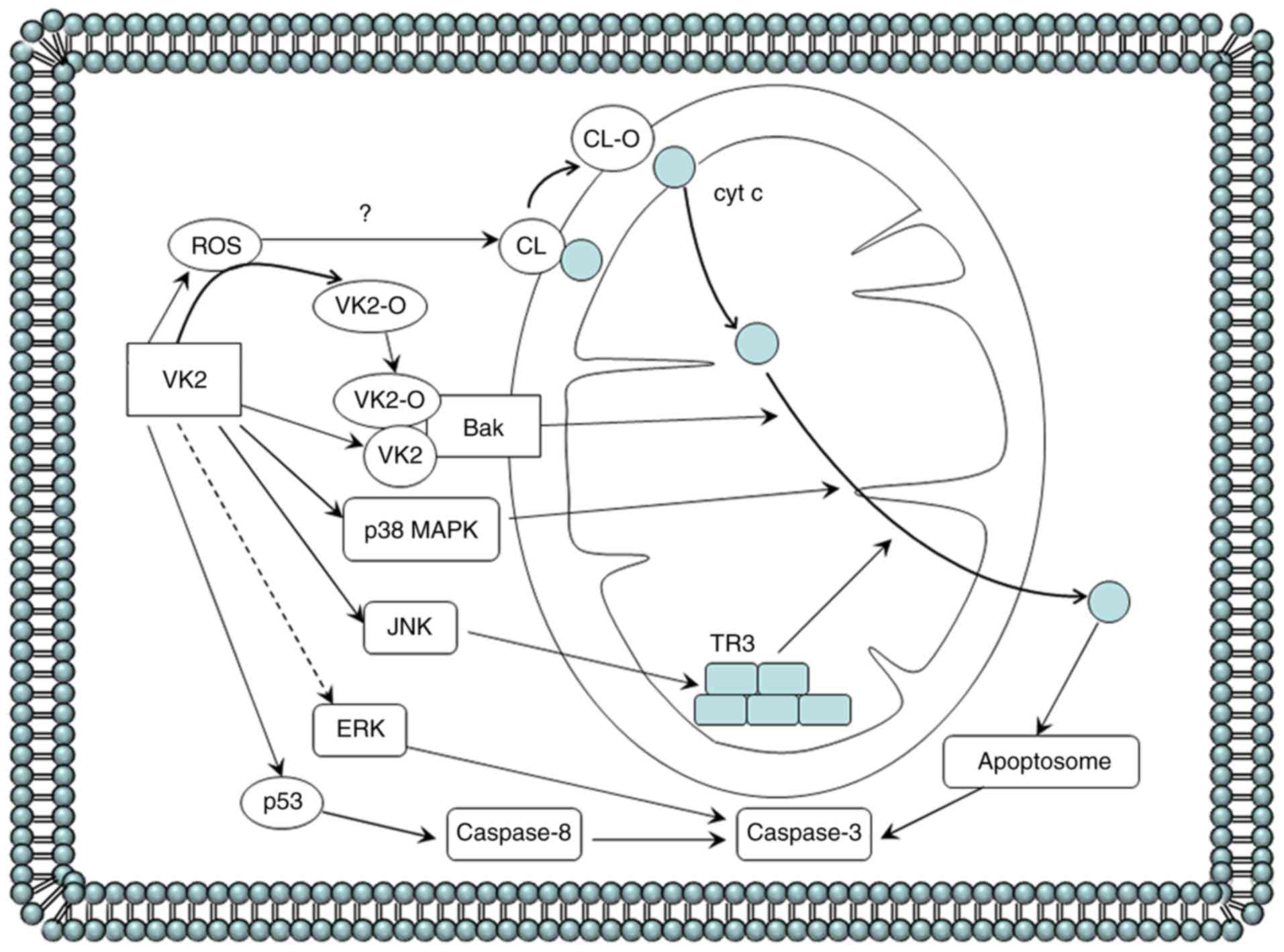 | Figure 2.Cell apoptosis induced by VK2 in
cancer cells. VK2 induces apoptosis in cancer cells by depolarizing
the mitochondrial membrane potential, followed by cytochrome c
release from the mitochondria into the cytosol to form apoptosomes,
which then activates caspase-3. The precise mechanism of
VK2-dependent initiation of the mitochondrial apoptosis pathway of
cancer cells is as follows. In HL-60 leukemia cells, VK2 and VK2-O
selectively binds to the mitochondrial protein Bak. VK2-induced ROS
generation prior to the induction of apoptosis possibly contributes
by converting VK2 to VK2-O. In myeloma cells, VK2 activates p38
MAPK to its phosphorylated form. VK2 exposure in PA-1 ovarian
cancer cells may activate JNK to phosphorylate TR3, also known as
Nur77 and neuron growth factor inducible factor I-B, and increase
TR3 levels in the mitochondria. The hypothesis that the release of
cytochrome c from the mitochondria partly results from the acidic
phospholipid CL being peroxidated by ROS is yet to be confirmed.
The role of ERK in the VK2-dependent activation of caspase-3 and
induction of apoptosis in hepatocellular carcinoma and pancreatic
cancer cells is contradictory, so this pathway is represented with
a dashed line. VK2 can inhibit ERK phosphorylation by suppressing
the Ras activation and subsequently suppressing the catalysis of
MEK, which causes apoptosis in HCC cells. Conversely, VK2-dependent
induction of pancreatic cancer cell apoptosis is primarily
associated with an increase in levels of phosphorylated ERK. In
addition, VK2 stimulates the extrinsic apoptosis pathway by
increasing p53 phosphorylation and then activating caspase-8 in
Smmc-7721 HCC cells. Bak, Bcl-2 antagonist killer 1; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2 associated X protein; CL, cardiolipin; HCC,
hepatocellular carcinoma; VK2, vitamin K2; VK2-O, VK2-2,3 epoxide;
ROS, reactive oxygen species; MAPK, mitogen-activated protein
kinase; JNK, c-Jun N-terminal kinase; MEK, MAPK kinase; ERK,
extracellular-signal-related kinase. |
Another previous study (6) reported that a large dose of VK2 could
induce apoptosis of Hep40 HCC cells by increasing the expression of
c-JUN and c-MYC; however, it did not identify the detailed
apoptosis pathway.
Induction of autophagy in cancer cells
by VK2
In different cancer cell lines, VK2 can inhibit the
growth of cancer cells by evoking autophagy. Autophagy is a
mechanism that aids cell survival in response to stresses, such as
nutrition starvation. It has been reported that autophagy is
essential for the inhibition of certain antitumor agents in cancer
(27,48). Owing to a lack of clarity regarding
the molecular mechanism that lead to autophagy, whether autophagy
protects or promotes cell death is debatable. Yokoyama et al
(48) reported that VK2 could
stimulate apoptosis and autophagy in leukemic cells simultaneously,
but autophagy is more dominant when Bcl-2 is highly expressed,
restraining apoptosis. Hence, Yokoyama et al (48) suggested that autophagy may act as an
alternative inducer of apoptosis. Another study that exposed
cholangiocellular carcinoma cell lines to VK2 identified the
effects of inducing apoptosis and cell-cycle arrest to be
inconspicuous; however, the autophagy induction exerted a maximal
effect in VK2 inhibiting cholangiocellular carcinoma cells
(27). Among these cells lines, the
TFK-1 cell line exhibited a higher expression of Bcl-2. On the
basis of these two studies, it can be inferred that the molecular
mechanism of apoptosis may lead to autophagy, which could be
regarded as the cell state prior to apoptosis. In addition, at
least in cholangiocellular carcinoma cells, Bcl-2 may be involved
in the determination of whether cells eventually undergo apoptosis
or autophagy. When Bcl-2 is highly expressed, apoptosis induction
may change to autophagy. In short, inducing autophagy is a
significant part in the mechanism of VK2-dependent inhibition of
cancer cells, although further details remain to be
investigated.
Invasion-inhibiting functions of VK2
in cancer cells
On the basis of the study conducted by Ide et
al (49), VK2 can restrain the
invasion of tumor cells mainly by downregulating the expression of
matrix metalloproteinases (MMPs) at the transcriptional level. MMPs
are a group of proteinases that degrade extracellular matrix
proteins, which are reportedly linked to tumor invasion and
metastasis; AP-1 and NF-κB are the common transcription factors of
the promoter regions of MMPs. AP-1 is the transcription complex
comprising proto-oncogene proteins c-FOS and c-JUN, and activity of
the MAPK pathway leads to the phosphorylation of specific threonine
and tyrosine residues of c-FOS and c-JUN (50). Ide et al (49) revealed that VK2 could inhibit MMP
expression by mediating NF-κB inhibition and downregulating AP-1 by
suppressing the ERK and JNK pathways, which restrained the invasion
of HCC cells.
Synergistic effect of VK2 in
combination with other chemotherapeutics
In several cases, the combination of VK2 with other
chemotherapy agents can produce stronger effects than the use of
either alone. The mechanisms associated with the synergistic
effects can also be classified into induction of the cell-cycle
arrest, differentiation, and apoptosis in cancer cells; however,
the specific details may slightly vary from the mechanism of action
of VK2 alone. VK2 pretreatment can restrain NF-kB activation and
increase cyclin D1 expression caused by 5-fluorouracil (5-FU),
promoting cell-cycle arrest and improving the 5-FU-dependent
inhibition of HCC cell growth (51).
In addition, VK2 can enhance the ability of cotylenin A to induce
cell differentiation in HL-60 leukemia cells, as VK2 can enhance
the increase of cyclin G2 expression stimulated by cotylenin A
(35). Cyclin G2 plays a beneficial
role in promoting and maintaining cell-cycle arrest, but treatment
with VK2 alone induces a non-significant cyclin G2 expression in
cancer cells (35). When treating
with VK2 and sorafenib together in HCC cells, VK2 can counteract
the decrease of p21 level and improve the inhibition of ERK
phosphorylation induced by sorafenib, which promotes cell-cycle
arrest and apoptosis (21).
VK2 can augment the efficacy of retinoids in cancer
cells (9,43). Retinoids suppress cell growth through
nuclear receptor retinoid X receptor (RXR), which can bind to the
RXR-responsive element located in the promoter regions of
retinoid-target genes. In HCC cells, the Ras/ERK pathway is
aberrantly activated, which causes accumulation of phosphorylated
RXRα and weakens the effects of retinoids (43). However, treatment with VK2 plus
retinoid can apparently decrease the level of phosphorylated ERK
and phosphorylated RXRα by cooperatively inhibiting the Ras/ERK
pathway, which contributes to retinoid binding of functional RXRα
and to the apoptosis of HCC cells (43). This mechanism is likely to be the
reason why the combination of acyclic retinoid and VK2 could
decrease the recurrence rate of HCC in clincal trials. Therefore,
VK2 alone and the combination of VK2 with other antitumor agents
requires further investigation.
Vitamin D3 is another micronutrient capable of
restricting the growth of cancer cells by inhibiting proliferation
and stimulating differentiation (52). However, an adverse effect of vitamin
D3 treatment is hypercalcemia, which can lead to vascular
calcification. VK2 regulates the calcium deposition between bone
tissue and other tissues, and inhibits the formation of vascular
calcified foci. The combination of vitamin D3 with VK2 on cancer
cells can synergistically improve the induction of cellular
differentiation and also significantly reduces the risk of
hypercalcemia and vascular calcification (52,53).
Discussion
The present review summarizes the effects of VK2 on
cancer in clinical, in vivo, and in vitro studies.
Clinical trials demonstrated that VK2 has the potential to improve
the prognosis of patients with cancer. In addition, evidence
indicates that VK2 treatment can prevent HCC in patients with
hepatic cirrhosis, and the dietary intake of VK2 can decrease the
risk of developing cancer, particularly prostate and lung cancer
(54). Furthermore, VK2 is confirmed
to restrain tumor cell growth in animal studies (1,7,20–22,55), with
cell-cycle arrest and apoptosis involved in this inhibition. In
vitro studies (1,23–27,35)
certified that VK2 could inhibit the growth of several cancer cell
lines. Although several detailed links remain to be investigated,
studies included in the present review (23–25,33–37,39,41,45,48,49)
indicated that induction of the cell-cycle arrest, cell
differentiation, apoptosis, and autophagy is crucial for
VK2-dependent suppression of cancer cell growth. Certain protein
kinases, such as PKA and PKC, signaling pathways, such as the MAPK
pathways, transcription factors, such as NF-κB and AP-2, and
essential proteins, such as Bak and Cx43, are involved in the
mechanism of VK2 activity against cancer cells (23,24,33,37,41).
The combination treatment of VK2 with other chemotherapeutics, such
as sorafenib, can exert a synergistic effect and reduce adverse
drug reactions.
In conclusion, VK2 can positively inhibit cancer
cells. VK2 appears to be an extremely promising agent with very
limited toxicity, which can be a useful option for prevention of
cancer and clinical therapy of cancer. However, the inhibition of
vitamin K and D in cancers indicated that vitamins might have
positive effects on the prevention and therapy of tumors.
Therefore, the effects of vitamins or minerals on tumors should be
investigated further.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Nature
Science Foundation of China (grant no. 30971065), the Science and
Technology Plan of Dalian (grant no. 2012E12SF074) and the
Education Fund Item of Liaoning Province (grant no. 2009 A
194).
Availability of data and materials
All data analyzed during this study are included in
this published article.
Authors' contributions
SL decided the topic of the manuscript. FX was a
major contributor in writing the manuscript. SL, FX, JC and LD
revised it critically for important intellectual content. JC and LD
analyzed and interpreted all the data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
All authors have read and approved the final
manuscript.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AML
|
acute myelocytic leukemia
|
|
Bak
|
Bal-2 antagonist killer 1
|
|
Bal-2
|
B-cell lymphoma 2
|
|
Bax
|
Bcl-2 associated X protein
|
|
Cdk
|
cyclin-dependent kinase
|
|
CL
|
cardiolipin
|
|
CREB
|
cAMP-response element binding
protein
|
|
Cx
|
connexin
|
|
HCC
|
hepatocellular carcinoma
|
|
HDGF
|
hepatoma-derived growth factor
|
|
IKK
|
IκB kinase
|
|
MDS
|
myelodysplastic syndrome
|
|
PKD1
|
protein kinase D1
|
|
RXR
|
retinoid X receptor
|
|
RXRE
|
retinoid X receptor responsive
element
|
|
SXR
|
steroid and xenobiotic receptor
|
|
VK2
|
vitamin K2
|
|
VK
|
vitamin K
|
|
VK2-O
|
VK2-2,3 epoxide
|
References
|
1
|
Ogawa M, Nakai S, Deguchi A, Nonomura T,
Masaki T, Uchida N, Yoshiji H and Kuriyama S: Vitamins K2, K3 and
K5 exert antitumor effects on established colorectal cancer in mice
by inducing apoptotic death of tumor cells. Int J Oncol.
31:323–331. 2007.PubMed/NCBI
|
|
2
|
Yoshida H, Shiratori Y, Kudo M, Shiina S,
Mizuta T, Kojiro M, Yamamoto K, Koike Y, Saito K, Koyanagi N, et
al: Effect of vitamin K2 on the recurrence of hepatocellular
carcinoma. Hepatology. 54:532–540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li ZQ, He FY, Stehle CJ, Wang Z, Kar S,
Finn FM and Carr BI: Vitamin K uptake in hepatocytes and hepatoma
cells. Life Sci. 70:2085–2100. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Iwamoto J, Takeda T and Sato Y: Effects of
vitamin K2 on osteoporosis. Curr Pharm Des. 10:2557–2576. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ushiroyama T, Ikeda A and Ueki M: Effect
of continuous combined therapy with vitamin K(2) and vitamin D(3)
on bone mineral density and coagulofibrinolysis function in
postmenopausal women. Maturitas. 41:211–221. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bouzahzah B, Nishikawa Y, Simon D and Carr
BI: Growth control and gene expression in a new hepatocellular
carcinoma cell line, Hep40: Inhibitory actions of vitamin K. J Cell
Physiol. 165:459–467. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hitomi M, Yokoyama F, Kita Y, Nonomura T,
Masaki T, Yoshiji H, Inoue H, Kinekawa F, Kurokohchi K, Uchida N,
et al: Antitumor effects of vitamins K1, K2 and K3 on
hepatocellular carcinoma in vitro and in vivo. Int J Oncol.
26:713–720. 2005.PubMed/NCBI
|
|
8
|
Takami A, Nakao S, Ontachi Y, Yamauchi H
and Matsuda T: Successful therapy of myelodysplastic syndrome with
menatetrenone, a vitamin K2 analog. Int J Hematol. 69:24–26.
1999.PubMed/NCBI
|
|
9
|
Fujita H, Tomiyama J and Tanaka T: Vitamin
K2 combined with all-trans retinoic acid induced complete remission
of relapsing acute promyelocytic leukaemia. Br J Haematol.
103:584–585. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshiji H, Noguchi R, Yamazaki M, Ikenaka
Y, Sawai M, Ishikawa M, Kawaratani H, Mashitani T, Kitade M, Kaji
K, et al: Combined treatment of vitamin K2 and
angiotensin-converting enzyme inhibitor ameliorates hepatic
dysplastic nodule in a patient with liver cirrhosis. World J
Gastroenterol. 13:3259–3261. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Otsuka T, Hagiwara S, Tojima H, Yoshida H,
Takahashi T, Nagasaka K, Tomioka S, Ando T, Takeuchi K, Kori T, et
al: Hepatocellular carcinoma with peritoneal dissemination which
was regressed during vitamin K2 and vitamin E administration.
Intern Med. 46:711–715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyazawa K, Nishimaki J, Ohyashiki K,
Enomoto S, Kuriya S, Fukuda R, Hotta T, Teramura M, Mizoguchi H,
Uchiyama T and Omine M: Vitamin K2 therapy for myelodysplastic
syndromes (MDS) and post-MDS acute myeloid leukemia: Information
through a questionnaire survey of multi-center pilot studies in
Japan. Leukemia. 14:1156–1157. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sada E, Abe Y, Ohba R, Tachikawa Y,
Nagasawa E, Shiratsuchi M and Takayanagi R: Vitamin K2 modulates
differentiation and apoptosis of both myeloid and erythroid
lineages. Eur J Haematol. 85:538–548. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Habu D, Shiomi S, Tamori A, Takeda T,
Tanaka T, Kubo S and Nishiguchi S: Role of vitamin K2 in the
development of hepatocellular carcinoma in women with viral
cirrhosis of the liver. JAMA. 292:358–361. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kojima K, Tamano M, Akima T, Hashimoto T,
Kuniyoshi T, Maeda C, Majima Y, Kusano K, Murohisa T, Iijima M and
Hiraishi H: Effect of vitamin K2 on the development of
hepatocellular carcinoma in type C cirrhosis.
Hepatogastroenterology. 57:1264–1267. 2010.PubMed/NCBI
|
|
16
|
Mizuta T, Ozaki I, Eguchi Y, Yasutake T,
Kawazoe S, Fujimoto K and Yamamoto K: The effect of menatetrenone,
a vitamin K2 analog, on disease recurrence and survival in patients
with hepatocellular carcinoma after curative treatment: A pilot
study. Cancer. 106:867–872. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kakizaki S, Sohara N, Sato K, Suzuki H,
Yanagisawa M, Nakajima H, Takagi H, Naganuma A, Otsuka T, Takahashi
H, et al: Preventive effects of vitamin K on recurrent disease in
patients with hepatocellular carcinoma arising from hepatitis C
viral infection. J Gastroenterol Hepatol. 22:518–522. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ishizuka M, Kubota K, Shimoda M, Kita J,
Kato M, Park KH and Shiraki T: Effect of menatetrenone, a vitamin
k2 analog, on recurrence of hepatocellular carcinoma after surgical
resection: A prospective randomized controlled trial. Anticancer
Res. 32:5415–5420. 2012.PubMed/NCBI
|
|
19
|
Zhong JH, Mo XS, Xiang BD, Yuan WP, Jiang
JF, Xie GS and Li LQ: Postoperative use of the chemopreventive
vitamin K2 analog in patients with hepatocellular carcinoma. PLoS
One. 8:580822013. View Article : Google Scholar
|
|
20
|
Yoshiji H, Kuriyama S, Noguchi R, Yoshii
J, Ikenaka Y, Yanase K, Namisaki T, Kitade M, Yamazaki M, Masaki T
and Fukui H: Combination of vitamin K2 and the
angiotensin-converting enzyme inhibitor, perindopril, attenuates
the liver enzyme-altered preneoplastic lesions in rats via
angiogenesis suppression. J Hepatol. 42:687–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Zhang B, Zhang A, Zhao Y, Zhao J,
Liu J, Gao J, Fang D and Rao Z: Synergistic growth inhibition by
sorafenib and vitamin K2 in human hepatocellular carcinoma cells.
Clinics. 67:1093–1099. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sakakima Y, Hayakawa A, Nagasaka T and
Nakao A: Prevention of hepatocarcinogenesis with
phosphatidylcholine and menaquinone-4: In vitro and in vivo
experiments. J Hepatol. 47:83–92. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ozaki I, Zhang H, Mizuta T, Ide Y, Eguchi
Y, Yasutake T, Sakamaki T, Pestell RG and Yamamoto K:
Menatetrenone, a vitamin K2 analogue, inhibits hepatocellular
carcinoma cell growth by suppressing cyclin D1 expression through
inhibition of nuclear factor kappaB activation. Clin Cancer Res.
13:2236–2245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xia J, Matsuhashi S, Hamajima H, Iwane S,
Takahashi H, Eguchi Y, Mizuta T, Fujimoto K, Kuroda S and Ozaki I:
The role of PKC isoforms in the inhibition of NF-kappaB activation
by vitamin K2 in human hepatocellular carcinoma cells. J Nutr
Biochem. 23:1668–1675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sibayama-Imazu T, Fujisawa Y, Masuda Y,
Aiuchi T, Nakajo S, Itabe H and Nakaya K: Induction of apoptosis in
PA-1 ovarian cancer cells by vitamin K2 is associated with an
increase in the level of TR3/Nur77 and its accumulation in
mitochondria and nuclei. J Cancer Res Clin Oncol. 134:803–812.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Showalter SL, Wang Z, Costantino CL,
Witkiewicz AK, Yeo CJ, Brody JR and Carr BI: Naturally occurring K
vitamins inhibit pancreatic cancer cell survival through a
caspase-dependent pathway. J Gastroenterol Hepatol. 25:738–744.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Enomoto M, Tsuchida A, Miyazawa K,
Yokoyama T, Kawakita H, Tokita H, Naito M, Itoh M, Ohyashiki K and
Aoki T: Vitamin K2-induced cell growth inhibition via autophagy
formation in cholangiocellular carcinoma cell lines. Int J Mol Med.
20:801–808. 2007.PubMed/NCBI
|
|
28
|
Miyazawa K, Yaguchi M, Funato K, Gotoh A,
Kawanishi Y, Nishizawa Y, You A and Ohyashiki K:
Apoptosis/differentiation-inducing effects of vitamin K2 on HL-60
cells: Dichotomous nature of vitamin K2 in leukemia cells.
Leukemia. 15:1111–1117. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Masaki T, Shiratori Y, Rengifo W, Igarashi
K, Yamagata M, Kurokohchi K, Uchida N, Miyauchi Y, Yoshiji H,
Watanabe S, et al: Cyclins and cyclin-dependent kinases:
Comparative study of hepatocellular carcinoma versus cirrhosis.
Hepatology. 37:534–543. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karin M and Delhase M: The I kappa B
kinase (IKK) and NF-kappa B: Key elements of proinflammatory
signalling. Semin Immunol. 12:85–98. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Holden NS, Squires PE, Kaur M, Bland R,
Jones CE and Newton R: Phorbol ester-stimulated NF-kappaB-dependent
transcription: Roles for isoforms of novel protein kinase C. Cell
Signal. 20:1338–1348. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Otsuka M, Kato N, Shao RX, Hoshida Y,
Ijichi H, Koike Y, Taniguchi H, Moriyama M, Shiratori Y, Kawabe T
and Omata M: Vitamin K2 inhibits the growth and invasiveness of
hepatocellular carcinoma cells via protein kinase A activation.
Hepatology. 40:243–251. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu W, Nakamura H, Yamamoto T, Ikeda N,
Saito M, Ohno M, Hara N, Imanishi H, Shimomura S, Yamamoto T, et
al: Vitamin K2 inhibits the proliferation of HepG2 cells by
up-regulating the transcription of p21 gene. Hepatol Res.
37:360–365. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maniwa Y, Kasukabe T and Kumakura S:
Vitamin K2 and cotylenin A synergistically induce monocytic
differentiation and growth arrest along with the suppression of
c-MYC expression and induction of cyclin G2 expression in human
leukemia HL-60 cells. Int J Oncol. 47:473–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamamoto T, Nakamura H, Liu W, Cao K,
Yoshikawa S, Enomoto H, Iwata Y, Koh N, Saito M, Imanishi H, et al:
Involvement of hepatoma-derived growth factor in the growth
inhibition of hepatocellular carcinoma cells by vitamin K(2). J
Gastroenterol. 44:228–235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaneda M, Zhang D, Bhattacharjee R,
Nakahama K, Arii S and Morita I: Vitamin K2 suppresses malignancy
of HuH7 hepatoma cells via inhibition of connexin 43. Cancer Lett.
263:53–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Trosko JE: The role of stem cells and gap
junctions as targets for cancer chemoprevention and chemotherapy.
Biomed Pharmacother. 59 Suppl 2:S326–S331. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Karasawa S, Azuma M, Kasama T, Sakamoto S,
Kabe Y, Imai T, Yamaguchi Y, Miyazawa K and Handa H: Vitamin K2
covalently binds to Bak and induces Bak-mediated apoptosis. Mol
Pharmacol. 83:613–620. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shibayama-Imazu T, Sonoda I, Sakairi S,
Aiuchi T, Ann WW, Nakajo S, Itabe H and Nakaya K: Production of
superoxide and dissipation of mitochondrial transmembrane potential
by vitamin K2 trigger apoptosis in human ovarian cancer TYK-nu
cells. Apoptosis. 11:1535–1543. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsujioka T, Miura Y, Otsuki T, Nishimura
Y, Hyodoh F, Wada H and Sugihara T: The mechanisms of vitamin
K2-induced apoptosis of myeloma cells. Haematologica. 91:613–619.
2006.PubMed/NCBI
|
|
42
|
Nishimaki J, Miyazawa K, Yaguchi M,
Katagiri T, Kawanishi Y, Toyama K, Ohyashiki K, Hashimoto S, Nakaya
K and Takiguchi T: Vitamin K2 induces apoptosis of a novel cell
line established from a patient with myelodysplastic syndrome in
blastic transformation. Leukemia. 13:1399–1405. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kanamori T, Shimizu M, Okuno M,
Matsushima-Nishiwaki R, Tsurumi H, Kojima S and Moriwaki H:
Synergistic growth inhibition by acyclic retinoid and vitamin K2 in
human hepatocellular carcinoma cells. Cancer Sci. 98:431–437. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Olson JM and Hallahan AR: p38 MAP kinase:
A convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li L, Qi Z, Qian J, Bi F, Lv J, Xu L,
Zhang L, Chen H and Jia R: Induction of apoptosis in hepatocellular
carcinoma Smmc-7721 cells by vitamin K(2) is associated with p53
and independent of the intrinsic apoptotic pathway. Mol Cell
Biochem. 342:125–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Meek DW: Tumour suppression by p53: A role
for the DNA damage response? Nat Rev Cancer. 9:714–723. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Muzio M, Stockwell BR, Stennicke HR,
Salvesen GS and Dixit VM: An induced proximity model for caspase-8
activation. J Biol Chem. 273:2926–2930. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yokoyama T, Miyazawa K, Naito M, Toyotake
J, Tauchi T, Itoh M, You A, Hayashi Y, Georgescu MM, Kondo Y, et
al: Vitamin K2 induces autophagy and apoptosis simultaneously in
leukemia cells. Autophagy. 4:629–640. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ide Y, Zhang H, Hamajima H, Kawaguchi Y,
Eguchi Y, Mizuta T, Yamamoto K, Fujimoto K and Ozaki I: Inhibition
of matrix metalloproteinase expression by menatetrenone, a vitamin
K2 analogue. Oncol Rep. 22:599–604. 2009.PubMed/NCBI
|
|
50
|
Karin M: The regulation of AP-1 activity
by mitogen-activated protein kinases. Philos Trans R Soc Lond B
Biol Sci. 351:127–134. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang H, Ozaki I, Hamajima H, Iwane S,
Takahashi H, Kawaguchi Y, Eguchi Y, Yamamoto K and Mizuta T:
Vitamin K2 augments 5-fluorouracil-induced growth inhibition of
human hepatocellular carcinoma cells by inhibiting NF-κB
activation. Oncol Rep. 25:159–166. 2011.PubMed/NCBI
|
|
52
|
Funato K, Miyazawa K, Yaguchi M, Gotoh A
and Ohyashiki K: Combination of 22-oxa-1,25-dihydroxyvitamin D(3),
a vitamin D(3) derivative, with vitamin K(2) (VK2) synergistically
enhances cell differentiation but suppresses VK2-inducing apoptosis
in HL-60 cells. Leukemia. 16:1519–1527. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Iguchi T, Miyazawa K, Asada M, Gotoh A,
Mizutani S and Ohyashiki K: Combined treatment of leukemia cells
with vitamin K2 and 1alpha,25-dihydroxy vitamin D3 enhances
monocytic differentiation along with becoming resistant to
apoptosis by induction of cytoplasmic p21CIP1. Int J Oncol.
27:893–900. 2005.PubMed/NCBI
|
|
54
|
Nimptsch K, Rohrmann S, Kaaks R and
Linseisen J: Dietary vitamin K intake in relation to cancer
incidence and mortality: Results from the Heidelberg cohort of the
European Prospective Investigation into cancer and nutrition
(EPIC-Heidelberg). Am J Clin Nutr. 91:1348–1358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yoshiji H, Kuriyama S, Noguchi R, Yoshii
J, Ikenaka Y, Yanase K, Namisaki T, Kitade M, Yamazaki M, Akahane
T, et al: Amelioration of carcinogenesis and tumor growth in the
rat liver by combination of vitamin K2 and angiotensin-converting
enzyme inhibitor via anti-angiogenic activities. Oncol Rep.
15:155–159. 2006.PubMed/NCBI
|














