Introduction
Liver kinase B1 (LKB1), also known as
serine/threonine kinase 11, is a well-established tumor suppressor.
Germline mutations in LKB1 are associated with Peutz-Jeghers
syndrome, a disorder characterized by benign hamartomas of the
gastrointestinal tract (1) and is
predisposed to certain types of cancer (2). LKB1 somatic mutations are frequently
identified in sporadic types of cancer, particularly in lung cancer
(20–30%) (3,4), ranking it as the third most frequently
mutated gene in lung adenocarcinoma following p53 and K-Ras
(5). As a serine/threonine protein
kinase, LKB1 acts as a master upstream kinase of a group of
adenosine monophosphate-activated protein kinases (AMPKs), and is
involved in a wide range of cellular functions (6,7).
AMPK, one of the major substrates of LKB1, is an
energy sensor and metabolic switch. AMPK is activated by LKB1 under
energy stresses, including adenosine triphosphate (ATP) depletion
induced by glycolysis inhibitors. 2-deoxyglucose (2-DG) is a
well-characterized glycolysis inhibitor (8). 2-DG is converted by hexokinase to
2-DG-P, which cannot be further metabolized, thus is trapped inside
the cell and inhibits hexokinase. The inhibition of glycolysis by
2-DG treatment induces a decrease in intracellular ATP
concentration and an increase in intracellular AMP concentration.
The increased AMP expression level binds to AMPK and alters its
conformation, resulting in AMPK activation by LKB1 via
phosphorylation of AMPK at Thr172 (9).
As a tumor suppressor, LKB1 was previously
demonstrated to arrest cell cycle and inhibit cancer cell growth by
inducing p21/cyclin dependent kinase inhibitor 1A (WAF1) (10–13).
However, the molecular mechanism underlying p21/WAF1 induction by
LKB1 is not well understood. Firstly, it remains an open question
whether LKB1-mediated p21/WAF1 induction is specific to certain
cancer cell lines or whether it is a general characteristic. In
addition, the substrate molecule downstream of LKB1 mediating
p21/WAF1 induction has not yet been identified. Finally, there have
been contradicting findings on whether functional p53, the key
transcription factor of p21/WAF1 (14), is required for p21/WAF1 induction by
LKB1. The present study built upon previous studies (10–12) by
confirming that ectopic LKB1-induced p21/WAF1 expression occurs in
lung and colon cancer cells. Mechanistically, it was revealed that
p53 was required for p21/WAF1 upregulation by LKB1. In addition,
the results of the present study suggested that AMPK may act
downstream of LKB1 to increase p21/WAF1 expression, possibly by
phosphorylating p53-Ser15. Taken together, the results
demonstrated that LKB1 acts via its substrate AMPK to induce
p21/WAF1 expression in a p53-dependent manner. Therefore, the
results of the present study have shed new light on the molecular
mechanism underlying the tumor suppressor role of LKB1.
Materials and methods
Materials
Mouse monoclonal antibody against LKB1 was purchased
from Abcam (Cambridge, UK). Antibodies against p21/WAF1,
phosphorylated (p)-p53-Ser15 and p-AMPK-Thr 172 were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Mouse anti-GAPDH antibody was purchased from Santa Cruz
Biotechonology, Inc. (Dallas, TX, USA). 2-DG was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). The pEGFP-C2,
pEGFP-LKB1 and pEGFP-LKB1-K78M plasmids were provided by Professor
Wei Zhou (The Winship Cancer Institute, Emory University School of
Medicine, Atlanta, GA, USA). The lentiviral LKB1 short hairpin RNA
(shRNA) construct and a negative control construct, which was
created in the same vector system (pLKO.1), were purchased from
Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Lentiviral
helper plasmids, pCMV-dR8.2 dvpr and pCMV-VSV-G, were obtained from
Addgene, Inc. (Cambridge, MA, USA).
Cell lines and cell culture
A549 and H460 lung cancer cell lines were purchased
from the American Type Culture Collection (Manassas, VA, USA). The
p53 wild-type HCT116 and the isogenic HCT116 p53−/−
colon cell lines were provided by Professor Wei Zhou (The Winship
Cancer Institute, Emory University School of Medicine). Cells were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (both from Gibco; Thermo Fisher Scientific Inc.) at 37°C in a
humidified atmosphere with 5% CO2.
Western blotting
Following treatment, cells were lysed on ice for 20
min using the radioimmunoprecipitation assay lysis and extraction
buffer (cat. no. 89900; Thermo Fisher Scientific, Inc.). The
protein concentration of each cell sample was determined using the
bicinchoninic acid protein assay reagent (Thermo Fisher Scientific,
Inc.). A total of 20 µg protein/lane was denatured in sodium
dodecyl sulfate sample buffer (cat. no. 9173; Takara Biotechnology
Co., Ltd., Dalian, China) and separated using SDS-PAGE (10% gel).
Subsequently, proteins were transferred onto polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). Followed
blocking with 5% skimmed milk for 1.5 h at room temperature, the
membranes were incubated with the recommended dilutions of primary
antibodies against LKB1 (cat. no. ab-15095; dilution, 1:1,000;
Santa Cruz Biotechnology, Inc.), p21/WAF1 (cat. no. 2947; dilution,
1:1,000; Cell Signaling Technology, Inc.), p-AMPK (cat. no. 8208;
dilution, 1:1,000; Cell Signaling Technology, Inc.), p-p53
Ser15 (cat. no. 9284; dilution, 1:1,000; Cell Signaling
Technology, Inc.), p53 (cat. no. 9282; dilution, 1:1,000; Cell
Signaling Technology, Inc.) and GAPDH (cat. no. sc-47724; dilution,
1:3,000; Santa Cruz Biotechnology, Inc.) at 4°C overnight and
rinsed twice with PBS. The membranes were subsequently incubated
with the following secondary antibodies: Horseradish peroxidase
(HRP)-conjugated anti-rabbit immunoglobulin G (IgG; cat. no.
ZB2301; dilution, 1:10,000; OriGene Technologies, Inc.) or the
HRP-conjugated anti-mouse IgG antibody (cat. no. ZB2305; dilution,
1:10,000; OriGene Technologies, Inc.) at room temperature for 2 h.
Peroxidase-labeled bands were visualized using an enhanced
chemiluminescence detection reagent (Pierce; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol.
Results were analyzed using Image software (version 1.5; National
Institutes of Health, Bethesda, MD, USA) to compare the relative
target protein expression.
Colony formation assay
A549 cells were plated at a density of
2×105 cells/well in six-well plates overnight at 37°C in
a humidified incubator containing 5% CO2. The following
day, cells were transfected with plasmids encoding wild-type LKB1
or mutant LKB1-K78M (kinase-deficient) in duplicate with
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 2 µg plasmid and 6 µl Lipofectamine
2000 were added to each well. Cells were selected using G418 (2
mg/ml) 72 h after transfection for 2 weeks at 37°C in a humidified
incubator containing 5% CO2. The medium was replaced
every 4 days. Finally, the cells were fixed with 10%
trichloroacetic acid solution for 20 min and stained with 0.5%
crystal violet for 30 min at room temperature. The stained cells
were washed and air-dried, then observed under a light microscope
(×40, magnification). Colony numbers were assessed visually and
colonies containing >50 normal-appearing cells were counted.
Statistical differences in colony numbers between the wild-type
LKB1 and LKB1-K78M plasmid-transfected cells were evaluated using
the two-tailed Student's t-test.
LKB1 stable knockdown using lentiviral
shRNA
Lentivirus stocks were prepared according to the
manufacturer's protocol, as previously described (15). Briefly, 1.5×106 293T cells
were plated in 10-cm dishes. Cells were co-transfected with shRNA
constructs (3 µg) together with pCMV-dR8.2 dvpr (3 µg) and
pCMV-VSV-G (0.3 µg) helper constructs. Following 2 days of
incubation at 37°C in a humidified atmosphere containing 5%
CO2, viral stocks were harvested from the culture
medium, which was filtered to remove non-adherent 293T cells. In
order to select the colon cancer cells that were stably expressing
shRNA constructs, HCT116 cells were plated at sub-confluent
densities, and infected with a cocktail of 1 ml virus-containing
RPMI medium, 3 ml RPMI-medium (no antibiotics, heat inactivated
serum) and 8 µg/ml polybrene at 37°C in a humidified atmosphere
with 5% CO2. Selection with 2 µg/ml puromycin was
initiated 48 h after lentivirus infection. Following ~4 weeks of
selection, monolayers of stably infected pooled clones were
harvested for use and cryopreserved.
RNA extraction and quantitative
polymerase chain reaction (qPCR)
Total RNA was isolated from HCT116-pLKO.1 and
HCT116-LKB1 isogenic colon cancer cell lines using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The first-strand cDNA was prepared using the PrimeScript
First strand cDNA Synthesis kit (Takara Bio, Inc.), according to
the manufacturer's protocol, using 1 µg total RNA. All qPCR
reactions were performed in a 20 µl mixture containing 1X SYBR
Green supermix (Takara Bio, Inc.), 0.2 µmol/l of each primer and 2
µl cDNA template. Primers for LKB1 were as follows: Forward,
5′-CTGGGGTCACCCTCTACAAC-3′; and reverse,
5′-ACTCAAGCATCCCTTTCAGC-3′. Primers for GAPDH were as follows:
Forward, 5′-GGAGTCAACGGATTTGGTCG-3′; and reverse,
5′-CTTGATTTTGGAGGGATCTCG-3′. qPCR was performed using the Applied
Biosystems 7500 system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) under the following cycling conditions: (Step 1)
95°C for 30 sec; (step 2) 40 cycles of 95°C for 5 sec; 60°C for 34
sec; the melting curve stage was followed. The relative expression
level of LKB1 was normalized to GAPDH. Relative mRNA concentrations
were determined using the 2−ΔΔCq method (16).
RNA interference
The LKB1 small interfering RNA (siRNA) sequence was
as follows: Sense, 5′-CCAACGUGAAGAAGGAAAUTT-3′ and antisense,
5′-AUUUCCUUCACGUUGGTT-3′. The control siRNA sequences was as
follows: Sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACACGUUCGGAGAATT-3′. All siRNAs were synthesized by
Shanghai Genechem Co., Ltd. (Shanghai, China). The HCT116 and
HCT116p53−/− cancer cell lines were used for RNA interference.
Cells were plated at a density of 2×105 cells/well in
6-well plates overnight at 37°C in a humidified incubator
containing 5% CO2. Cells were subsequently transfected
with 100 nmol/l of LKB1 siRNA or negative control siRNA using the
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific Inc.), according to the manufacturer's protocol. For
every 105 cells, 0.5 µg LKB1 siRNA and control siRNA were diluted
and mixed with 3 µl transfection reagent. After incubation for 30
min at room temperature, the transfection mixture was added to the
cells. Following 6 h of incubation at 37°C in a humidified
incubator containing 5% CO2, the RPMI-1640 medium was
replaced with serum-enriched 1640-RPMI medium (RPMI-1640 medium
supplemented with 10% fetal bovine serum) and the cells were
cultured for an additional 48 h at 37°C in a humidified incubator
containing 5% CO2. Subsequently, the transfected cells
were collected.
Statistical analysis
SPSS software (version 17; IBM Corporation, NY, USA)
was used for statistical analysis of the results. The majority of
the results are representative ≥3 independent experiments and are
presented as the mean ± standard deviation of triplicate samples.
Error bars represent standard deviations between experiments.
One-way analysis of variance and Student's t-test (≤2 groups) were
used to determine statistical significance. Fisher's least
significant difference post-hoc test was used to compare the
differences between ≥3 groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
Restoring LKB1 activity in lung cancer
cells induces growth suppression
To investigate whether the kinase activity of LKB1
is required for growth inhibition in lung cancer cells, LKB1 mutant
A549 cells were transfected with plasmids encoding wild-type LKB1
or mutant LKB1-K78M (kinase-deficient). Subsequently, these
transfections were subjected to G418 (2 mg/ml) selection for 14
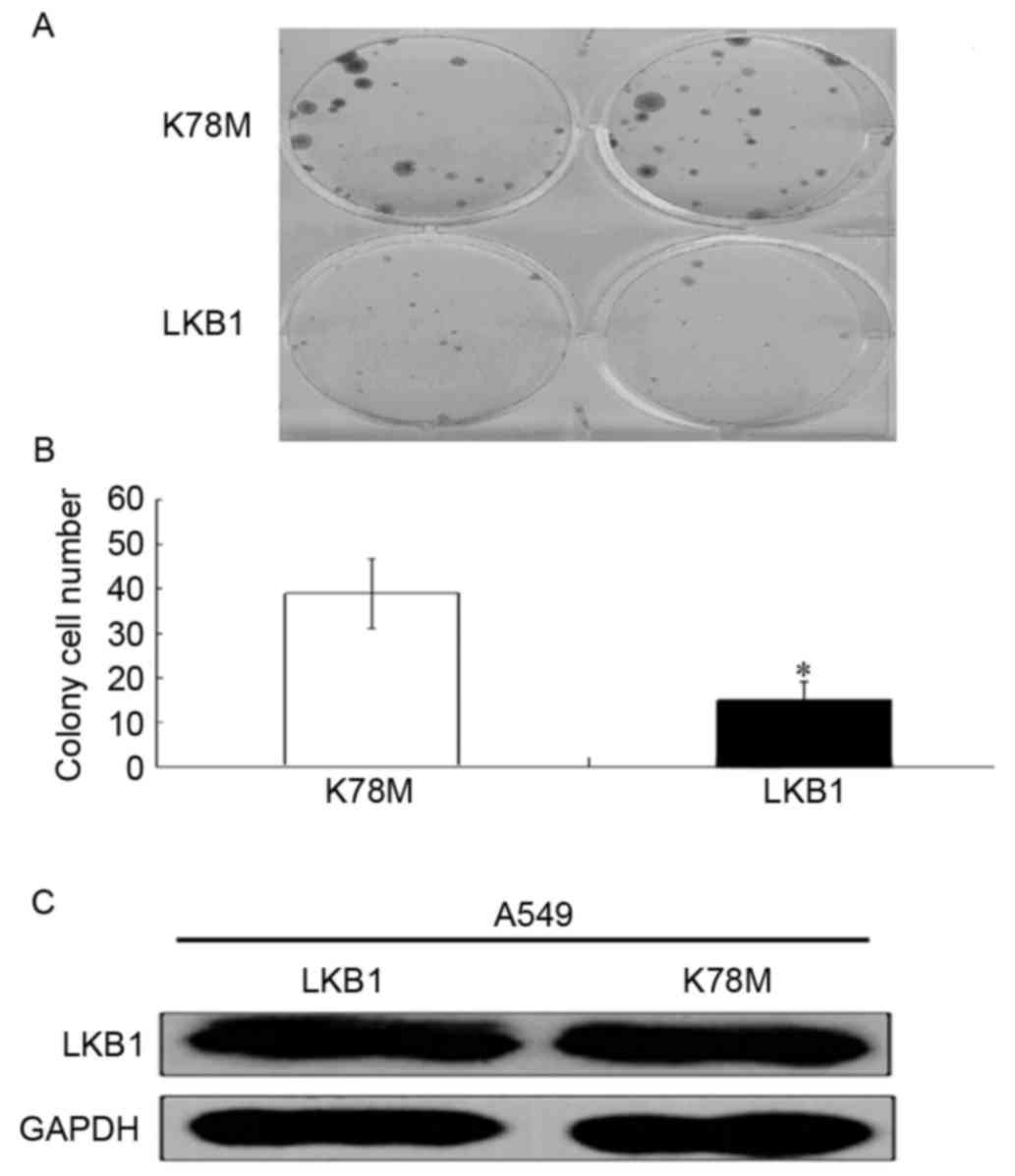
days. As presented in Fig. 1A and B,
ectopic expression of wild-type LKB1 resulted in a significantly
reduced number of colonies compared with those transfected with
LKB1-K78M. In order to evaluate the transfection efficiency of
LKB1, the protein expression level of LKB1 was evaluated using
western blotting. As presented in Fig.
1C, the expression level of wild-type LKB1 and the mutant were
similar, suggesting equal transfection efficiencies. This result
suggests that the kinase activity of LKB1 is required for the
suppression of lung cancer cell proliferation.
LKB1 upregulates p21/WAF1 expression
in lung and colon cancer cells
p21/WAF1 serves an essential role in arresting cell
cycle and inducing growth suppression; therefore, the present study
suggested that p21/WAF1 may be functionally important for the tumor
suppressive effects of LKB1 in lung cancer cells. To evaluate
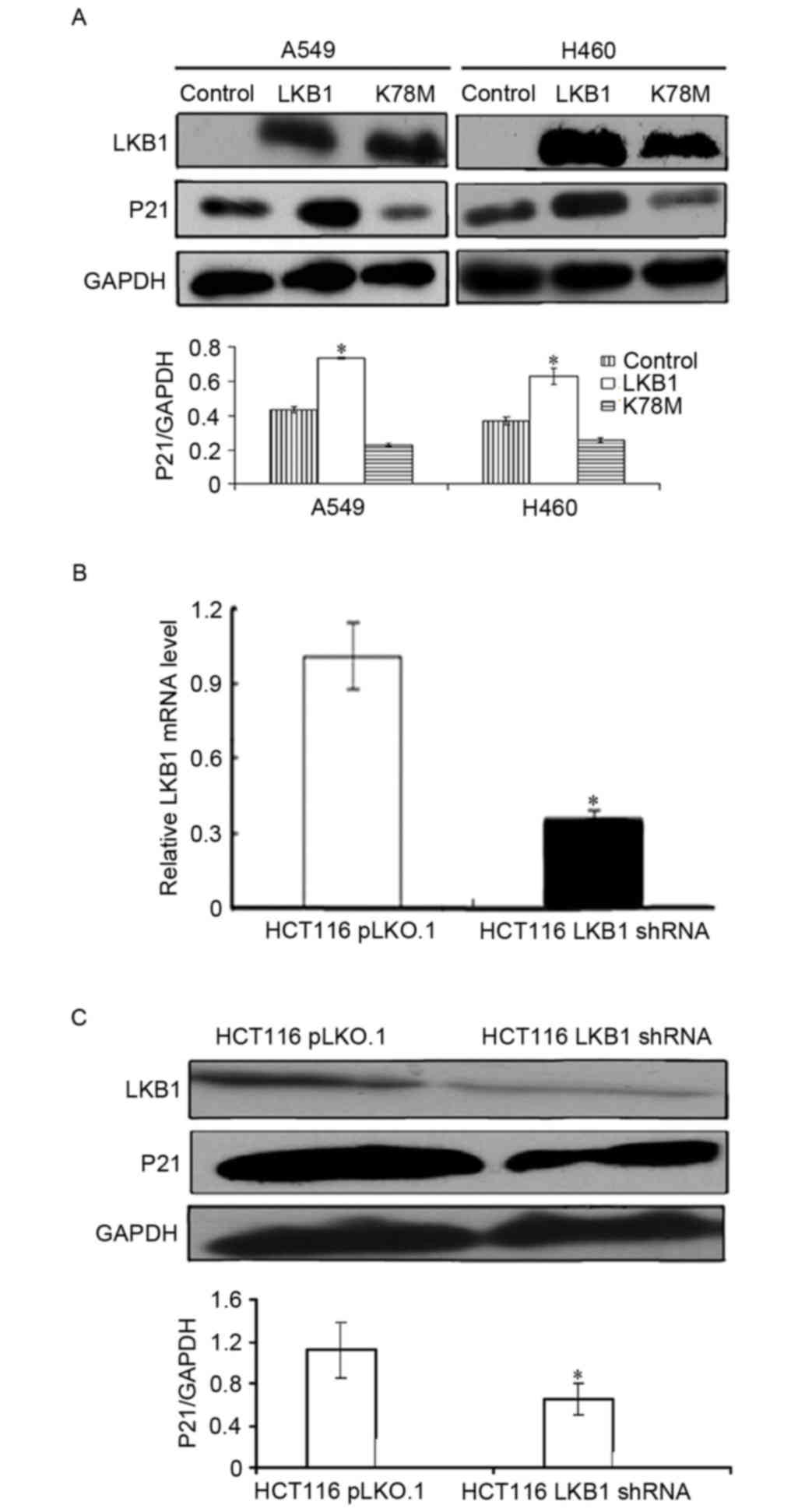
whether LKB1 regulates p21/WAF1 expression in NSCLC cells, LKB1
mutant A549 and H460 cells were used. According to our previous
study, H460 and A549 cells harbor a nonsense mutation at codon 37
of LKB1, thus LKB1 protein is not expressed in these cells lines
(15). The cells were transfected
with plasmids encoding wild-type LKB1, LKB1 K78M or vector, and the
protein expression level of p21/WAF1 was determined through western
blotting. As presented in Fig. 2A,
compared with the control group, ectopic LKB1 significantly
increased p21/WAF1 protein expression level in both cell lines.
Conversely, kinase deficient LKB1-K78M did not have a significant
effect on p21/WAF1 expression level. These results suggest that the
kinase activity of LKB1 is required to upregulate p21/WAF1
expression in lung cancer cells.
In order to validate this observation in other types
of cancer cells, the LKB1 wild-type colon cancer cell line HCT116
was used. Using the lentivirus system, isogenic HCT116-pLKO.1 and
HCT 116-LKB1 shRNA cell lines were established. As presented in
Fig. 2B and C, compared with the
control group, LKB1 mRNA and protein expression levels were
significantly suppressed in HCT 116 cells stably expressing LKB1
shRNA. Additionally, it was revealed that LKB1 knockdown
significantly decreased the expression level of p21/WAF1 (Fig. 2C), confirming that LKB1 positively
regulated p21/WAF1 expression level in colon cancer cells.
AMPK acts downstream of LKB1 to
regulate p21/WAF1 expression level
It has been reported that deficiency of AMPK-α1, a
canonical substrate of LKB1 as described above, reduces p21/WAF1
expression level in mouse embryonic fibroblasts (17). Thus, the present study suggested that
LKB1 acts indirectly to induce p21/WAF1 expression via its
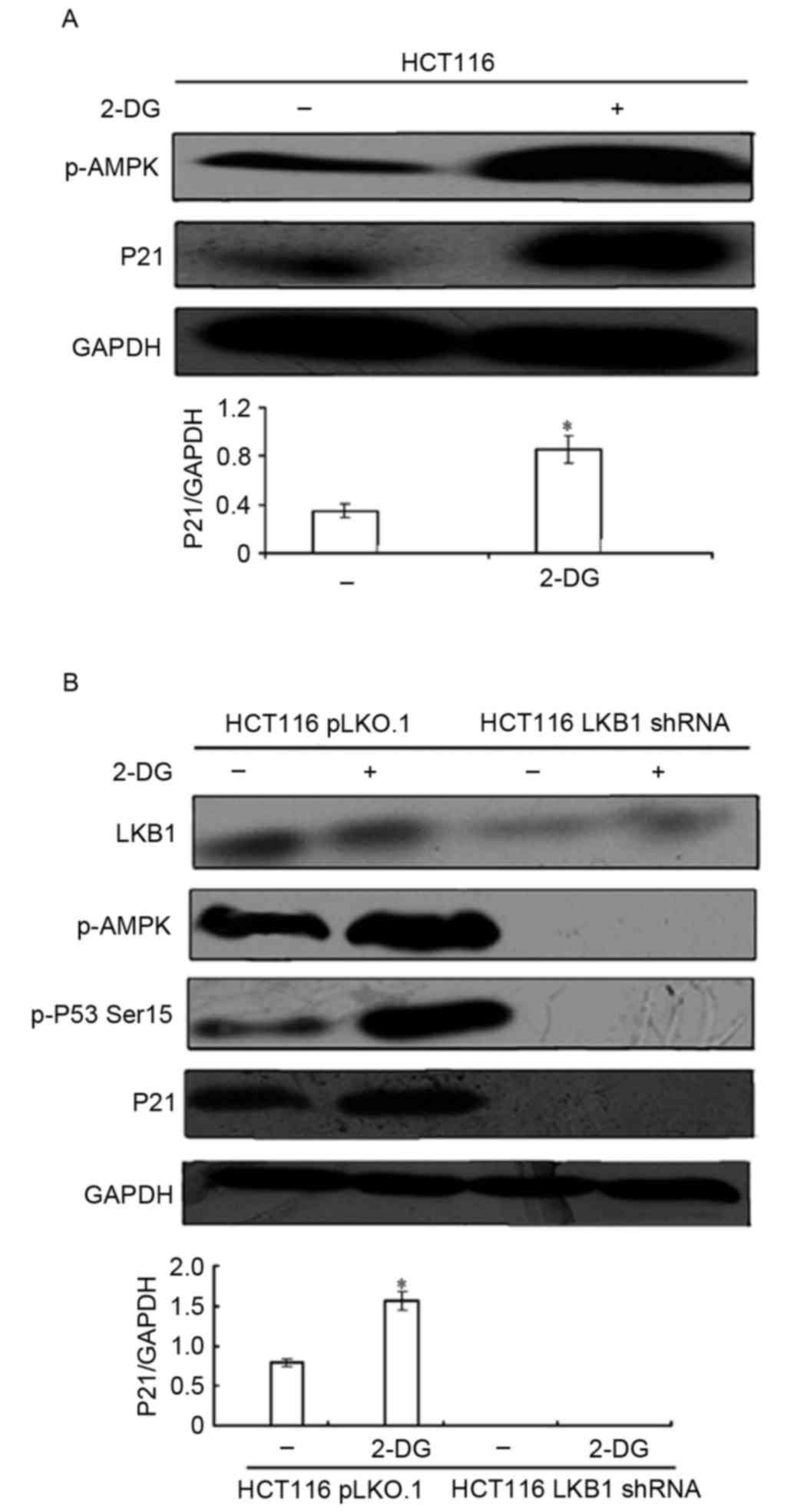
substrate AMPK. To investigate this hypothesis, 2-DG, a glycolysis
inhibitor and AMPK activator, was used. HCT116 cells were treated
with 25 mM 2-DG for 24 h, the expression levels of p-AMPK Thr172
and p21/WAF1 were evaluated using western blotting. As presented in
Fig. 3A, 2-DG treatment induced AMPK
phosphorylation and significantly enhanced p21/WAF1 expression,
suggesting that AMPK activation serves a role in p21/WAF1
induction.
2-DG also demonstrated an off-target effect, which
is independent of LKB1/AMPK signaling. To confirm that 2-DG induced
p21/WAF1 expression mediated by LKB1/AMPK signaling, the isogenic
HCT116-pLKO.1 and HCT116-LKB1 shRNA cell lines were treated with 25
mM 2-DG for 24 h. As presented in Fig.
3B, 2-DG activated AMPK and significantly increased p21/WAF1
expression in HCT116-pLKO.1 cells. In contrast, in HCT116-LKB1
shRNA cells, 2-DG failed to induce AMPK activation, and LKB1
depletion diminished the 2-DG-inducing effects on p21/WAF1. In
addition, it was revealed that the phosphorylation of
p53-Ser15 was increased following treatment with 2-DG,
which was significantly attenuated by LKB1 loss. These results
suggest that AMPK acts downstream of LKB1 to induce p21/WAF1
expression.
LKB1/AMPK requires p53 to regulate
p21/WAF1
The phosphorylation of p53 Ser15 is
important for the stabilization of p53 protein (18), thus the present study suggested that
p53 may be involved in p21/WAF1 induction by LKB1/AMPK. To
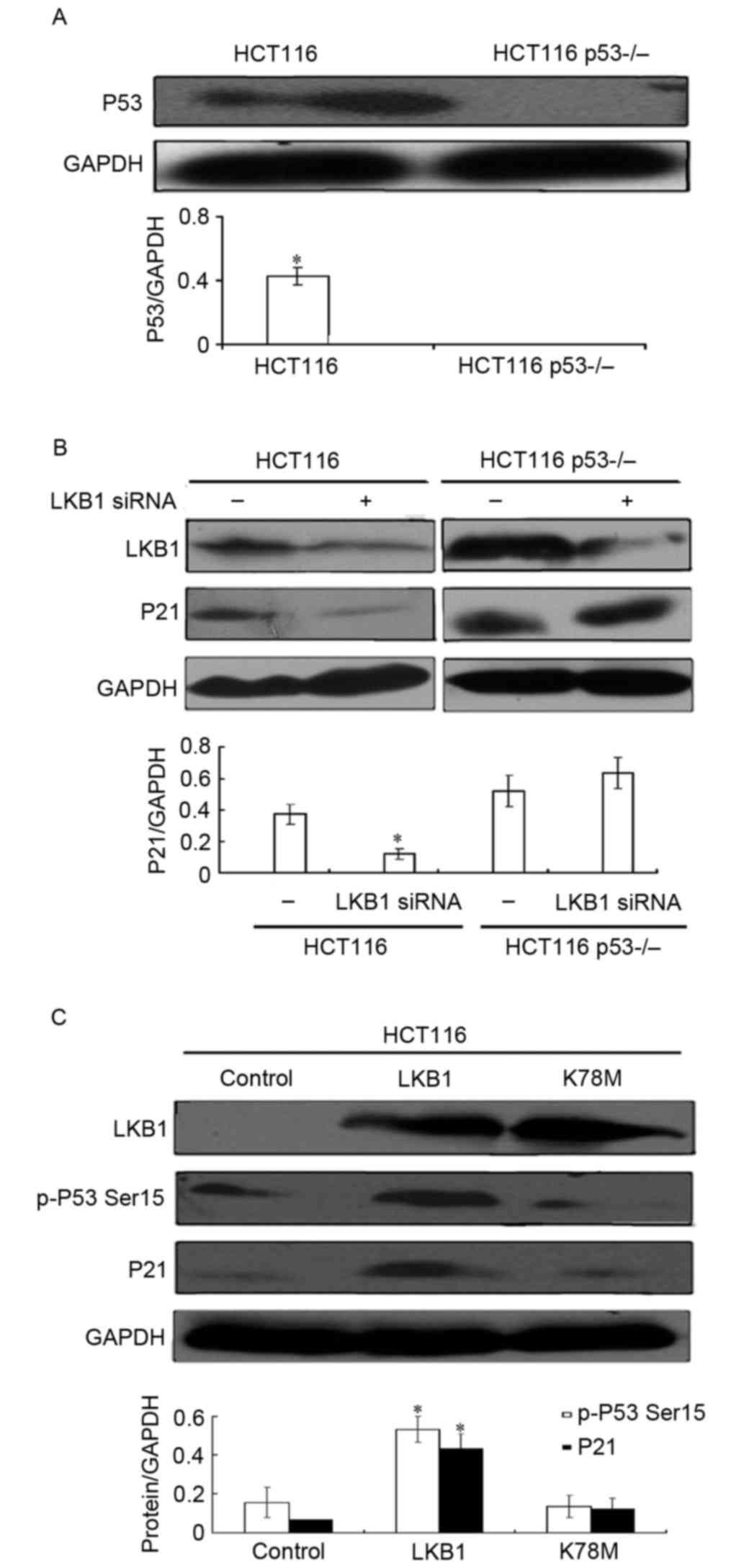
determine whether p53 is required for LKB1-mediated p21/WAF1
induction, the p53 wide-type HCT116 colon cell line and its
isogenic clones with p53 depletion (HCT116 p53−/−) were
used. As presented in Fig. 4A, the
protein expression level of p53 was almost undetectable in HCT116
p53−/− cells. These cells were then transiently
transfected with control short interfering (si)RNA or LKB1 siRNA,
and the expression level of p21/WAF1 was analyzed by western
blotting. As presented in Fig. 4B, it
was revealed that LKB1 depletion by siRNA in p53 wide-type HCT116
cells induced a lower expression level of p21/WAF1. Conversely, in
p53-absent HCT116 p53−/− cells, LKB1 transient knockdown
did not significantly alter the p21/WAF1 expression level.
Therefore, these results indicate that LKB1-mediated p21/WAF1
induction is p53-dependent.
To directly evaluate whether LKB1 was involved in
p53-Ser15 phosphorylation, HCT116 cells were transfected
with plasmids encoding wild-type LKB1, LKB1 K78M or vector, and
western blot analysis was performed to analyze p-p53
Ser15 and p21/WAF1 expression levels. As presented in
Fig. 4C, wild-type LKB1 significantly
increased the expression levels of p-p53 Ser15 and
p21/WAF1 compared with the control, whereas kinase-deficient LKB1
did not have any significant effects. These results suggest that
LKB1 induces the phosphorylation of p53-Ser15 in a
kinase-dependent manner, which may contribute to the upregulation
of p21/WAF1 expression.
Discussion
Numerous previous studies provide evidence that LKB1
serves an essential role as a tumor suppressor. Tiainen et
al (10) demonstrated that LKB1
induced cell cycle arrest and inhibited cell growth by upregulating
the p21/WAF1 expression level in LKB1 deficient cervical (HeLa),
S3, and melanoma (G361) cancer cell lines. However, it remains
unclear whether LKB1 regulates p21/WAF1 expression in other types
of cancer cells. The results of the present study confirmed and
extended previous observations in lung cancer and colon cancer
cells (10–12). Firstly, the present study demonstrated
that ectopic LKB1 increased p21/WAF1 expression level in LKB1
mutant NSCLC cells in a kinase-dependent manner. Furthermore, by
establishing an isogenic LKB1 stable knockdown colon cell line, it
was revealed that LKB1 depletion significantly reduced p21/WAF1
protein expression level. Human LKB1 is a nuclear and cytoplasmic
protein. There have been contradicting studies investigating the
effect of LKB1 cellular localization on p21/WAF1 expression levels
(11–13). Future investigations are required to
identify which part of LKB1, localized in the cytoplasm or in the
nucleus, serves a more significant role in p21/WAF1 induction.
AMPK, one of the key substrates of LKB1, is
considered as the ‘cellular fuel gauge’ in sensing and modulating
metabolic processes (9). AMPK is a
heterotrimeric enzyme composed of two regulatory β and γ subunits
and a catalytic α subunit (α1 and α2) (19). Xu et al (17) demonstrated that AMPKα1 deficiency
induced p21/WAF1 reduction in mouse embryonic fibroblasts.
Consistent with their findings, the present study revealed that
pharmacological activation of AMPK by 2-DG significantly increased
the p21/WAF1 expression level, and depletion of LKB1/AMPK impaired
the ability of 2-DG to induce p21/WAF1. Thus, the present study
identified AMPK as a potential downstream molecule of LKB1 involved
in the mediation of p21/WAF1 induction. We previously revealed that
2-DG treatment potently inhibits the proliferation of LKB1
wild-type lung cancer cells (8).
Given the importance of p21/WAF1 in growth arrest, it has been
suggested that 2-DG-mediated p21/WAF1 induction may contribute to
the inhibitory effect of the compound. Upregulation of p21/WAF1 by
LKB1/AMPK may represent the mechanism underlying growth arrest when
cancer cells are exposed to energy stresses with ATP depletion.
Contradicting findings have been reported regarding
the role of transcription factor p53 in p21/WAF1 induction mediated
by LKB1. Tiainen et al (11)
and Zeng et al (12) reported
that LKB1 required p53 to induce p21/WAF1 expression. In contrast,
a previous study by Setogawa et al (20) demonstrated that LKB1 has the potential
to induce p21/WAF1 expression in collaboration with LIM domain only
4, GATA binding protein 6 and LIM domain binding, 1 in the
p53-deficient HelaS3 cell line (20),
suggesting a p53-independent mechanism. The present study confirmed
that p53 was required for LKB1-mediated p21/WAF1 induction, as p53
depletion in colon cancer cells were able to inhibit p21/WAF1
regulation by LKB1. Furthermore, the present study revealed that
the phosphorylation of endogenous p53-Ser15 was
increased by LKB1 overexpression or AMPK activation, suggesting
that p53-Ser15 phosphorylation, a modification essential
for p53 stabilization, may be involved in p21/WAF1 upregulation.
The results of the present study are consistent with previous
reports indicating that AMPK induces phosphorylation of p53-Ser15
in hepatoma HepG2 cells (21), mouse
embryonic fibroblasts (22), human
aortic smooth muscle cells and rabbit aortic strips (23). Other kinases have been demonstrated to
phosphorylate p53 at Ser15, including ATM and ATR
serine/threonine kinases, as they also target p53 at this site
(24). Thus, some phosphorylation
does occur, despite the fact that the results of the present study
demonstrated that LKB1 depletion induced a significant reduction in
the expression levels of p-p53. Further investigations are required
to confirm that LKB1/AMPK-mediated p53-Ser15 contributes
to p21/WAF1 induction.
In conclusion, the results of the present study
demonstrated that in lung and colon cancer cells, LKB1 acts via
AMPK to induce p21/WAF1 expression in a p53-dependent manner.
Therefore, the present study provides novel molecular insights into
the tumor suppressor gene, LKB1.
Acknowledgements
The authors would like to thank Dr. Wei Zhou (The
Winship Cancer Institute, Emory University School of Medicine,
Atlanta, GA, USA) for providing the plasmids.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81572268), Natural
Science Foundation of Tianjin (grant no. 17JCYBJC25500), Natural
Science Foundation of Tianjin (grant no. 16JCYBJC24400), National
Natural Science Foundation of China (grant no. 31301160).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DZ was responsible for study conception; QM, PX, LS
designed and performed the research; QM, PX, LS, and JW analyzed
the data; PX, LS and DZ wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hemminki A, Markie D, Tomlinson I,
Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M,
Höglund P, et al: A serine/threonine kinase gene defective in
Peutz-Jeghers syndrome. Nature. 391:184–187. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hearle N, Schumacher V, Menko FH,
Olschwang S, Boardman LA, Gille JJ, Keller JJ, Westerman AM, Scott
RJ, Lim W, et al: Frequency and spectrum of cancers in the
Peutz–Jeghers syndrome. Clin Cancer Res. 12:3209–3215. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sanchez-Cespedes M, Parrella P, Esteller
M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG and Sidransky
D: Inactivation of LKB1/STK11 is a common event in adenocarcinomas
of the lung. Cancer Res. 62:3659–3662. 2002.PubMed/NCBI
|
|
4
|
Matsumoto S, Iwakawa R, Takahashi K, Kohno
T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna
JD and Yokota J: Prevalence and specificity of LKB1 genetic
alteration in lung cancer. Oncogene. 26:5911–5918. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhong D, Guo L, de Aguirre I, Liu X, Lamb
N, Sun SY, Gal AA, Vertino PM and Zhou W: LKB1 mutation in large
cell carcinoma of the lung. Lung Cancer. 53:285–294. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao Y, Ge G and Ji H: LKB1 in lung
cancerigenesis: A serine/threonine kinase as tumor suppressor.
Protein Cell. 2:99–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Alessi DR, Sakamoto K and Bayascas JR:
LKB1-dependent signaling pathways. Annu Rev Biochem. 75:137–163.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dong LX, Sun LL, Zhang X, Pan L, Lian LJ,
Chen Z and Zhong DS: Negative regulation of mTOR activity by
LKB1-AMPK signaling in non-small cell lung cancer cells. Acta
Pharmacol Sin. 34:314–318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woods A, Johnstone SR, Dickerson K, Leiper
FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M and
Carling D: LKB1 is the upstream kinase in the AMP-activated protein
kinase cascade. Curr Biol. 13:2004–2008. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tiainen M, Ylikorkala A and Makela TP:
Growth suppression by LKB1 is mediated by a G1 cell cycle arrest.
Proc Natl Acad Sci USA. 96:9248–9251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tiainen M, Vaahtomeri K, Ylikorkala A and
Makela TP: Growth arrest by the LKB1 tumor suppressor: Induction of
p21(WAF1/CIP1). Hum Mol Genet. 11:1497–1504. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng PY and Berger SL: LKB1 is recruited
to the p21/WAF1 promoter by p53 to mediate transcriptional
activation. Cancer Res. 66:10701–10708. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhong DS, Sun LL and Dong LX: Molecular
mechanisms of LKB1 induced cell cycle arrest. Thorac Cancer.
4:229–233. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sun LL, Zhong DS, Wu S, Bai H and Chen Z:
Establishment and gene expression profiling of LKB1 stable
knockdown lung cancer cell line. Chin Med J (Engl). 124:2028–2032.
2011.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xu H, Zhou Y, Coughlan KA, Ding Y, Wang S,
Wu Y, Song P and Zou MH: AMPKα1 deficiency promotes cellular
proliferation and DNA damage via p21/WAF1 reduction in mouse
embryonic fibroblasts. Biochim Biophys Acta. 1853:65–73. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shieh SY, Ikeda M, Taya Y and Prives C:
DNA damage-induced phosphorylation of p53 alleviates inhibition by
MDM2. Cell. 91:325–334. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hardie DG and Alessi DR: LKB1 and AMPK and
the cancer-metabolism link-ten years after. BMC Biol. 11:362013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Setogawa T, Shinozaki-Yabana S, Masuda T,
Matsuura K and Akiyama T: The tumor suppressor LKB1 induces p21
expression in collaboration with LMO4, GATA-6, and Ldb1. Biochem
Biophys Res Commun. 343:1186–1190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Imamura K1, Ogura T, Kishimoto A,
Kaminishi M and Esumi H: Cell cycle regulation via p53
phosphorylation by a 5′-AMP activated protein kinase activator,
5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside, in a human
hepatocellular carcinoma cell line. Biochem Biophys Res Commun.
287:562–567. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jones RG, Plas DR, Kubek S, Buzzai M, Mu
J, Xu Y, Birnbaum MJ and Thompson CB: AMP-activated protein kinase
induces a p53-dependent metabolic checkpoint. Mol Cell. 18:283–293.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Igata M, Motoshima H, Tsuruzoe K, Kojima
K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D,
et al: Adenosine monophosphate-activated protein kinase suppresses
vascular smooth muscle cell proliferation through the inhibition of
cell cycle progression. Circ Res. 97:837–844. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun B, Ross SM, Rowley S, Adeleye Y and
Clewell RA: Contribution of ATM and ATR kinase pathways to
p53mediated response in etoposide and methyl methanesulfonate
induced DNA damage. Environ Mol Mutagen. 58:72–83. 2017. View Article : Google Scholar : PubMed/NCBI
|