Introduction
Osteosarcoma (OS) is a common bone malignant tumor
with a high degree of malignancy (1).
Of patients with OS, ~75% are between 15 and 25 years old, and the
primary treatment options include surgical section and
chemoradiotherapy (2). However, the
prognosis of patients with OS is poor, and the long-term survival
rate of patients with metastasis or recurrence is <20% (3). Therefore, investigating the molecular
mechanisms underlying OS progression may contribute to the
development of novel prognostic biomarkers and targeted
therapies.
A recent study has indicated that epigenetic
modulation serves important roles in various physiological and
pathological processes (4). RNA
binding proteins (RBPs) and microRNAs (miRNAs) are essential
epigenetic modulators (5,6). RBPs specifically bind to and enhance
mRNA stability, thus increasing mRNA expression (5). miRNAs, a type of non-coding and
single-strand RNA containing 18–22 nucleotides, regulate mRNAs at
the post-transcriptional level via binding to complementary
sequences in untranslated regions (UTRs) of target mRNAs, inducing
their degradation or repressing translation (6). Human antigen R (HuR), as a RBP, has been
demonstrated to facilitate the progression of various tumor types
(7), and binds to mRNA 3′UTR, thus
repressing the inhibition of miRNAs on mRNA expression (8). However, the roles and associated
underlying mechanisms of HuR in OS progression are unclear.
miR-142-3p could function as a potential tumor
suppressor in OS via targeting High mobility Group AT-Hook 1
(HMGA1) (9). Long non-coding RNA
MALAT1 promotes OS progression by regulating HMGB1 expression via
miR-142-3p and miR-129 (10). In the
present study, bioinformatics suggested that HuR binds to HMGA1.
Notably, HMGA1 may promote thyroid cancer and colon cancer
proliferation and invasion (11,12). Thus,
it was hypothesized that HuR promotes OS progression via binding to
and enhancing HMGA1 mRNA expression, and whether HuR could promote
HMGA1 expression via miR-142-3p was further explored.
The results of the present study demonstrated that
HuR directly binds to HMGA1, and enhances HMGA1 mRNA stability and
expression. The promotion of HuR in OS progression and HMGA1
expression may be attenuated via miR-142 overexpression. Notably,
HMGA1 3′UTR with a mutated miR-142-3p binding site did not respond
to HuR overexpression. Collectively, these results elucidated the
roles and associated underlying mechanisms of HuR in OS
progression.
Materials and methods
Clinical tissues, cells culture and
bioinformatics analysis
The human OS cell line MG63 was purchased from the
American Type Culture Collection (Manassas, VA, USA). MG63 cells
were cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal
bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.),
penicillin and streptomycin at 37°C under a humidified atmosphere
containing 5% CO2. A total of 32 OS tumor samples,
paired with adjacent normal tissues, were obtained from 12 females
and 20 males patients aged 19–61 who underwent surgery at Shanghai
Putuo District Central Hospital (Shanghai, China) between February
2014 and January 2017. Approval from the Institute Research Ethics
Committee of Shanghai Putuo District Central Hospital was obtained
for the use of these clinical materials for research purposes and
written informed consent was obtained from patients prior to
surgery. Bioinformatics analysis (http://starbase.sysu.edu.cn/index.php) was used to
predict the potential targets of RNA binding protein HuR.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
mRNA expression was confirmed via RT-qPCR analysis.
Briefly, total RNA was extracted from the tissues and the MG63 cell
line using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). The first strand cDNA was synthesized using the
RevertAid™ First Strand cDNA Synthesis kit (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
The mRNA expression levels were determined on the ABI Prism 7500
Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with Universal SYBR® Green mix (Vazyme,
Piscataway, NJ, USA). The denaturing process was 95°C for 5 min,
the annealing process was 58°C for 30 sec and the elongation
process was 72°C for 30 sec. There were 35 cycles of this RT-qPCR
performed. Relative mRNA expression was calculated with the
2−∆∆Cq method (13). The
primers for RT-qPCR were indicated in Table I. Melting curve analysis and 1%
agarose gel electrophoresis were used to check amplification
specificity and length of PCR products. GAPDH and U6 snRNA served
as an internal control for mRNA and miRNA expression,
respectively.
 | Table I.PCR primer sequences. |
Table I.
PCR primer sequences.
| Gene | Sequence (5′-3′) |
|---|
| HuR RT-qPCR
forward |
ATTGTATGTGGTCTCCGCTGTTTG |
| HuR RT-qPCR
reverse |
TTTCTTTTGGGTTGAGCCTTTTTT |
| HMGA1 RT-qPCR
forward |
CCCGCCCACCCACGCATACACACA |
| HMGA1 RT-qPCR
reverse |
GCCCCCAAACCAAAAGCCCAGAGA |
| Ki67 RT-qPCR
forward |
GTGCTCAACAACTTCATTTCCAAC |
| Ki67 RT-qPCR
reverse |
AACACATTTCCTCCAAAACTCTCT |
| E-cadherin RT-qPCR
forward |
ATGGCTTCCCTCTTTCATCTCCTG |
| E-cadherin RT-qPCR
reverse |
TTCATAGTTCCGCTCTGTCTTTGG |
| Vimentin RT-qPCR
forward |
TCAGAATATGAAGGAGGAAATGGC |
| Vimentin RT-qPCR
reverse |
TCAGGGAGGAAAAGTTTGGAAGAG |
| GAPDH RT-qPCR
forward |
CGGAGTCAACGGATTTGGTCGTAT |
| GAPDH RT-qPCR
reverse |
AGCCTTCTCCATGGTGGTGAAGAC |
| Luc-HMGA1-wt
forward |
AAGAAAAACCTTCCCGGTGCAATCG |
| Luc-HMGA1-wt
reverse |
CAAGTAACTGCAAATAGGAAACCAG |
| Lenti-HuR
forward |
ATGTCTAATGGTTATGAAGACCACA |
| Lenti-HuR
reverse |
TTATTTGTGGGACTTGTTGGTTTTG |
| Lenti-HMGA1
forward |
ATGAGTGAGTCGAGCTCGAAGTCCA |
| Lenti-HMGA1
reverse |
TCACTGCTCCTCCTCCGAGGACTCC |
Western blot analysis
Following different treatments, proteins were
extracted using the protein extraction kit (Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China). Protein concentration was examined via
the Bradford assay. The detailed procedure was referred to in the
previous study (9). A total of 30 µg
of protein was analyzed on 10% SDS-PAGE and electrotransferred to
PVDF membranes (Millipore, Bedford, Massachusetts). The membranes
were blocked in 5% non-fat dried milk for 60 min at room
temperature. Primary antibodies against HuR (cat. no. ab136542),
HMGA1 (cat. no. ab129153), Cleaved-caspase3 (cat. no. ab49822),
caspase3 (cat. no. ab13847), epithelial (E)-cadherin (cat. no.
ab1416), Vimentin (cat. no. ab8978) and β-actin (cat. no. ab8227)
were purchased from Abcam (Cambridge, UK), and the dilution ratio
of all antibodies was 1:5,000. Following incubating with primary
antibodies, blots were washed with tris-buffered saline with 0.5%
Tween 20 and incubated with a peroxidase-conjugated secondary Goat
Anti-Rabbit (cat. no., A0208, Beyotime Institute of Biotechnology;
dilution rate: 1:5,000) and Goat Anti-Mouse antibody (cat. no.,
A0216, Beyotime Institute of Biotechnology; dilution rate:
1:5,000), and chemiluminescence was detected using an enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc.) followed by
exposure in Bio-Rad ChemiDoc™ MP system (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The protein expression
level was normalized to β-actin.
Cell viability analysis
Cell Counting Kit-8 (CCK-8; Shanghai Yeasen
Biotechnology Co., Ltd., Shanghai, China) was used to analyze the
cell viability rate of cells with different treatments, in which
cells without HuR knockdown were used as control. In brief, cells
were suspended and 3,000 cells/well were seeded into 96-well
plates, following culturing with DMEM medium with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) for 24, 48 and 72 h. CCK-8 was
added into the DMEM, following the manufacturer's protocol. The
absorbance was detected at 450 nm. The cell viability rate was
presented with the value relative to the control group. Experiments
were repeated at least three times.
Cell apoptosis assay
Cell apoptotic rate was analyzed using the Annexin
V-FITC and propidium iodide (PI) kit (Beyotime Institute of
Biotechnology, Haimen, China) via flow cytometry. Cells with
different treatments were stained with Annexin V-FITC and PI,
followed with flow cytometry analysis using a BD FACSCanto II (BD
Biosciences, Franklin Lakes, NJ, USA), following the manufacturer's
protocol. FlowJo software (version 10.0.7; FlowJo LLC, Ashland, OR,
USA) was used to analyze the data which were expressed as cell
percentage.
Luciferase reporter assay
HMGA1 3′UTR sequences were inserted into the
PMIR-Reporter plasmid (cat. no., AM5795; Thermo Fisher Scientific,
Inc.), denoted as Luc-HMGA1-wt. PCR primers for Luc-HMGA1-wt were
indicated in Table I. PMIR-Reporter
plasmid holding HMGA1 3′UTR sequences with the mutated binding site
of miR-142-3p was obtained with the Site-directed Gene Mutagenesis
kit (Beyotime Institute of Biotechnology) and noted as
Luc-HMGA1-mut. These aforementioned plasmids were co-transfected
with β-gal control plasmid plus Lenti-HuR infection into MG63 cells
using Lipofectamine® 2000 reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
A total of 48 h later, cells were lysed with Reporter lysis buffer
(cat. no, E397A; Promega Corporation, Madison, WI, USA) and
luciferase activity was measured with VivoGlo Luciferin kit (cat.
no., P1041; Promega Corporation) using a luminometer (Thermo Fisher
Scientific, Inc.). The luciferase activity was normalized to β-gal
activity.
Lentivirus package
shRNAs against HuR and a scramble non-targeting
shRNA were purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). The shRNA sequences were inserted into pLKO.1
(Sigma-Aldrich, Merck KGaA), termed as Lenti-HuR-shRNA.
Additionally, HMGA1 and HuR sequences were inserted into
pLVX–IRES-ZsGreen1, termed as Lenti-HMGA1 and Lenti-HuR,
respectively. PCR primers for plasmids construction were mentioned
in Table I. Lentivirus were packaged
in HEK293T cells via co-transfecting Lenti-HuR-shRNA or Lenti-HMGA1
or Lenti-HuR with pCMV-dR8.2 and pMD2.G constructs using
Lipofectamine 2000. Following 72 h, the supernatants were collected
and ultrafiltrated. The virus supernatants (10 µl in 108
TU/ml) were added to MG63 cells with 2 µg/ml Polybrene
(Genomeditech, Shanghai, China).
mRNA stability assay
HuR was knocked down via infecting with
Lenti-HuR-shRNA for 48 h at 37°C. Then, 5 µg/ml ActD (ApexBio,
Houston, TX, USA) was added to inhibit the de novo RNA
synthesis. Total RNA was collected at 2, 4 and 6 h and mRNA
expression was determined via RT-qPCR analysis under the same
conditions as previously described. The mRNA half-life was
determined by comparing to the mRNA level prior to adding ActD.
RNA immunoprecipitation (RIP)
assay
MG63 cells with or without HuR knockdown were lysed
with 25 mM Tris-HCl buffer (pH 7.5) and 100 U/ml RNase inhibitor
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), and then incubated
with protein-A Sepharose beads (Genescript, Nanjing, China)
precoated with 2 µg anti-HuR antibody (as previously stated,
dilution rate, 1:100), or control rabbit IgG (cat no. ab191867,
Abcam, dilution rate, 1:100) was used for negative control for 3 h
at 4°C. The RNA-protein complexes were pulled-down by protein A/G
agarose beads (Genescript) and RNA was extracted with TRIzol,
followed by detecting the HMGA1 expression level with RT-qPCR
assay, as previously described. This experiments were repeated at
least three times.
Statistical analysis
All data were obtained from at least three
independent experiments (n≥3), and presented as the mean ± standard
deviation. Datasets with only two groups were analyzed using
Student's t-test. Differences among multiple groups were analyzed
using one-way analysis of variance with the Tukey's post-hoc test,
and P<0.05 was considered to indicate a statistically
significant difference.
Results
HuR expression level is upregulated in
OS tissues compared with normal adjacent tissues
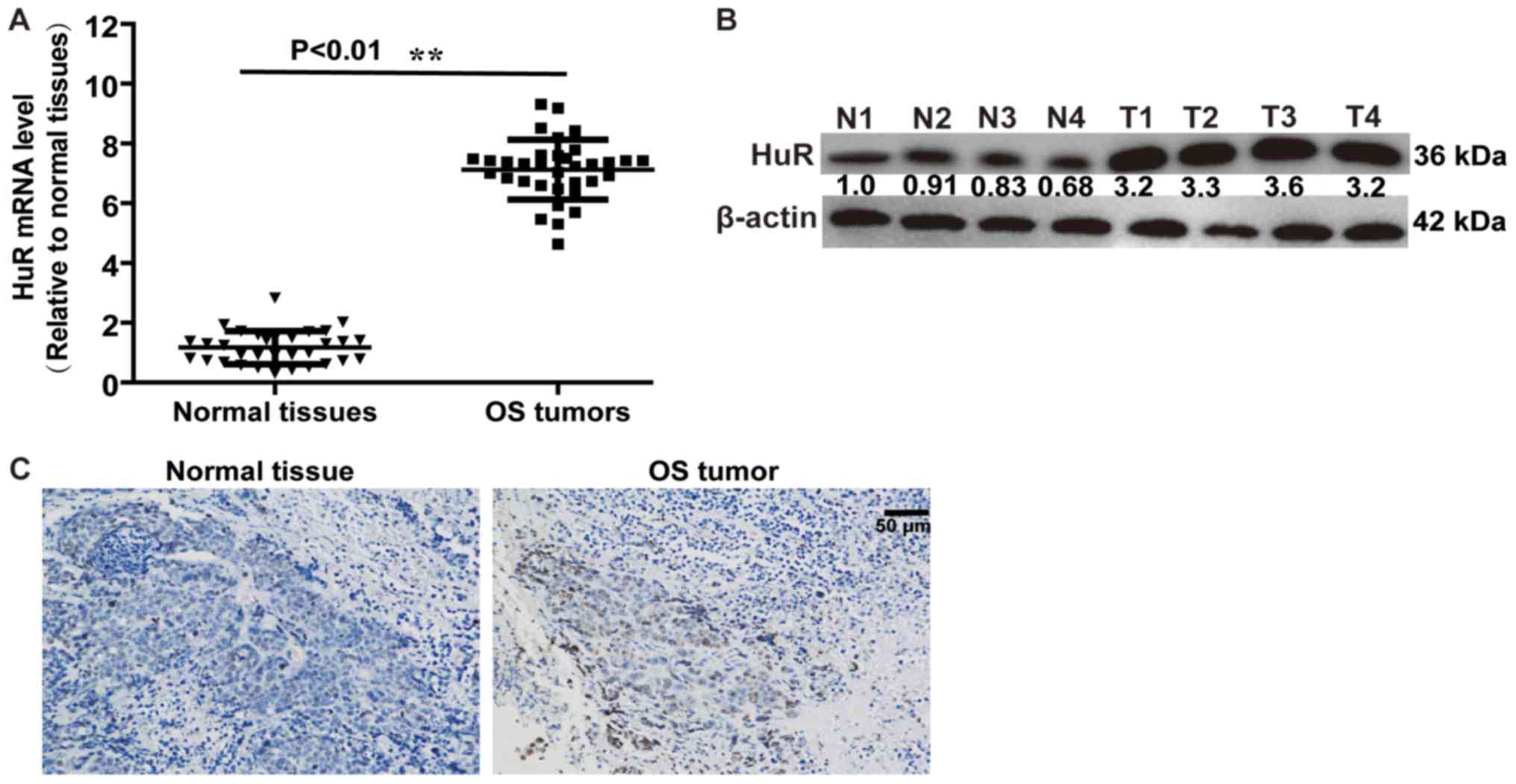
Firstly, the expression level of HuR in OS tissues
and normal adjacent tissues was detected. RT-qPCR and western blot
analysis demonstrated that HuR expression was increased in OS
tissues, compared with normal adjacent tissues (Fig. 1A and B). An identical result was
obtained in a further immunohistochemical assay (Fig. 1C). These results indicated that HuR
promoted OS progression.
Knockdown of HuR enhances OS cell
apoptosis, and inhibits cell viability and the
epithelial-mesenchymal transition (EMT) process
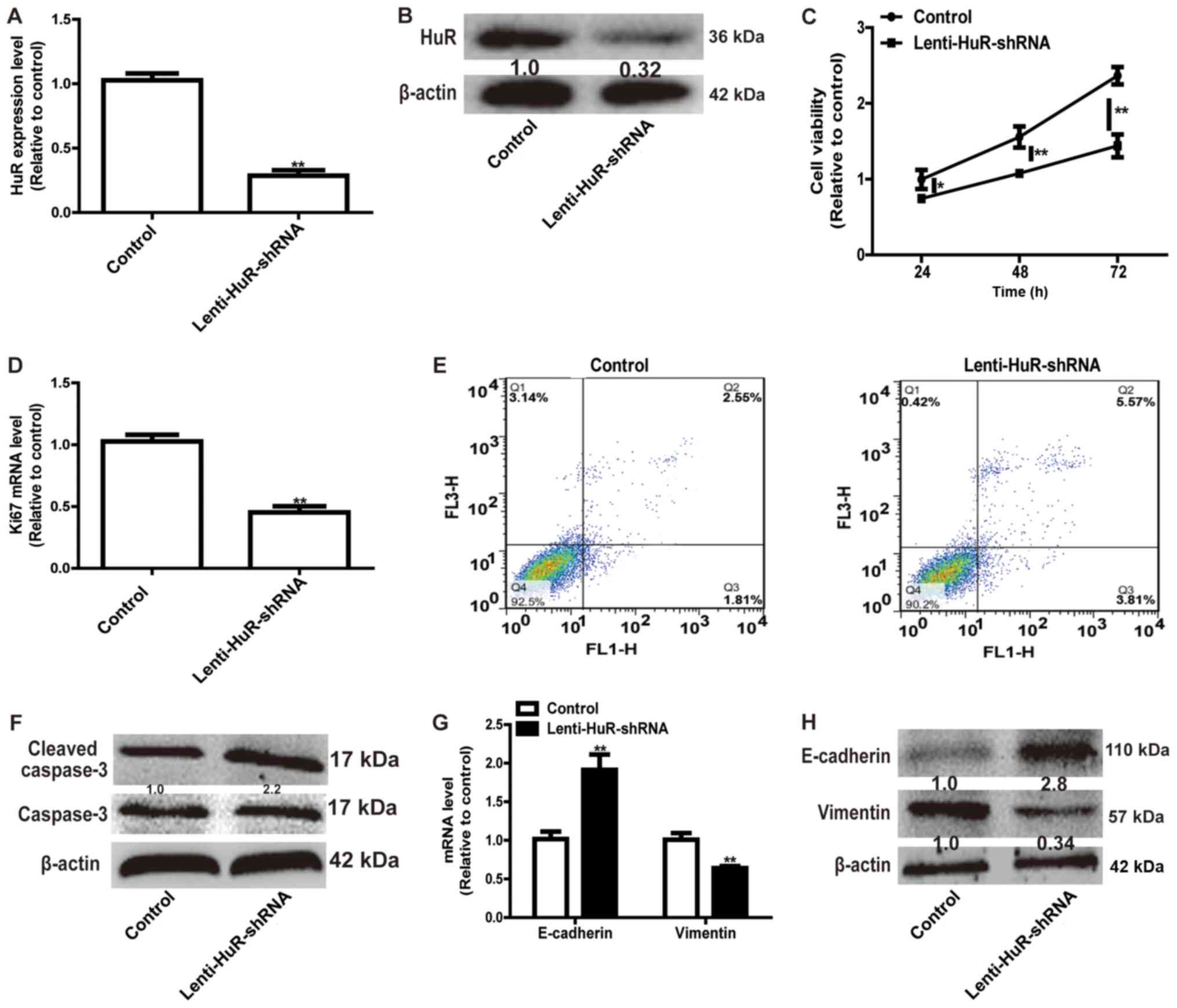
Since the expression level of HuR was upregulated in
OS tissues, HuR expression was knocked down in OS cells. RT-qPCR
and western blot analysis confirmed the knockdown efficiency of
Lenti-HuR-shRNA (Fig. 2A and B).
CCK-8 analysis indicated that HuR knockdown significantly inhibited
MG63 cell viability (Fig. 2C) and the
expression of proliferation marker Ki67 (Fig. 2D). A further cell apoptosis assay
indicated that the knockdown of HuR facilitated cell apoptosis (Q2
and Q3) in MG63 cells (Fig. 2E) and
promoted the expression of the apoptosis executor, cleaved caspase
3 (Fig. 2F). Additionally, HuR
knockdown notably suppressed the EMT process in MG63 cells,
characterized by the downregulation of the expression of the
mesenchymal marker Vimentin and upregulation of the expression of
the epithelial marker E-cadherin (Fig. 2G
and H). These results demonstrated that HuR knockdown inhibited
OS progression.
HuR represses the inhibition of
miR-142-3p on HMGA1 expression
As HuR belongs to the group of RBPs, which bind to
mRNA, and enhance mRNA stability and expression (7), targets were identified via
bioinformatics analysis (http://starbase.sysu.edu.cn/index.php). HMGA1 has been
identified to promote the progression of OS (11). RT-qPCR and western blot analysis
demonstrated that the knockdown of HuR significantly decreased
HMGA1 expression in MG63 cells (Fig. 3A
and B). Furthermore, HMGA1 mRNA stability was reduced with HuR
knockdown (Fig. 3C). Thus, it was
speculated that HuR may directly bind to HMGA1 in MG63 cells. HuR
expression was induced in MG63 cells, with the HuR-binding complex
knocked down via the HuR antibody, followed by examination of the
bound mRNAs via RT-qPCR. RIP results indicated that HuR had a
significantly greater association with HMGA1 compared with the
control (Fig. 3D). Since the
miR-142-3p/HMGA1 axis has been confirmed in OS cells (9), whether HuR was involved in the
miR-142-3p/HMGA1 regulatory pathway in MG63 cells was further
investigated. As depicted in Fig. 3E and
F, overexpression of miR-142-3p significantly attenuated the
promoter effect of HuR on HMGA1 expression. By contrast, knockdown
of miR-142-3p reversed the inhibitory effects of HuR knockdown on
HMGA1 expression (Fig. 3G and H).
Additionally, mutation of the miR-142-3p binding site on HMGA1
3′UTR partially reversed the enhancement of HuR on the luciferase
activity of Luc-HMGA1-wt (Fig. 3I).
Overall, these results indicated that HuR promoted HMGA1 expression
via competitively binding to HMGA1 3′UTR with miR-142-3p.
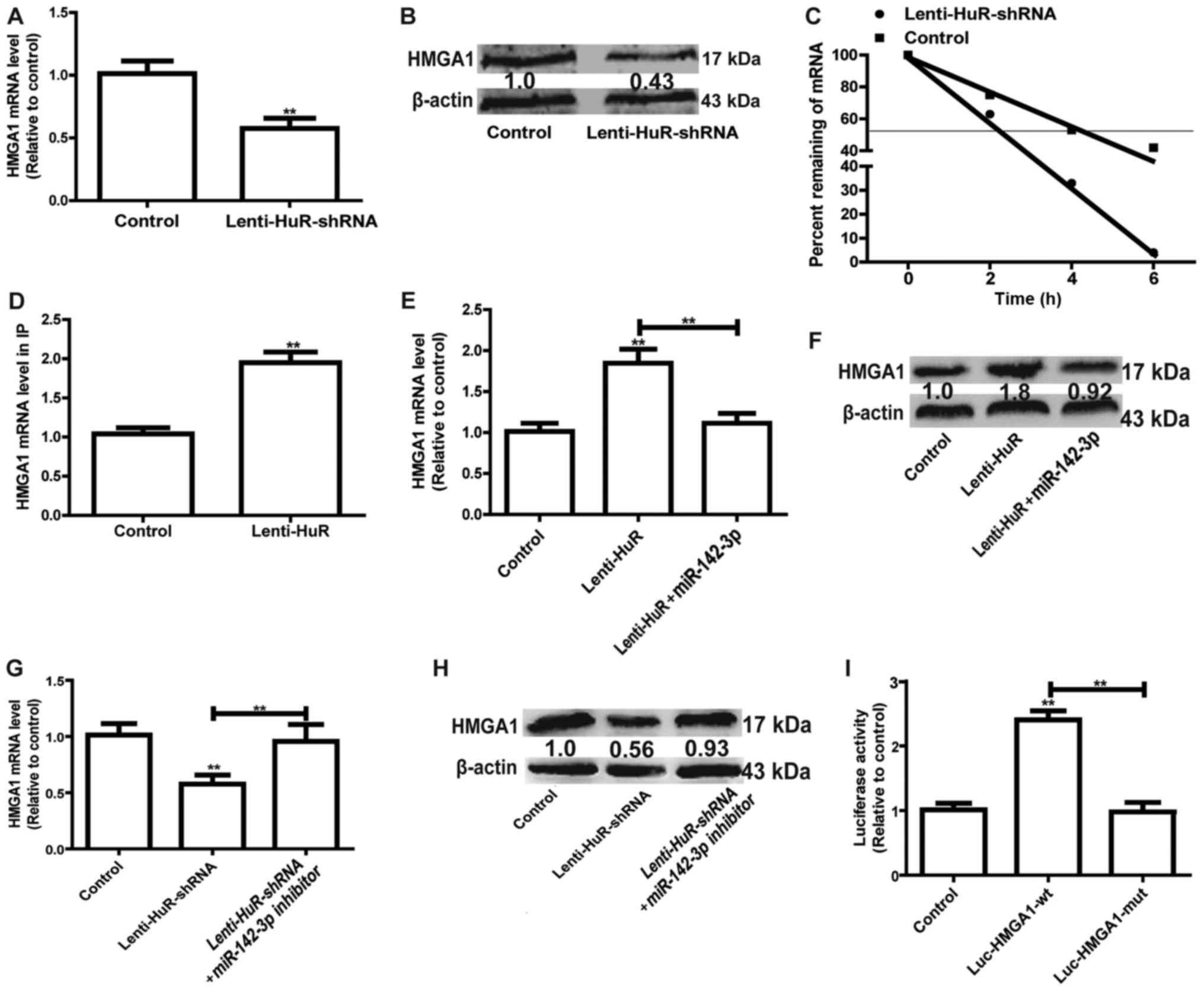 | Figure 3.HuR represses the inhibition of
miR-142-3p on HMGA1 expression. (A) The mRNA expression level of
HMGA1 was detected in cells with or without Lin28A. (B) The protein
expression level of HMGA1 was examined in the cells depicted in
Fig. 3A. (C) The mRNA expression
level of HMGA1 was measured at the indicated times with the
addition of ActD (5 µg/ml) in the cells depicted in Fig. 3A. (D) RT-qPCR was used to measure
HMGA1 abundance presented in the HuR-IP materials following the RIP
assay in cells with or without HuR overexpression. (E) mRNA and (F)
protein expression levels of HMGA1 were detected in cells with HuR
overexpression, and with or without miR-142-3p overexpression. (G)
mRNA and (H) protein expression levels of HMGA1 were detected in
cells with HuR knockdown, and with or without miR-142-3p knockdown.
(I) A luciferase reporter assay was performed to detect the effect
of HuR overexpression on luciferase activities of the control
vector, Luc-HMGA1-wt and Luc-HMGA1-mut. **P<0.01 vs. control
unless indicated otherwise. Lenti-HuR-shRNA, shRNA sequences
inserted into pLKO.1; HuR, human antigen R; HMGA1, High Mobility
Group AT-Hook 1; RIP, RNA immunoprecipitation; Lenti-HuR, HuR
sequences inserted into pLVX-IRES-ZsGreen1; Luc-HMGA1-wt, wild type
HMGA1 3′UTR sequences inserted into the PMIR-Reporter plasmid;
Luc-HMGA1-mut, Luc-HMGA1-wt with mutated miR-142-3p binding
site. |
HuR promotes OS progression in a
miR-142-3p/HMGA1 axis-dependent manner
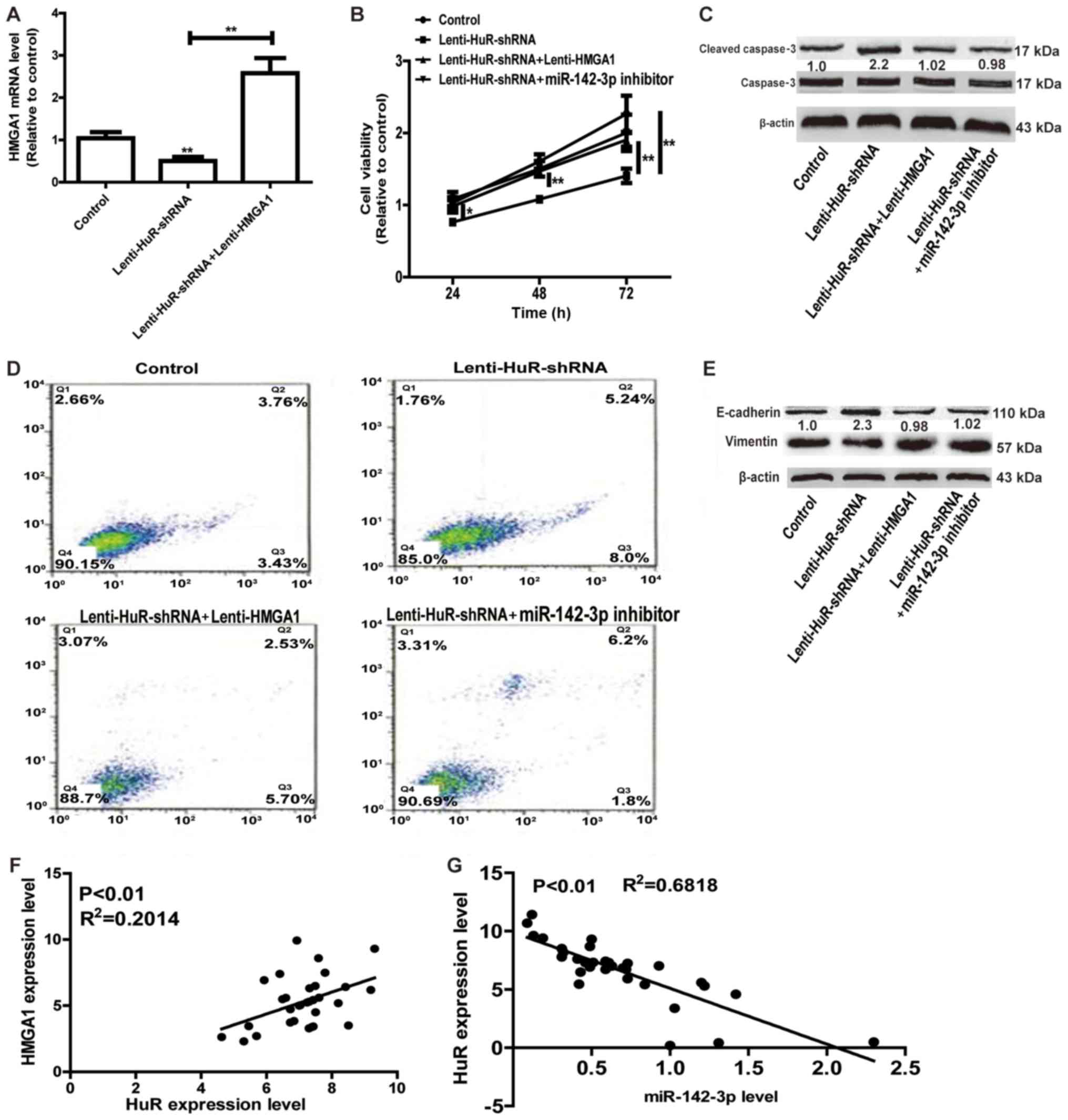
Whether miR-142-3p/HMGA1 axis was involved in the
promotion of HuR in OS progression was further investigated. As
depicted in Fig. 4A, infection with
Lenti-HMGA1 significantly upregulated the HuR expression level,
reversing the suppressive effects of HuR knockdown on HMGA1
expression. Cell viability and apoptosis assays indicated that
overexpression of HMGA1 or transfection with miR-142-3p inhibitor
attenuated the inhibition of HuR knockdown on cell proliferation
and promotion on cell apoptosis (Fig.
4B-D). Additionally, HMGA1 overexpression or miR-142-3p
knockdown notably attenuated or even reversed EMT process inhibited
by HuR knockdown (Fig. 4E). Notably,
HuR expression was positively associated with HMGA1 expression, and
negatively associated with miR-142-3p expression in OS tissues
(Fig. 4F and G). Therefore, the
results of the present study results indicated that HuR promoted OS
progression at least partly through the miR-142-3p/HMGA1 axis.
Discussion
HuR is overexpressed in a number of cancer types,
including malignant brain tumors (8)
and pancreatic cancer cells (14).
However, its role in OS remains unclear. In the present study, it
was elucidated that HuR contributed to cell viability and inhibited
cell apoptosis in MG63 cells.
Although HuR have been frequently investigated, the
majority of its functions are via its targets (15). A previous study indicated that RBPs
bind to mRNA 3′UTR competitively with miRNAs, including transformer
2β and miR-204, regulating apoptosis through competitive binding to
3′UTR of B-cell lymphoma-2 mRNA (16). Additionally, DND microRNA-mediated
repression inhibitor 1 promotes breast cancer apoptosis via
stabilizing Bim mRNA in a miR-221 binding site (17). These results demonstrate that HuR may
facilitate OS progression via competitively binding to mRNA with
miRNAs. A bioinformatics assay demonstrated that HMGA1 was a
potential target of HuR. Previous studies have indicated the
promotion of HMGA1 in OS progression (9), and that HMGA1 modulates autophagy in
cancer cells (18). These results
confirmed the oncogenic roles of HMGA1. Notably, the results of the
present study indicated that HMGA1 overexpression attenuated the
inhibition of HuR knockdown on OS progression, indicating that HuR
exerts its effects at least partly through HMGA1. Furthermore, as
the miR-142-3p/HMGA1 axis has been associated with OS progression
(9), and it was determined that
mutation of the binding site of miR-142-3p on HMGA1 sequences
prevented the promotion of HuR via HMGA1, these results indicated
that miR-142-3p is involved in the interaction between HuR and
HMGA1, and HuR may competitively bind to HMGA1 3′UTR via
miR-142-3p.
To the best of our knowledge, this is the first
study demonstrating the roles and associated mechanisms underlying
HuR in OS progression. As HMGA1 may be targeted by other RBPs,
including insulin growth factor 2 (19), there may be other RBPs, which are
involved in the regulation of HMGA1 in OS progression, which
require further investigation. However, since the oncogenic roles
of HuR have been identified in other tumor types, it was
hypothesized that analyzing other functions, and more detailed
mechanisms underlying HuR/HMGA1 axis during OS development may
provide significant insights into gene regulatory networks and
their clinical implications for OS.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during the current
study are included in this published article.
Authors' contributions
WP and ZW designed the study. WP, JP and LZ analyzed
the data. WP, JP, BJ, YC and CL performed the experiments. WP and
LZ wrote the manuscript.
Ethics approval and consent to
participate
Approval from the Institute Research Ethics
Committee of Shanghai Putuo District Central Hospital was obtained
for the use of these clinical materials for research purposes and
written informed consent was obtained from patients prior to the
study start.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
References
|
1
|
Picci P: Osteosarcoma (osteogenic
sarcoma). Orphanet J Rare Dis. 2:62007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heare T, Hensley MA and Dell'Orfano S:
Bone tumors: Osteosarcoma and Ewing's sarcoma. Curr Opin Pediatr.
21:365–372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weeden S, Grimer RJ, Cannon SR, Taminiau
AH and Uscinska BM; European Osteosarcoma Intergroup: The effect of
local recurrence on survival in resected osteosarcoma. Eur J
Cancer. 37:39–46. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu J, Li X, Guo X, Guo Q, Xiang C, Zhang
Z, Xing Y, Xi T and Zheng L: CCR2 3′UTR functions as a competing
endogenous RNA to inhibit breast cancer metastasis. J Cell Sci.
130:3399–3413. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nahas GR, Murthy RG, Patel SA, Ganta T,
Greco SJ and Rameshwar P: The RNA-binding protein Musashi 1
stabilizes the oncotachykinin 1 mRNA in breast cancer cells to
promote cell growth. FASEB J. 30:149–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang L, Du WW, Yang X, Chen K, Ghanekar A,
Levy G, Yang W, Yee AJ, Lu WY, Xuan JW, et al: Versican
3′-untranslated region (3′-UTR) functions as a ceRNA in inducing
the development of hepatocellular carcinoma by regulating miRNA
activity. FASEB J. 27:907–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang W, Vreeland AC and Noy N:
RNA-binding protein HuR regulates nuclear import of protein. J Cell
Sci. 129:4025–4033. 2016.PubMed/NCBI
|
|
8
|
Steinmeyer N, Doller A, Biyanee A, Brauss
T, Schmid T, Pfeilschifter J and Eberhardt W: Lymphotoxin α, a
novel target of posttranscriptional gene regulation by HuR in HepG2
cells. FEBS Lett. 589:1943–1950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu G, Wang J, Jia Y, Shen F, Han W and
Kang Y: MiR-142-3p functions as a potential tumor suppressor in
human osteosarcoma by targeting HMGA1. Cell Physiol Biochem.
33:1329–1339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu K, Huang J, Ni J, Song D, Ding M, Wang
J, Huang X and Li W: MALAT1 promotes osteosarcoma development by
regulation of HMGB1 via miR-142-3p and miR-129-5p. Cell Cycle.
16:578–587. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhong J, Liu C, Zhang QH, Chen L, Shen YY,
Chen YJ, Zeng X, Zu XY and Cao RX: TGF-β1 induces HMGA1 expression:
The role of HMGA1 in thyroid cancer proliferation and invasion. Int
J Oncol. 50:1567–1578. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang Q, Wang X, Tang C, Chen X and He J:
H19 promotes the migration and invasion of colon cancer by sponging
miR-138 to upregulate the expression of HMGA1. Int J Oncol.
50:1567–1578. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Scmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Romeo C, Weber MC, Zarei M, DeCicco D,
Chand SN, Lobo AD, Winter JM, Sawicki JA, Sachs JN, Meisner-Kober
N, et al: HuR contributes to TRAIL resistance by restricting death
receptor 4 expression in pancreatic cancer cells. Mol Cancer Res.
14:599–611. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Muralidharan R, Panneerselvam J, Chen A,
Zhao YD, Munshi A and Ramesh R: HuR-targeted nanotherapy in
combination with AMD3100 suppresses CXCR4 expression, cell growth,
migration and invasion in lung cancer. Cancer Gene Ther.
22:581–590. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kuwano Y, Nishida K, Kajita K, Satake Y,
Akaike Y, Fujita K, Kano S, Masuda K and Rokutan K: Transformer 2β
and miR-204 regulate apoptosis through competitive binding to 3′
UTR of BCL2 mRNA. Cell Death Differ. 22:815–825. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng F, Pan Y, Lu YM, Zhu L and Chen S:
RNA-binding protein Dnd1 promotes breast cancer apoptosis by
stabilizing the Bim mRNA in a miR-221 binding site. Biomed Res Int.
2017:95961522017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Conte A, Paladino S, Bianco G, Fasano D,
Gerlini R, Tornincasa M, Renna M, Fusco A, Tramontano D and
Pierantoni GM: High mobility group A1 protein modulates autophagy
in cancer cells. Cell Death Differ. 24:1948–1962. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dai N, Ji F, Wright J, Minichiello L,
Sadreyev R and Avruch J: IGF2 mRNA binding protein-2 is a tumor
promoter that drives cancer proliferation through its client mRNAs
IGF2 and HMGA1. Elife. 6:e271552017. View Article : Google Scholar : PubMed/NCBI
|