Introduction
Breast cancer is the most frequent malignant
disorder in women. Patients with ERα-positive tumors are amenable
for therapy with tamoxifen and achieve an overall survival of
approximately 81.3% after 5 years (1). But 10–15% of all breast cancer cases are
designated triple-negative breast cancer (TNBC) as they neither
expresses estrogen receptor ERα nor progesterone receptors and they
do not overexpress Her-2. As a consequence, there is no successful
targeted therapy available for TNBC patients and mortality of
patients with TNBC is twice as high as for patients with ERα
positive tumors (2). Mutations of the
BRCA1 gene were identified as possible therapeutic target in TNBC,
making these tumors particularly sensitive to platinum-compounds
(3). In addition, the receptor for
epidermal growth factor receptor (EGFR) is overexpressed in 30–52%
of TNBC (4). A combination of
anti-EGFR antibody Cetuximab and platinum compounds in the
treatment of TNBC increased overall survival from 9.4 to 12.9 month
(5).
A most recently discovered candidate for targeted
therapy of TNBC is the membrane-bound estrogen receptor, G
protein-coupled estrogen receptor (GPER). This heterotrimeric
G-protein coupled receptor has a lower affinity for 17β-estradiol
and is responsible for its nongenomic effects in various tissues.
GPER mediates estrogen-induced signaling and proliferation in human
breast epithelial cells and normal and malignant breast (6). An immunohistochemical analysis of tissue
sections from TNBC tumors revealed a positive staining of 94% of
TNBC samples. In particular, TNBC with high GPER expression was
associated with younger age of patients. Recurrence rate of
GPER-positive tumors was essentially higher than in a GPER-negative
cohort (7). Our recent observation,
that 17β-estradiol stimulates growth of TNBC cell lines, despite
the lack of ERα expression, points to an involvement of GPER in
malignant transformation of TNBC. A knock-down of GPER using
specific siRNA completely prevented this growth stimulation of TNBC
by 17β-estradiol (8). A
pharmacological inhibition of GPER by the specifically developed
inhibitor (G15) or with estriol was also successful in TNBC cell
lines but super-physiologically high concentrations of these
compounds were needed to achieve a sufficient inhibition of growth
(9).
Alternatively, instead of a pharmacological
inhibition of GPER, a reduction of GPER expression would lead to a
lower activation of signaling pathways depending on GPER. A number
of factors are established regulating GPER expression. Expression
of GPER has been reported to correlate with over-expression of the
receptor for EGF (10). In
approximately 50% of TNBC cases EGFR was strongly expressed
predicting short survival of patients carrying triple negative
breast tumors (11). We have recently
analyzed the impact of the tyrosine-kinase inhibitor gefitinib on
the expression of GPER in TNBC cell lines. Treatment of TNBC cell
lines HCC1806 and HCC70 with 200 nM gefitinib for four days reduced
GPER expression by 70%. Activation of c-src and EGFR by
17β-estradiol was almost completely prevented in cells pretreated
with gefitinib (12).
The growth hormone (GH)/insulin-like growth factor
axis has been implicated in breast cancer progression and growth of
MCF-7 xenografts was successfully prevented by the growth hormone
receptor (GHR) antagonist Somavert (pegvisomant) (13). In addition, antagonists of
GH-releasing hormone were shown to suppress in vivo growth
of TNBC (14). This fact led us to
the assumption that GH is a further factor involved in the
regulation of GPER expression. To our knowledge the impact of a
direct inhibition of GH-receptor on the expression of GPER has not
yet been analyzed. Somavert (Pegvisomant) is a specific inhibitor
of GH-receptor. It is a peptide of 191 amino acids with
sequence-homology to GH. Solely, amino acid Gly120 is
substituted in the original sequence by Lys or Arg and the peptide
is chemically modified by the addition of PEG at five positions to
increase solubility and stability of the compound (15).
Somavert has already been clinically applied for
several years in treatment of acromegaly, a disease, caused in most
cases by a pituitary adenoma leading to an over-production of GH
responsible for the clinical features of acromegaly (16).
According to the above mentioned facts, it is
plausible that reducing transcription of GPER by inhibition of the
GHR is a promising procedure for the prevention of 17β-estradiol
dependent growth stimulation of TNBC. In this study we analyzed
whether expression of GPER in TNBC cell lines is down-regulated
following inhibition of GHR using Somavert as competitive
inhibitor. After reduction of GPER expression in TNBC cells using
Somavert the consequences of this inhibition on the signaling of
GPER were analyzed and the impact of the reduced GPER expression on
the induction of proliferation by 17β-estradiol was measured. Since
inhibition of GPER was shown to suppress expression of CCN family
member 1 (CCN1; cysteine-rich angiogenic inducer 61, CYR61), a
factor involved in tumor cell invasion (17), we also analyzed the impact of GPER
downregulation by Somavert on expression of CCN1.
Materials and methods
Reagents
Somavert™ (Pegvisomant) was a generous
gift from Pfizer (New York, NY, USA). 17β-estradiol (E2), insulin
and transferrin were purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany).
Cell lines
TNBC cell lines HCC1806, HCC70 and MDA-MB-453 were
purchased from ATCC (Manassas, VA, USA) and maintained in DMEM
containing 10% fetal bovine serum (both Biochrom, Berlin, Germany),
supplemented with 2 mM glutamine, 6 ng/ml insulin, 10 ng/ml
transferrin, penicillin (50 U/ml), streptomycin (50 µg/ml) from
Gibco; Thermo Fisher Scientific, Inc. (Paisley, UK).
Treatment of cells
To analyze the effect of Somavert on expression of
GPER, four million cells of each cell line were grown in 2 ml DMEM
in 25 ml tissue flasks. Cells were either treated with 1 µM
Somavert, the concentration clinically applied in treatment of
acromegaly, for 48 or 96 h.
For analysis of the impact of Somavert treatment on
signal transduction of 17β-estradiol in TNBC cells, culture medium
was replaced by phenolred-free culture medium without serum 24 h
before stimulation of the cells with 10−8 M
17β-estradiol for 15 min. Cells were harvested in 1 mM EDTA/PBS,
centrifuged at 400 × g and lysed in 50 µl Cell Lytic M supplemented
with protease- and phosphatase-inhibitors (Sigma-Aldrich; Merck
KGaA).
Western blots
Lysates of cells were centrifuged at 15,000 × g for
5 min and protein concentration was measured using the method of
Bradford. 20 µg of each sample were loaded on a 7.5% polyacrylamide
gel, run for one hour at 100 V. Proteins were blotted on
PVDF-membrane and sequentially detected with a series of rabbit
primary antibodies: Anti-phospho-Src and anti-c-Src both from Cell
Signaling Technology, Inc. (Danvers, MA, USA), anti-phospho
Tyr1173 EGFR delivered by Calbiochem (Darmstadt,
Germany), anti-EGFR antibody from Epitomics (Hamburg, Germany) and
anti-actin from Sigma-Aldrich; Merck KGaA. After four washes in
TBST, blots were incubated with a 1:20,000 dilution of
goat-anti-rabbit antibody conjugated with horseradish peroxidase
(ECL; GE Healthcare Europe, GmbH, Freiburg, Germany). After further
washing, blots were incubated with chemoluminescence reagent Femto
(Thermo Fisher Scientific, Inc.) for 5 min and protein bands were
detected on a LiCor chemoluminescence detector (Licor, Lincoln, NE,
USA). Densitometric evaluation of the blots was performed with
Image Studio Digits program from Licor. Expression values of the
detected proteins were normalized to actin.
Proliferation assays
The proliferation assays for 17β-estradiol were
performed in phenolred-free medium supplemented with charcoal
depleted serum as previously described (18).
103 cells seeded in 100 µl phenolred-free
MEM supplemented with 2% charcoal depleted serum into 96-well
plates. Somavert was added in 50 µl to achieve a final
concentration of 1 µM. For stimulation 50 µl of either vehicle or
4×10−8 M 17β-estradiol were added to four
replicates.
Cells were grown for 7 days at 37°C, 5%
CO2 and saturated humidity. Cell number was determined
by a colorimetric method as previously described (18).
Proliferation assays were performed at least three
times with different passages. Means and standard deviations of the
optical density (OD) of the replicates were calculated.
Reverse
transcription-semi-quantitative polymerase chain reaction
(RT-sqPCRs)
RNA was purified from TNBC cells after pretreatment
with 1 µM Somavert and stimulation with 17β-estradiol using
RNeasy-kit (Qiagen, Hilden, Germany).
Reverse transcription polymerase chain reaction was
performed as previously described (8).
PCR-products were separated in a 2% agarose gel
(Type IV, special high EEO; Sigma-Aldrich; Merck KGaA) and ethidium
bromide stained gels were photographed using a CDS camera
(Biometra, Göttingen, Germany).
Densitometric evaluation of
PCR-products
The band intensities of the PCR-products were
evaluated by the Digital science 1D-software (Kodak, Rochester, NY,
USA). Values of the RT-PCR products were normalized to the
ribosomal protein L7.
Statistical analysis
The data were tested for significant differences by
one-way analysis of variance using GraphPad Prism 6.01-Software
(GraphPad Software, Inc., La Jolla, CA, USA) followed by
Student-Newman-Keuls'test for comparison of individual groups,
after a Bartlett test had shown that variances were homogenous.
Results
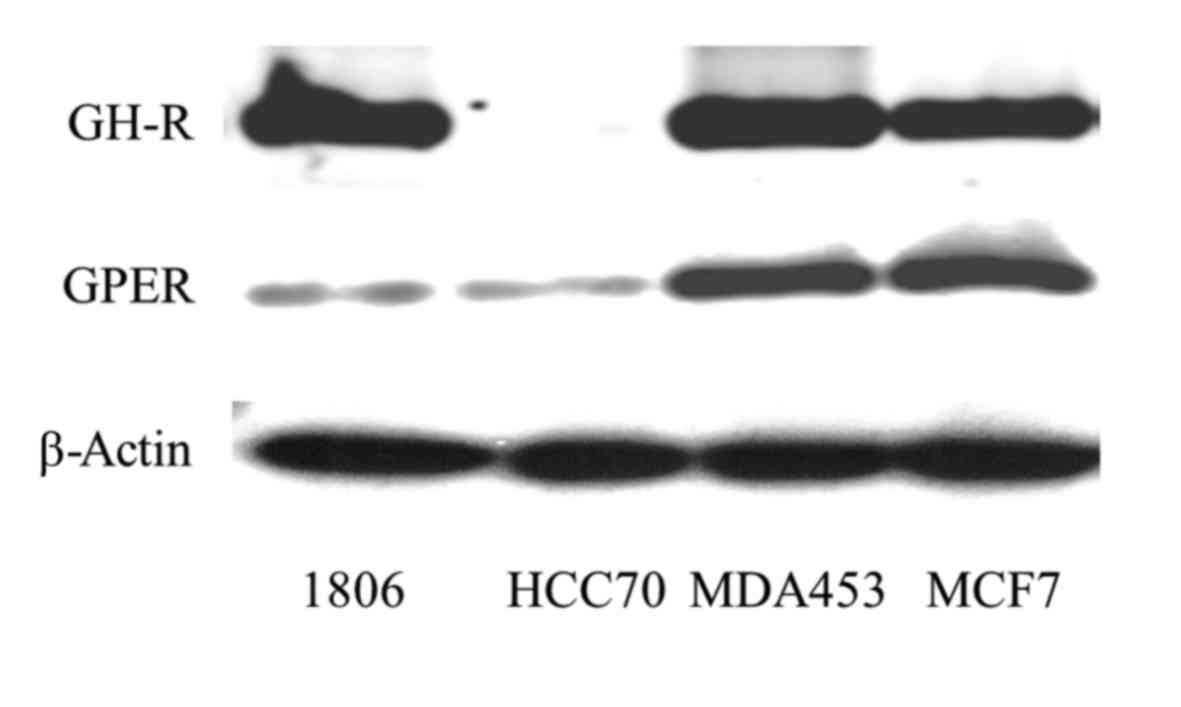
Expression of GHR in TNBC
Expression of GHR of three TNBC-cell lines (HCC1806,
HCC70 and MDA-MB-453) was compared to expression in ERα-positive
breast cancer cell line MCF-7 on western blotting. GHR was
expressed strongest in HCC1806 and MDA-MB-453. HCC1806 cells
expressed 118±24% and MDA-MB-453 cells 136±18% of the amount of GHR
expressed in MCF-7. HCC70 cells contained exceptionally low amounts
of GHR, approximately 2.4±0.8% (P<0.01) of the amount detected
in HCC1806 cells.
GPER expression was also highest in MDA-MB-453
cells. The amount of GPER detected in MDA-MB-453 was approximately
133±22% of the amount expressed in MCF-7 cells. GPER expression was
lowest in HCC70 cells, being only 7.5±0.3% of GPER amount detected
in MDA-MB-453 and correlated with the minimal amount of
growth-hormone receptor found in this cell line (Fig. 1, lane 2). These results suggest that
GPER expression might be regulated depending on GH-receptor.
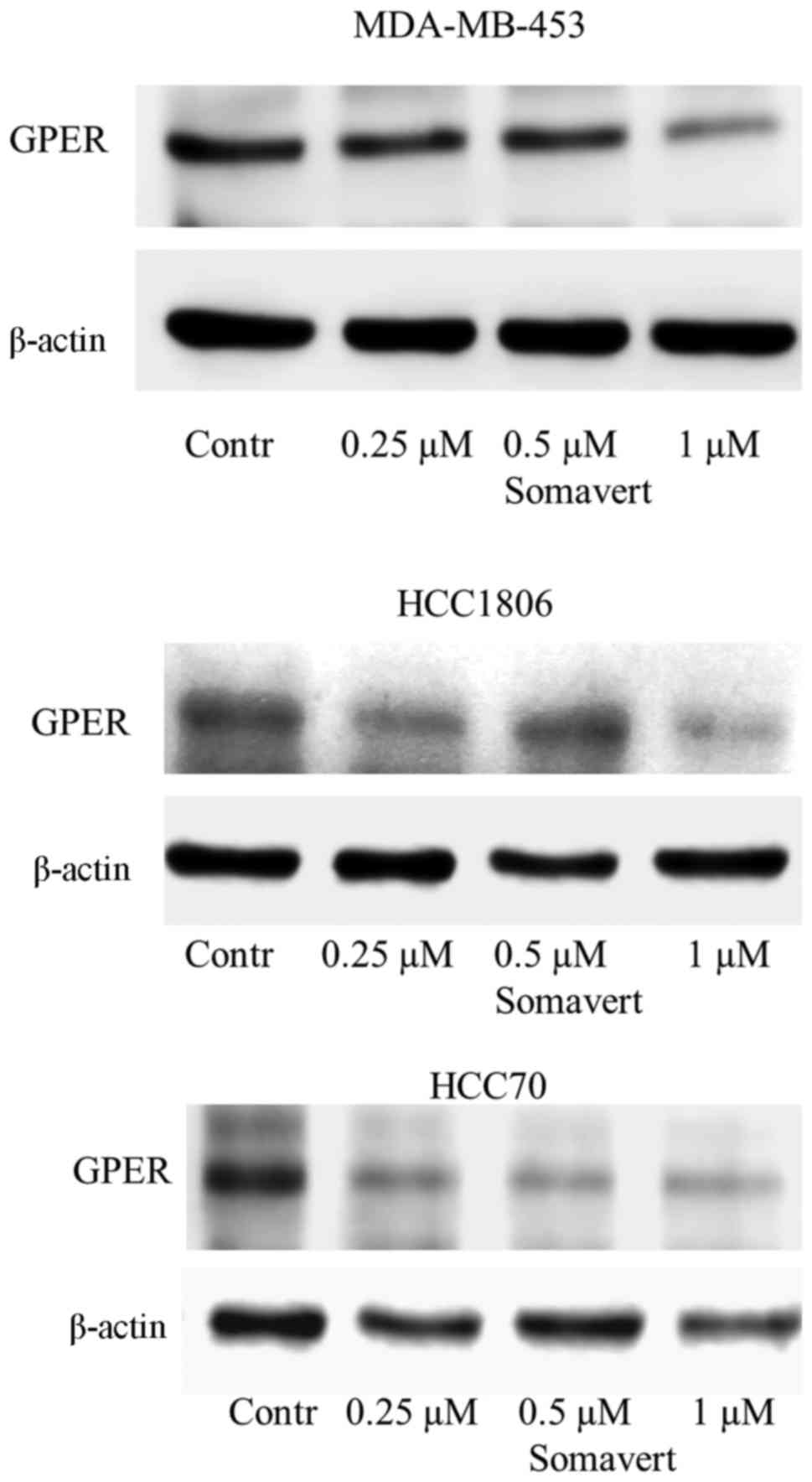
Inhibition of growth-hormone receptor
by Somavert reduces GPER expression
To prove this hypothesis the impact of inhibition of
GH-receptor by the specific GH-antagonist Somavert on GPER
expression was analyzed in the TNBC cell lines. GPER expression was
lowered concentration-dependently. As we have previously show for
the reduction of GPER expression after inhibition of EGFR in
HCC1806 using 500 nM gefitinib, 96 h of treatment were needed to
achieve maximally a reduction to 26±18% (12). To analyze the effect of an inhibition
of GH-receptor on GPER expression the impact of various
concentrations of Somavert were analyzed after 96 h of treatment.
TNBC cells were treated with increasing concentrations (0.25-1 µM)
of Somavert for 96 h. GPER expression was determined on western
blotting of cellular lysates. In MDA-MB-453 cells, maximal
reduction of GPER expression to 46±7% of control (P<0.05) was
observed after treatment with 1 µM Somavert for 96 h. To show the
inhibitory effect of Somavert on GPER expression in HCC1806 and
HCC70 cells, expressing only low amounts of GPER according to
Fig. 1, 100 µg of cell lysates were
loaded on Western blots to get a sufficient signal. In HCC1806
treated with 1 µM for 96 h GPER expression was lowered to 56±5%
(P<0.01) of the amount detected in non-treated cells. In a third
TNBC cell line, and in HCC70 cells GPER expression reached 58±6%
(P<0.05) of untreated control cells under these conditions
(Fig. 2).
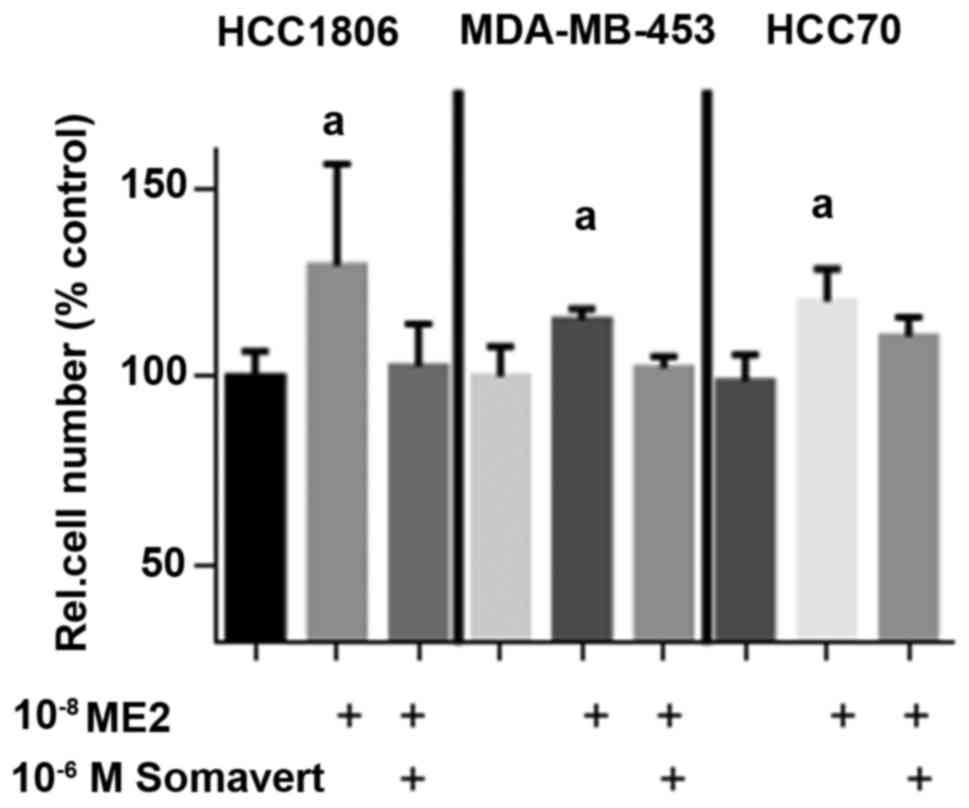
Somavert prevents stimulation of cell
proliferation by 17β-estradiol in TNBC cells
Due to the lower affinity of GPER to 17β-estradiol
compared to ERα the induction of proliferation of TNBC cell lines
was compared at 10−8 M 17β-estradiol in the absence or
in the presence of 1 µM Somavert (Fig.
3).
Stimulation of cell growth of HCC1806 cells by
10−8 M 17β-estradiol increased cell number within 7 days
of culture to 130±26% (P<0.05) of controls. As reported earlier
(8) this growth stimulation is
dependent on the presence of GPER in TNBC. 10−8 M
17β-estradiol were necessary for maximal stimulation of GPER
(9). If HCC1806 cells were
additionally treated with 1 µM Somavert the increase of cell number
by 17β-estradiol was prevented and cell number remained at 103±11%.
10−8 M 17β-estradiol increased proliferation of
MDA-MB-453 cells by 15% and cotreatment with 1 µM Somavert
completely prevented the effects of 17β-estradiol on cell number.
In HCC70 cells an increase of cell number to 120±9% (P<0.05) was
achieved by 10−8 M 17β-estradiol and after co-treatment
with 1 µM Somavert cell number still increased to 111±5% after 7
days of treatment (Fig. 3). This
failure of Somavert to totally prevent induction of proliferation
is in agreement with the smaller reduction of GPER expression due
to lower amount of GHR in HCC70 cells (Fig. 2).
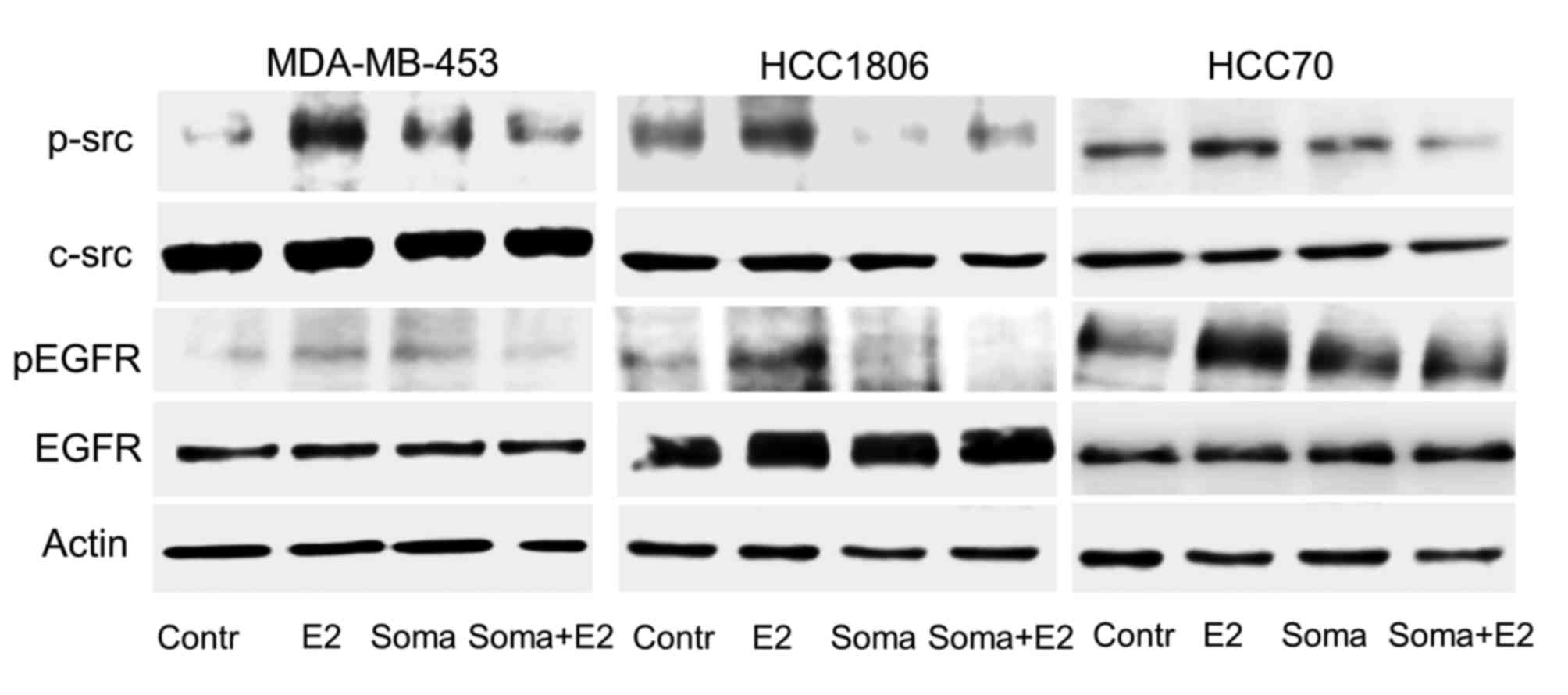
Reduction of GPER expression by
Somavert inhibits phosphorylation of c-src and activation of EGFR
by 17β-estradiol
Following stimulation of GPER with 17β-estradiol
c-src and EGFR are activated by phosphorylation (8,19). As
reported earlier (8) this growth
stimulation is dependent on the presence of GPER in TNBC. Next, we
analyzed activation of c-src and EGFR by 10−8 M
17β-estradiol in TNBC cells after GPER expression was reduced after
pretreatment by Somavert. Cells of the TNBC cell lines, HCC1806,
HCC70 and MDA-MB453 were treated with 1 µM Somavert for four days
and stimulated with 17β-estradiol. Phosphorylation of c-src and
EGFR was compared to control cells, not treated with Somavert on
western blots (Fig. 4).
In serum starved MDA-MB-453 cells phosphorylation of
c-src at Tyr416 was very weak. A 15 min stimulation with
10−8 M 17β-estradiol lead to a 7.3-fold increase of
c-src phosphorylation. After pretreatment of the cells with 1 µM
Somavert, 17β-estradiol only led to a 4-fold activation of c-src.
Even in the serum-starved TNBC cells of HCC70 and HCC1806 a basal
phosphorylation of c-src was already quite strong (Fig. 4, lane 1). In HCC1806 cells the
activation of c-src by the treatment with 17β-estradiol amounted to
177±49% (P<0.01; Fig. 4, lane 2).
A stimulation of HCC70 cells with 10−8 M 17β-estradiol
lead to an increase of p-src to 125±32% of control (P<0.05). In
MDA-MB-453 cells and HCC1806 cells expressing high amounts of GHR
the reduction of GPER expression following treatment with 1 µM
Somavert for 96 h prevented the increase of c-src phosphorylation
by 17β-estradiol (Fig. 4, lane 4). In
HCC70 cells wherein GPER expression was less strongly reduced by
Somavert treatment 17β-estradiol was still able to induce c-src
phosphorylation although weaker than in non-treated HCC70 cells to
110±9% (P<0.05; Fig. 4, lane
4).
Phosphorylation of src subsequently leads to release
of heparin-bound EGF from extracellular matrix by
matrixmetalloproteases. The released EGF initiates
autophosphorylation of the cytosolic domain of the EGFR. In all
three TNBC cell lines Tyr1173 phosphorylation of the
EGFR was analyzed after stimulation with 10−8 M
17β-estradiol (Fig. 4, lane 2). In
MDA-MB-453 cells phosphorylation of EGFR increased to 136±19% of
non-stimulated control (P<0.05). Stimulation of HCC1806 cells
elevated p-EGFR level to 216±24% of control (P<0.01) and in
HCC70 cells to 202±112% (P<0.05; Fig.
4, lane 2).
In MDA-MB-453- and HCC1806-cells phosphorylation of
Tyr1173 by 17β-estradiol was completely prevented in
cells pretreated with 1 µM Somavert (Fig.
4, lane 4). But in HCC70 cells expressing lower amounts of GHR
Somavert was less effective in preventing the induction of EGFR
phosphorylation. In HCC70 cells pretreated with 1 µM Somavert
17β-estradiol was still able to increase EGFR phosphorylation to
166±59% of control (P<0.05; Fig.
4, lane 4). This observation provides further evidence that
GPER expression is regulated at least in part by the GHR.
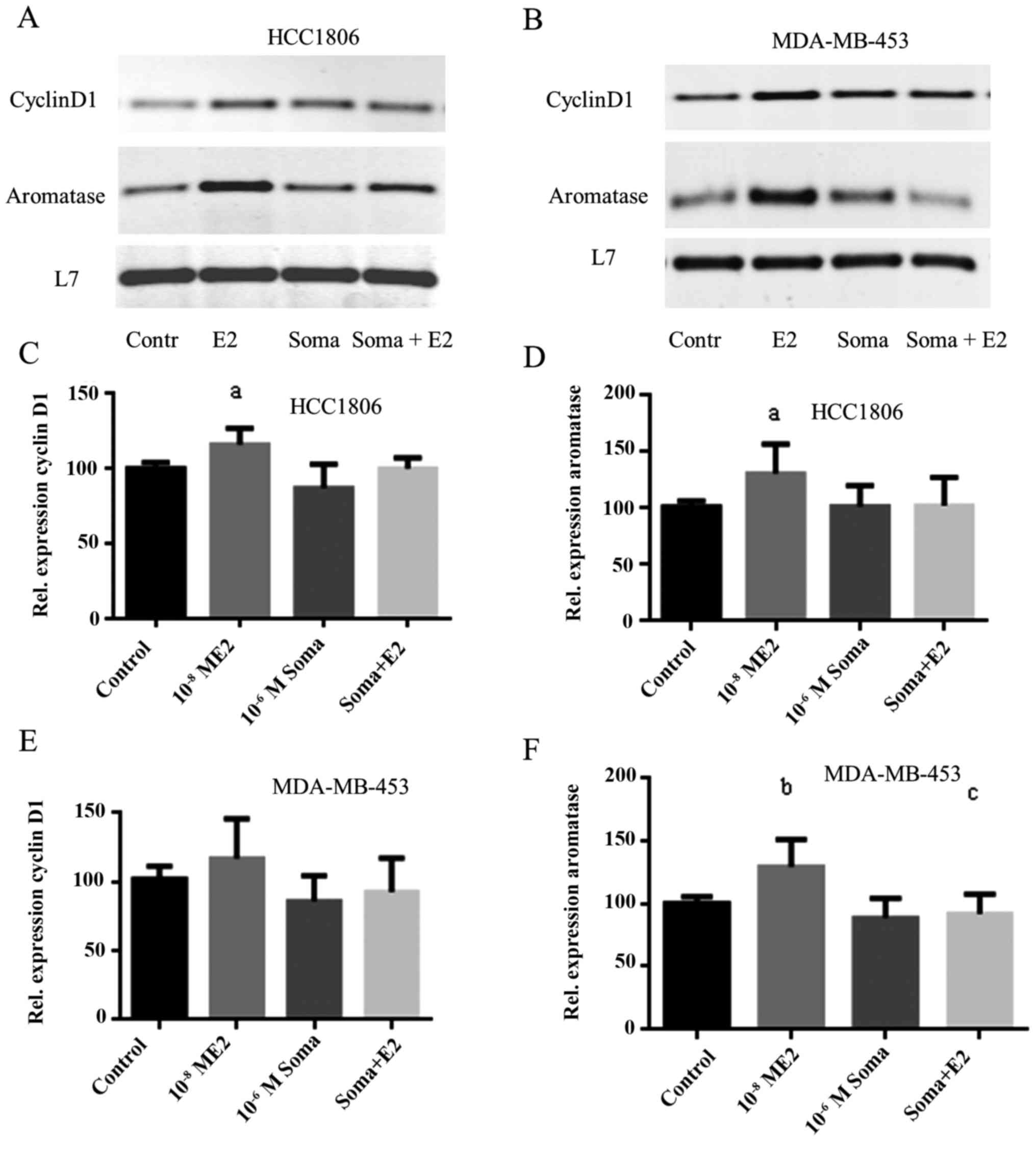
Somavert inhibits induction of cyclin
D1 expression by 17β-estradiol
Cyclin D1 regulates of the transition from G1-phase
to S-phase of the cell cycle. As we reported earlier expression of
cyclin D1 and aromatase is induced by 17β-estradiol in a GPER
dependent manner (8). Expression of
cyclin D1 was analyzed in Somavert treated TNBC cells by RT-PCR
after stimulation with 10−8 M 17β-estradiol (Fig. 5, first panel).
In HCC1806 cells expression of cyclin D1
significantly increased to 116±11% of control (P<0.05) after 30
min stimulation with 17β-estradiol. In HCC1806 cells treated with
Somavert cyclin D1 expression did not increase after stimulation
with 17β-estradiol for 30 min (Fig.
5C).
In control cells of MDA-MB-453 cyclin D1 was more
strongly expressed than in HCC1806 (Fig.
5B, lane 1). Stimulation of these cells with 17β-estradiol
increased cyclin D1 expression only to 117±29% of control.
Pretreatment of MDA-MB-453 cells with 1 µM Somavert completely
prevented induction of cyclin D1 expression by 17β-estradiol
(Fig. 5E).
Aromatase expression in TNBC
cells
The aromatase gene essential for an autocrine
biosynthesis of 17β-estradiol in TNBC is upregulated via GPER
(9). Stimulation of HCC1806 cells
with 17β-estradiol increased aromatase expression to 131±26% of
control (P<0.05). Reduction of GPER expression by Somavert
reduced the induction of aromatase expression by 17β-estradiol only
marginally to 121±51% (Fig. 5D). In
MDA-MB-453 cells 17β-estradiol increased aromatase expression only
to 110±36% of control. If MDA-MB-453 cells were pretreated with
10−6 M Somavert for 96 h induction of aromatase
expression was lowered below control level (Fig. 5F).
CCN1 expression is not induced by
17β-estradiol nor is it reduced by Somavert treatment
CCN1 (Cyr61) is a secreted protein that plays divers
roles in cellular proliferation, survival and migration (17). Estrogen is a powerful inducer of CCN1
in breast cancer cells. In order to prove, whether CCN1 induction
in TNBC cells is dependent on expression of GPER, expression of
CCN1 was analyzed after stimulation with 17β-estradiol in TNBC
cells with and without pretreatment with Somavert (data not shown).
In HCC1806- and MDA-MB-453 cells 17β-estradiol was not able to
increase CCN1 expression, neither on mRNA level nor on protein
level (data not shown).
Discussion
Triple negative breast tumors lack of the expression
of estrogen receptor α (ERα) and of progesterone receptors and do
not overexpress Her-2, the target of Trastuzumab. Therefore,
therapies using the antiestrogen Tamoxifen or antibody therapy with
Trastuzumab are no rationale. GPER, a membrane-bound receptor for
estrogens, initiates fast non-genomic effects of 17β-estradiol that
are independent of ERα. GPER might become a promising target in
treatment of TNBC. Most tumors of TNBCs were shown to express GPER
by immunohistochemical staining. High GPER expression predicted a
higher recurrence rate of TNBC (7).
Therefore, we assume that stimulation of GPER by circulating
17β-estradiol contributes to the malignant behavior of TNBC. We
were already able to show that 17β-estradiol increases
proliferation of TNBC cells (8,9,12). On the other hand, antiestrogens, like
tamoxifen or fulvestrant, that inhibit ERα, are agonists of GPER
and therefor these antiestrogens are not clinically applicable
against triple negative breast tumors (20).
A pharmacological inhibition of GPER by estriol and
G15, a compound selective for GPER, inhibited signal transduction
of GPER and reduced proliferation of TNBC cells only at extremely
high non-physiological concentrations (9).
Due to this disadvantage we decided to follow a
different path to prevent growth stimulation of TNBC cells by
17β-estradiol via GPER. A reduction of GPER expression might have
similar effects as pharmacological inhibition of GPER. It has been
shown, that in TNBC expression of GPER correlates with expression
of EGFR (10). Inhibition of EGFR
with tyrosine-kinase inhibitor gefitinib reduced GPER expression by
up to 85% (12).
Recently, Perez et al reported that growth of
TNBC tumors is stimulated autocrinely by the GH releasing hormone
(GHRH) (14). For this reason we
analyzed in this report, whether GPER expression is regulated by
the GH. A comparison of GPER expression and expression of GHR in a
number of TNBC cell lines showed that in three of four cell lines
GPER correlated with GHR. Somavert is a competitive inhibitor of
the GHR. As further proof, that GPER is regulated by GH we observed
that inhibition of GHR using 1 µM Somavert, a concentration
clinically applied in the treatment of acromegaly, led to a
reduction of GPER expression in all TNBC cell lines tested for up
to 54%.
As a consequence, treatment of TNBC cell lines
MDA-MB-453 and HCC1806 with 1 µM Somavert for 96 h prevented the
17β-estradiol dependent activation of c-src completely. In the TNBC
cell line HCC70 inhibition of c-src activation by Somavert was less
pronounced than in the other two TNBC cell lines tested, probably
due to the fact that this cell line expresses less GHR and GPER
expression in this cell line was less sensitive to treatment with
Somavert. This observation is in concert with our previous finding
that in HCC70 cells GPER expression is more strongly dependent on
stimulation of the EGFR (12).
In the signaling pathway of GPER downstream of c-src
the activation of EGFR following stimulation of the TNBC cell lines
MDA-MB-453 and HCC1806 with 17β-estradiol was also reduced in cells
pretreated with Somavert. In contrast, in HCC70 cells activation of
EGFR was also less strongly prevented by Somavert for the same
reasons as described for c-src.
The induction of the genes relevant for
proliferation, c-fos, and Cyclin D1, after stimulation of the TNBC
cells with 17β-estradiol was subsequently prevented in the cells
pretreated with Somavert. This observation additionally proves that
reduction of GPER by Somavert leads to a specific downregulation of
cell growth.
A further gene, reported to be regulated by
17β-estradiol in ERα-negative breast cancer cells, is CCN1 (Cyr61).
CCN1 plays divers roles in cellular proliferation, survival and
migration. Estrogen has been shown to be an inducer of CCN1 in
MDA-MB-231 breast cancer cells (17).
However, in MDA-MB-453 and HCC1806 TNBC cells 17β-estradiol did not
increase CCN1 expression.
A complex interaction between estrogens and GH in
the regulation of breast cancer growth has formerly been described.
In ovariectomized rats supplementation of estrogen increased the
level of GH and increased expression of GHR at least on
osteosarcoma and liver cells (21,22). On
the other hand, estrogens were shown to inhibit signaling of
GH-receptors by suppressing GH-dependent JAK phosphorylation. This
effect is exerted by induction of SOCS expression by estradiol, a
known negative regulator of signaling of several cytokine receptors
(23). But vice versa GH also
influences effects of estrogens via GPER expression as shown in the
present report.
Somavert is clinically applied for the treatment of
acromegaly, a disorder observed in patients suffering from a tumor
in the pituitary, secreting high amounts of GH (16). In patient suffering from acromegaly
treatment with Somavert was experienced as effective and safe with
only minor side effects (15).
Therefore, application of Somavert in the treatment of patients
with triple negative breast cancer would be of low risk.
Acknowledgements
The authors would like to thank Mrs Sonja Blume and
Mr. Matthias Läsche for their technical assistance.
Funding
This study was supported by a grant from the German
Research Foundation (grant no. GR 1895/10-1).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RG and CG together developed the conception of the
project. RG carried out all experiments, performed data analysis
and drafted the manuscript. CG participated in the design of the
study and the statistical analysis and he supervised the drafting
of the manuscript. GE critically revised the manuscript and
approved the final version.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dowsett M, Cuzick J, Ingle J, Coates A,
Forbes J, Bliss J, Buyse M, Baum M, Buzdar A, Colleoni M, et al:
Meta-analysis of breast cancer outcomes in adjuvant trials of
aromatase inhibitors versus tamoxifen. J Clin Oncol. 28:509–518.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carey LA, Dees EC, Sawyer L, Gatti L,
Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML and Perou
CM: The triple negative paradox: Primary tumor chemosensitivity of
breast cancer subtypes. Clin Cancer Res. 13:2329–2334. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Silver DP, Richardson AL, Eklund AC, Wang
ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A, et
al: Efficacy of neoadjuvant Cisplatin in triple-negative breast
cancer. J Clin Oncol. 28:1145–1153. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reis-Filho JS and Tutt AN: Triple negative
tumours: A critical review. Histopathology. 52:108–118. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baselga J, Gómez P, Greil R, Braga S,
Climent MA, Wardley AM, Kaufman B, Stemmer SM, Pego A, Chan A, et
al: Randomized phase II study of the anti-epidermal growth factor
receptor monoclonal antibody cetuximab with cisplatin versus
cisplatin alone in patients with metastatic triple-negative breast
cancer. J Clin Oncol. 31:2586–2592. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scaling AL, Prossnitz ER and Hathaway HJ:
GPER mediates estrogen-induced signaling and proliferation in human
breast epithelial cells and normal and malignant breast. Horm
Cancer. 5:146–160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Steiman J, Peralta EA, Louis S and Kamel
O: Biology of the estrogen receptor, GPR30, in triple negative
breast cancer. Am J Surg. 206:698–703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Girgert R, Emons G and Gründker C:
Inactivation of GPR30 reduces growth of triple-negative breast
cancer cells: Possible application in targeted therapy. Breast
Cancer Res Treat. 134:199–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Girgert R, Emons G and Gründker C:
Inhibition of GPR30 by estriol prevents growth stimulation of
triple-negative breast cancer cells by 17β-estradiol. BMC Cancer.
14:9352014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vivacqua A, Lappano R, De Marco P, Sisci
D, Aquila S, De Amicis F, Fuqua SA, Andó S and Maggiolini M: G
protein-coupled receptor 30 expression is up-regulated by EGF and
TGF alpha in estrogen receptor alpha-positive cancer cells. Mol
Endocrinol. 23:1815–1826. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nielsen TO, Hsu FD, Jensen K, Cheang M,
Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler
L, et al: Immunohistochemical and clinical characterization of the
basal-like subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Girgert R, Emons G and Gründker C:
17β-estradiol-induced growth of triple-negative breast cancer cells
is prevented by the reduction of GPER expression after treatment
with gefitinib. Oncol Rep. 37:1212–1218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Divisova J, Kuiatse I, Lazard Z, Weiss H,
Vreeland F, Hadsell DL, Schiff R, Osborne CK and Lee AV: The growth
hormone receptor antagonist pegvisomant blocks both mammary gland
development and MCF-7 breast cancer xenograft growth. Breast Cancer
Res Treat. 98:315–327. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Perez R, Schally AV, Vidaurre I, Rincon R,
Block NL and Rick FG: Antagonists of growth hormone-releasing
hormone suppress in vivo tumor growth and gene expression in triple
negative breast cancers. Oncotarget. 3:988–997. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Neggers SJ, Franck SE, de Rooij FW,
Dallenga AH, Poublon RM, Feelders RA, Janssen JA, Buchfelder M,
Hofland LJ, Jorgensen JO, et al: Long-term efficacy and safety of
pegvisomant in combination with long-acting somatostatin analogs in
acromegaly. J Clin Endocrinol Metab. 99:3644–3652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Neggers SJ and van der Lely AJ:
Somatostatin analog and pegvisomant combination therapy for
acromegaly. Nat Rev Endocrinol. 5:546–552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kireeva ML, Mo FE, Yang GP and Lau LF:
Cyr61, a product of a growth factor-inducible immediate-early gene,
promotes cell proliferation, migration, and adhesion. Mol Cell
Biol. 16:1326–1334. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Girgert R, Bartsch C, Hill SM, Kreienberg
R and Hanf V: Tracking the elusive antiestrogenic effect of
melatonin: A new methodological approach. Neuro Endocrinol Lett.
24:440–444. 2003.PubMed/NCBI
|
|
19
|
Filardo EJ, Quinn JA, Frackelton AR Jr and
Bland KI: Estrogen action via the G protein-coupled receptor,
GPR30: Stimulation of adenylyl cyclase and cAMP-mediated
attenuation of the epidermal growth factor receptor-to-MAPK
signaling axis. Mol Endocrinol. 16:70–84. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ignatov A, Ignatov T, Weissenborn C,
Eggemann H, Bischoff J, Semczuk A, Roessner A, Costa SD and
Kalinski T: G-protein-coupled estrogen receptor GPR30 and tamoxifen
resistance in breast cancer. Breast Cancer Res Treat. 128:457–466.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Contreras B and Talamantes F: Growth
hormone (GH) and 17beta-estradiol regulation of the expression of
mouse GH receptor and GH-binding protein in cultured mouse
hepatocytes. Endocrinology. 140:4725–4731. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Slootweg MC, Swolin D, Netelenbos JC,
Isaksson OG and Ohlsson C: Estrogen enhances growth hormone
receptor expression and growth hormone action in rat osteosarcoma
cells and human osteoblast-like cells. J Endocrinol. 155:159–164.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Birzniece V, Sata A and Ho KK: Growth
hormone receptor modulators. Rev Endocr Metab Disord. 10:145–156.
2009. View Article : Google Scholar : PubMed/NCBI
|