Introduction
Lung cancer is the leading cause of
cancer-associated mortality worldwide, accounting for 1.3 million
mortalities annually (World Health Organization, 2008) (1). It has the highest mortality rate of all
types of cancer in women and men globally, with its mortality rate
exceeding the combined mortality rates of breast, prostate,
colorectal and pancreatic cancer (1).
Non-small cell lung cancer (NSCLC) accounts for ~80% of all lung
cancer cases globally, with ~75% of patients being diagnosed in the
middle-late stages, and the 5-year survival rate of NSCLC is poor
(mean, 9–11 months) (2). NSCLC
includes squamous cell carcinoma, adenocarcinoma and large cell
carcinoma subtyes (3); and NSCLC
cells divide more slowly and spread relatively late compared with
small-cell carcinoma cells. At present, the lack of knowledge
concerning the molecular mechanisms of NSCLC progression has
limited the development of novel treatment strategies. However, in
combination with a large number of applications involving
bioinformatics available for clinical studies, a large volume of
disease-associated bioinformatics data has been produced. Obtaining
detailed biological information from these resources is valuable
for the study and development of therapeutic strategies for
NSCLC.
High-throughput bioinformatics platforms may promote
the analysis of differential gene expression, including
microarrays, and have a wide range of applications in medical
oncology, particularly in searching for disease-associated
biomarkers (4), alternative splicing
(5) and gene function prediction
(6). Numerous previous studies have
generated a large volume of microarray data, and a number of gene
expression profiling studies on NSCLC have identified
differentially expressed genes (DEGs) in various pathways,
molecular functions and biological processes.
Therefore, in the present study, the original data
(GSE33532) was downloaded from the Gene Expression Omnibus (GEO;
http://www.ncbi.nlm.nih.gov/geo/), which
contains variations in gene expression profiles in stage I and II
(Tumor-Node-Metastasis classification of malignant tumors)
(7) NSCLC tissues (8), and in normal tissues. The gene
expression profiling of NSCLC tissues has resulted in the
establishment of several prognostic and predictive gene signatures
with little overlap (8).
Subsequently, R software (version 3.4.1; https://www.r-project.org/) was used to compare the
expression profiles of NSCLC tissues with those of normal tissues
in order to identify DEGs. Subsequent to obtaining the DEGs,
biological function enrichment and integrated protein-protein
interaction network (PPI) analyses were performed to establish the
complete characterization of the DEGS for NSCLC and obtain further
understanding into the mechanism underlying NSCLC. By analyzing the
biological function of the DEGs, certain potential biomarkers were
identified for additional study.
Materials and methods
Microarray data
The GSE33532 microarray expression dataset was
downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). This dataset was
based on the Affymetrix GPL570 platform (Affymetrix Human Genome
U133 Plus 2.0 Array), submitted by Meister et al (8). The GSE33532 dataset contained 100
samples, including 80 NSCLC tissue samples and 20 normal tissue
samples.
Identification of DEGs
The raw data used for analysis contained CEL files
(GPL570 platform), and the results were obtained using Affy package
(version 1.52.0) (9), Limma package
(version 3.0.1) (10) and Gplot
package (version 3.3.2; https://cran.r-project.org/web/packages/gplots/). A
hierarchical clustering method was applied to classify these data
into either the NSCLC or normal group. The quality control was
indicated by using the ‘Robust Multiarray Averaging’ (RMA) function
in the Affy packages. Subsequently, the adjust method ‘BH’ in the
Limma R package was used to identify DEGs with log |fold
change|>1 and adj. P<0.05 as cut-off levels for statistically
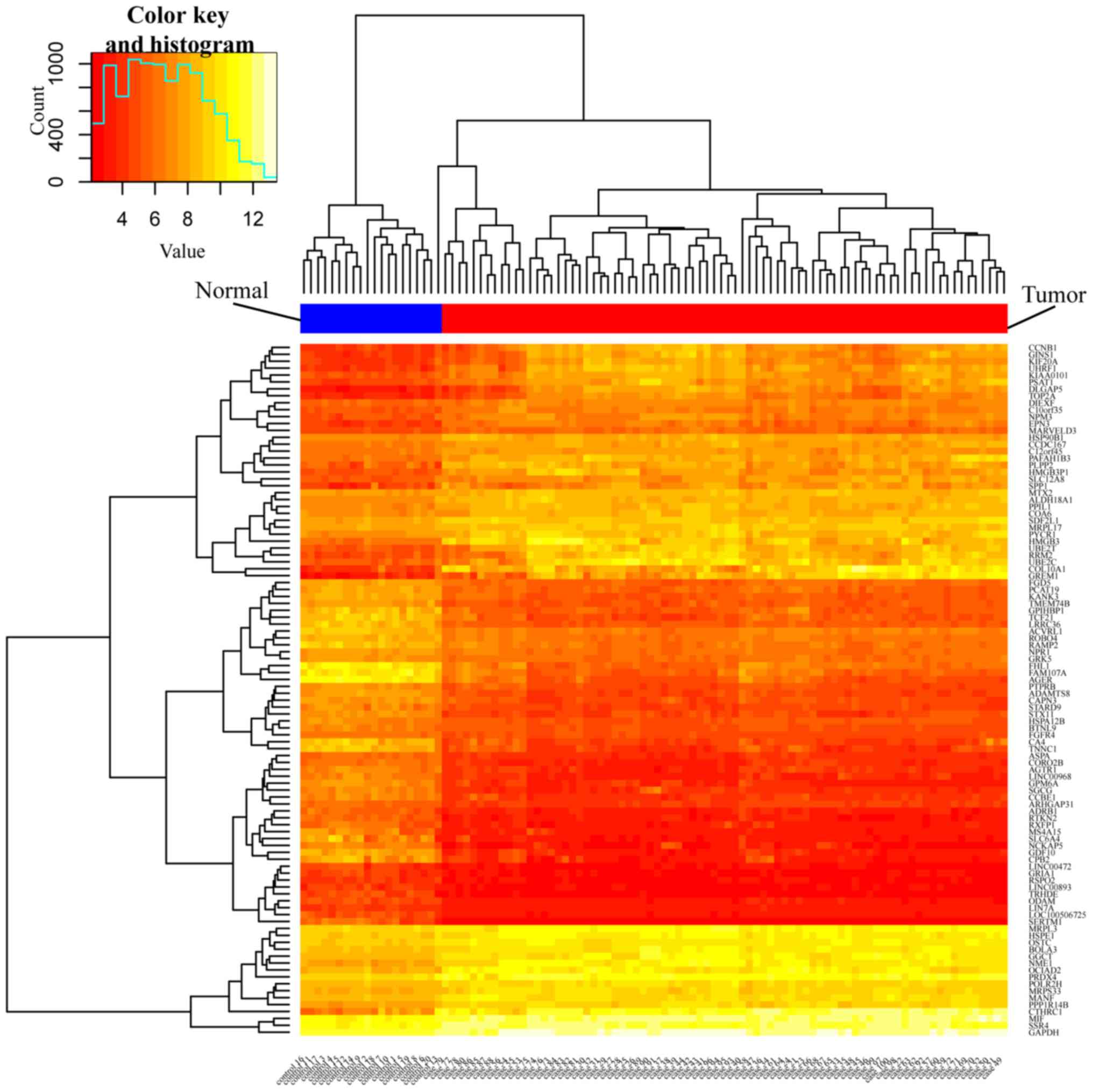
significant candidate genes. Following this, a heatmap was
constructed to indicate the differential expression levels of the
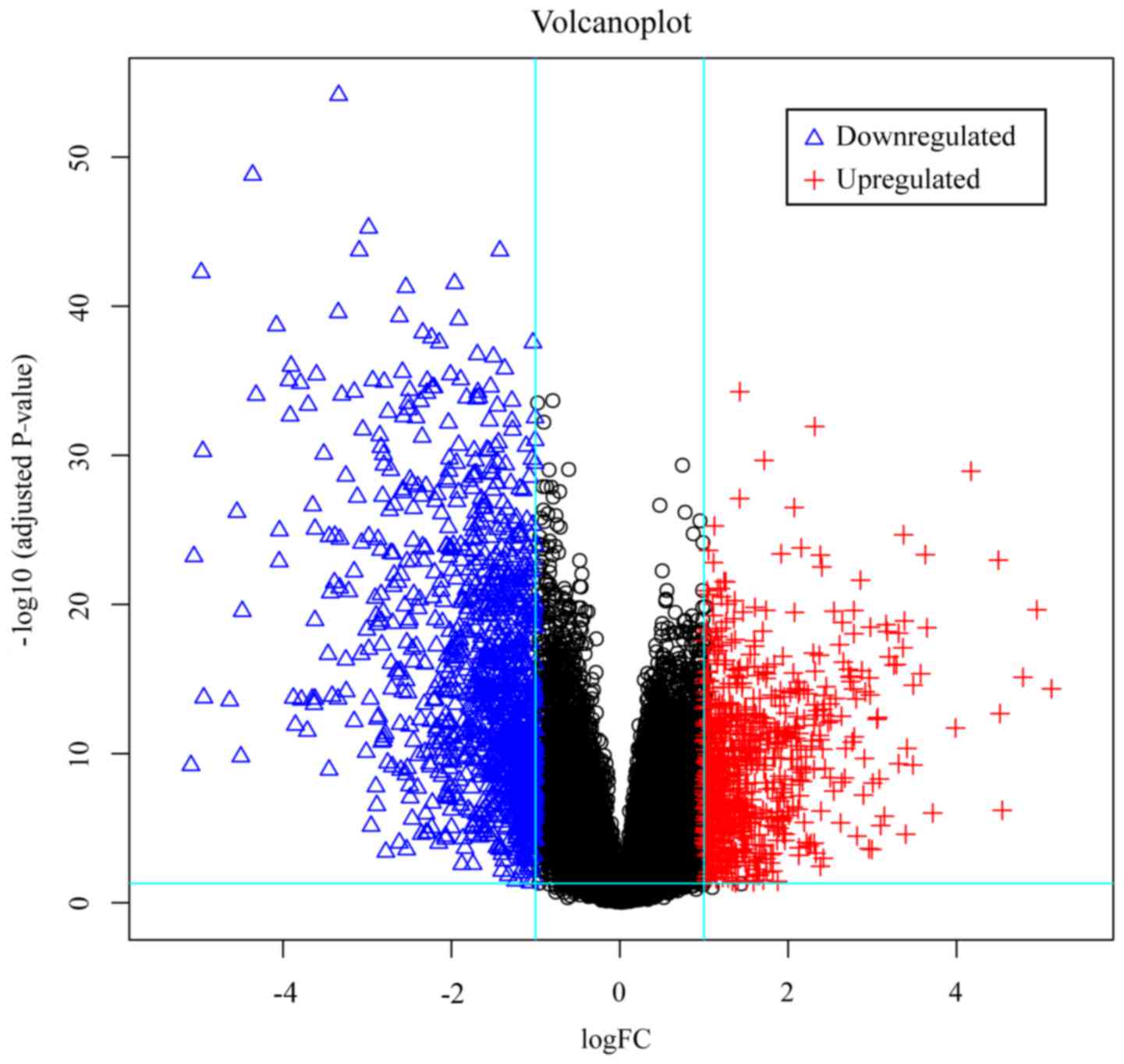
top 100 DEGs (50 upregulated and 50 downregulated), and a volcano
plot was produced to map all DEGs in this dataset.
Gene Ontology (GO) and kyoto
encyclopedia of genes and genomes (KEGG) analyses
In the field of molecular biology, GO is the most
developed and widely-used ontology. Through the GO method, it is
also possible to characterize biological concepts with different
specificity levels, from general to precise concepts (11). KEGG (http://www.genome.ad.jp/kegg/) is a collection of
databases and associated software for understanding and simulating
higher-order functional behaviors of cells or organisms from their
genomic information (12). In
addition, analyzing DEGs using the Database for Annotation,
Visualization and Integrated Discovery (DAVID; http://david.ncifcrf.gov/) is an important means of
identifying the relevant biological functions for any
high-throughput gene functional analysis (13). The DEGs of the present study were
analyzed to identify their biological function using the results
from the GO and KEGG pathway analyses and the DAVID online tool.
P<0.05 was considered to indicate a statistically significant
difference.
Disease module creation using the
integration of PPIs
The Search Tool for the Retrieval of Interacting
Genes (STRING) database includes 5,214,234 proteins from 1,133
organisms. It identified the PPIs of the DEGs identified in the
present study. To evaluate their interactive associations, all DEGs
were mapped to this database, in order to get an improved result,
interactions with the highest confidence score (score >0.9) in
the STRING database were selected. Subsequently, the PPIs were
analyzed by Cytoscape software (version 3.2.1; National Resource
for Network Biology) (14) to obtain
the PPI network. The criteria of disease module searching were set
as follows: Molecular COmplex DEtection (MCODE) score >3, and
each module must have >4 nodes. P<0.05 was considered to
indicate a statistically significant difference.
Pathway crosstalk
The regulation of biological pathways is complex,
yet it underlies the functional coordination of cells (15). Cancer is a disease that is
characterized by unregulated cell proliferation, driven by
underlying pathway deregulation (16). This pathway deregulation occurs within
and between pathways (17). Pathway
crosstalk analysis may assist in identifying the interactions among
pathways enriched by DEGs. The present study used the pathway
information of DEGs from the KEGG database to conduct a pathway
crosstalk analysis in NSCLC. The principle of pathway crosstalk is
defined by >3 overlapping genes in 2 pathways (each pathway must
have ≤5 genes) (17). To measure the
interaction of crosstalk, 2 novel variables were introduced: The
Jaccard Coefficient (JC) JC=|A∩BA∪B| and the Overlap Coefficient (OC)
OC=|A∩B|min(|A|,|B|),
where A and B are the lists of genes included in the 2 analyzed
pathways. The rank value (RV) was calculated by JC+OC2 (17). Subsequently, Cytoscape software was
used to map the interaction between pathways and use RV as the
interaction type in order to display the weight of the
crosstalk.
Survival analysis
The Kaplan-Meier plotter (KMplot, http://www.kmplot.com/analysis) is capable of
assessing the effect of 54,675 genes on survival using 10,293
cancer samples. These include 5,143 breast, 1,648 ovarian, 2,437
lung and 1,065 gastric cancer samples, with mean follow-up periods
of 69, 40, 49 and 33 months, respectively. The primary purpose of
this tool is to conduct meta-analysis-based biomarker assessments
(18). The top 9 hub genes of disease
module in the present study were entered into the KMplot database
to examine the association between these genes and the 5-year
survival rates of patients.
Results
Identification of DEGs
The dataset of the present study contained 100
samples; 20 normal tissue samples and 80 NSCLC tissue samples. Each
sample from the chip was analyzed by Affy and Limma packages of R
software, respectively. Based on the Affy package, using the RMA
method to pre-process the dataset, then using the lmFit function of
the Limma package to screen differentially expressed genes with
P<0.05 and fold control (Log|FC|)>1 criteria to obtain DEGs
from the dataset, a total of 1,795 genes were identified. Of these,
729 were upregulated and 1,066 were downregulated. The Gplots
package (R software) was used to obtain the top 50 upregulated and
the top 50 downregulated DEGs, and to produce an expression heatmap
(Fig. 1) and a volcano plot of the
DEG distribution (Fig. 2).
GO enrichment analysis
Enriched GO categories and KEGG pathways were
identified by uploading all DEGs to DAVID. The results of the GO
analysis indicated that the upregulated DEGs were significantly
enriched in ‘biological processes’ (BP), which included ‘cell
cycle’ and ‘nuclear division’ (Table
I); downregulated DEGs were also significantly enriched in BP,
including ‘response to wounding’, ‘anatomical structure
morphogenesis’ and ‘response to stimulus’ (Table I). For ‘molecular function’, the
upregulated DEGs were enriched in ‘microtubule motor activity’,
‘protein binding’ and ‘structural molecule activity’, and the
downregulated DEGs were enriched in ‘calcium ion binding’, ‘protein
binding’ and ‘growth factor binding’ (Table I). Concurrently, the GO ‘cell
component’ analysis also revealed that the upregulated DEGs were
significantly enriched in ‘chromosome’ and ‘centromeric region’,
and that the downregulated DEGs were enriched in ‘plasma membrane
part’, ‘extracellular region part’ and ‘cell periphery’ (Table I).
 | Table I.Gene Ontology analysis of DEGs
associated with non-small cell lung cancer. |
Table I.
Gene Ontology analysis of DEGs
associated with non-small cell lung cancer.
| A, Upregulated
DEGs |
|---|
|
|---|
| Category | Term/gene
function | Gene count | P-value |
|---|
| BP | GO:0022403; cell
cycle phase | 117 |
3.56×10−30 |
| BP | GO:0000278; mitotic
cell cycle | 106 |
3.67×10−27 |
| BP | GO:0022402; cell
cycle process | 124 |
3.65×10−26 |
| BP | GO:0000087; M phase
of mitotic cell cycle | 71 |
4.22×10−26 |
| BP | GO:0000280; nuclear
division | 69 |
1.79×10−25 |
| MF | GO:0003777;
microtubule motor activity | 13 |
4.16×10−6 |
| MF | GO:0005515; protein
binding | 323 |
1.48×10−5 |
| MF | GO:0005198;
structural molecule activity | 45 |
1.59×10−5 |
| MF | GO:0016538;
cyclin-dependent protein kinase regulator activity | 6 |
4.69×10−5 |
| MF | GO:0003678; DNA
helicase activity | 8 |
2.00×10−4 |
| CC | GO:0044427;
chromosomal part | 64 |
2.90×10−17 |
| CC | GO:0005694;
chromosome | 70 |
5.28×10−17 |
| CC | GO:0000793;
condensed chromosome | 34 |
2.21×10−16 |
| CC | O:0000775;
chromosome, centromeric region | 32 |
2.73×10−16 |
| CC | GO:0000779;
condensed chromosome, centromeric region | 25 |
5.44×10−16 |
|
| B, Downregulated
DEGs |
|
|
Category | Term/gene
function | Gene
count | P-value |
|
| BP | GO:0009611;
response to wounding | 130 |
1.29×10−18 |
| BP | GO:0009653;
anatomical structure morphogenesis | 198 |
1.74×10−18 |
| BP | GO:0050896;
response to stimulus | 468 |
3.25×10−18 |
| BP | GO:0042221;
response to chemical stimulus | 239 |
5.30×10−17 |
| BP | GO:0044707;
single-multicellular organism process | 406 |
2.16×10−16 |
| MF | GO:0005509; calcium
ion binding | 65 |
1.64×10−7 |
| MF | GO:0005515; protein
binding | 434 |
2.35×10−7 |
| MF | GO:0019838; growth
factor binding | 20 |
2.36×10−7 |
| MF | GO:0005102;
receptor binding | 99 |
5.28×10−7 |
| MF | GO:0097367;
carbohydrate derivative binding | 27 |
7.64×10−7 |
| CC | GO:0044459; plasma
membrane part | 200 |
2.10×10−24 |
| CC | GO:0044421;
extracellular region part | 139 |
2.28×10−24 |
| CC | GO:0071944; cell
periphery | 346 |
2.98×10−21 |
| CC | GO:0005886; plasma
membrane | 338 |
2.20×10−20 |
| CC | GO:0005615;
extracellular space | 105 |
1.81×10−17 |
KEGG pathway analysis
Table II indicates
the KEGG analysis result of the most significantly enriched
pathways (top 5 upregulated and downregulated) in the DEGs of
NSCLC. The upregulated DEGs were significantly enriched in ‘cell
cycle’, ‘DNA replication’, ‘tumor protein 53 (p53) signaling
pathway’, ‘Extracellular matrix (ECM)-receptor interaction’ and
‘Protein digestion and absorption’, while the downregulated DEGs
were significantly enriched in ‘Complement and coagulation
cascades’, ‘Malaria’, ‘Cell adhesion molecules (CAMs)’, ‘Axon
guidance’ and ‘Renin secretion’.
| Table II.Kyoto Encyclopedia of Genes and
Genomes pathway analysis of DEGs associated with non-small cell
lung cancer. |
Table II.
Kyoto Encyclopedia of Genes and
Genomes pathway analysis of DEGs associated with non-small cell
lung cancer.
| A, Upregulated
DEGs |
|---|
|
|---|
| Pathway ID | Name | Count | P-value | Genes |
|---|
| hsa04110 | Cell cycle | 25 |
8.4×10−11 | CDK1, CDC6, E2F3,
DBF4, TTK, ESPL1, CDC20, CHEK1, MCM2, PTTG1, SFN, MCM4, CCNB1,
CCNE2, CCNE1, CDC45, CDKN2A, CCNB2, MAD2L1, PLK1, PCNA, BUB1B,
ORC6, ORC1, CCNA2 |
| hsa03030 | DNA
replication | 10 |
8.8×10−6 | PRIM1, DNA2, RFC4,
POLE2, PCNA, MCM2, RNASEH2A, MCM4, FEN1, RPA3 |
| hsa04115 | p53 signaling
pathway | 13 |
1.2×10−5 | CDK1, CHEK1, SFN,
PMAIP1, GTSE1, CCNB1, CCNE2, CCNE1, CDKN2A, CCNB2, SERPINB5, RRM2,
IGFBP3 |
| hsa04512 | Extracellular
matrix-receptor interaction | 11 |
2.0×10−3 | IBSP, COMP, COL3A1,
COL1A2, COL1A1, COL11A1, THBS2, COL5A2, COL5A1, SPP1, HMMR |
| hsa04974 | Protein digestion
and absorption | 11 |
3.0×10−3 | KCNN4, COL17A1,
COL7A1, PRSS2, COL3A1, COL1A2, COL1A1, COL11A1, COL5A2, COL5A1,
COL10A1 |
|
| B, Downregulated
DEGs |
|
| Pathway
ID | Name | Count | P-value | Genes |
|
| hsa04610 | Complement and
coagulation cascades | 16 |
4.2×10−6 | C7, C5AR1, C6, F8,
SERPING1, C4BPA, C1QA, C8B, C1QB, VWF, CD55, THBD, SERPIND1, CFD,
CPB2, PROS1 |
| hsa05144 | Malaria | 12 |
5.9×10−5 | CSF3, GYPC, ICAM1,
ITGAL, SELP, IL6, CD36, PECAM1, ACKR1, TLR4, HBB, SELE |
| hsa04514 | Cell adhesion
molecules | 20 |
3.1×10−4 | ICAM1, ITGAL, SELP,
CLDN18, OCLN, PTPRM, CADM1, ICAM2, CLDN5, NECTIN3, HLA-DMA, CDH5,
SIGLEC1, CD34, ITGA8, PECAM1, ESAM, JAM2, SELE, NEGR1 |
| hsa04360 | Axon guidance | 18 |
6.3×10−4 | ABLIM1, PLXNA2,
ABLIM3, EFNB2, NTN4, DPYSL2, CXCL12, SLIT2, SLIT3, SEMA5A, SEMA6A,
RND1, SEMA6D, FYN, SEMA3G, CFL2, SEMA3E, ROBO2 |
| hsa04924 | Renin
secretion | 12 |
7.1×10−4 | AGTR1, ACE, ADRB2,
ADRB1, PLCB4, PTGER4, GUCY1A2, GUCY1A3, NPR1, AQP1, CACNA1D,
ITPR1 |
Integration of PPIs create disease
module
All DEGs of NSCLC were loaded into the STRING
database, to obtain the PPI data among them, and PPIs with highest
interaction score (confidence >0.9) were selected. Subsequently,
Cytoscape was used to identify the 9 hub nodes with the highest
degrees, which included aurora kinase B (AURKB), centromere protein
A (CENPA), cyclin dependent kinase 1 (CDK1), proliferating cell
nuclear antigen (PCNA), BUB1 mitotic checkpoint serine/threonine
kinase B (BUB1B), cell division cycle 20 (CDC20), baculoviral
Inhibitor of apoptosis (IAP) repeat containing 5 (BIRC5), MAD2
mitotic arrest deficient-like 1 (MAD2L1) and polo like kinase 1
(PLK1). Of these hub genes (Table
III), CDK1 revealed the highest node degree (degree=58). In
addition, a total of 684 nodes and 2,134 edges were analyzed using
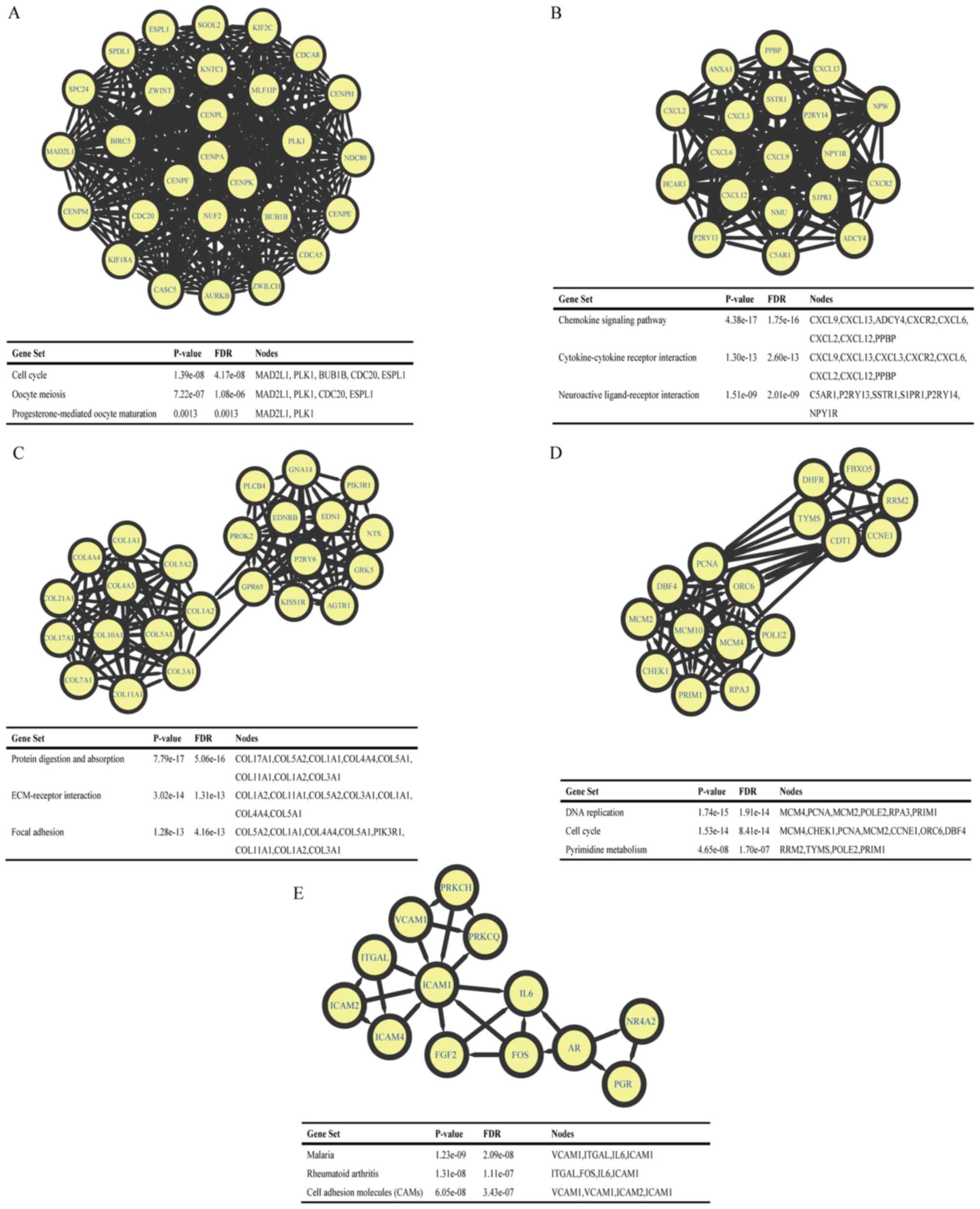
MCODE in Cytoscape software. The top 5 largest size modules were
identified, and the functional annotations of the genes within them
were isolated (Fig. 3). KEGG
enrichment analysis of these modules demonstrated that the genes in
modules 1–5 were primarily associated with ‘cell cycle’, ‘chemokine
signaling pathway’, ‘protein digestion and absorption’, ‘DNA
replication’ and ‘malaria’.
| Table III.Hub genes and rank of
degreesa. |
Table III.
Hub genes and rank of
degreesa.
| Gene symbol | Full name | Degree |
|---|
| CDK1 | Cyclin dependent
kinase 1 | 58 |
| PLK1 | Polo-like kinase
1 | 53 |
| AURKB | Aurora kinase
B | 46 |
| CDC20 | Cell division cycle
20 | 42 |
| BIRC5 | Baculoviral
initiator of apoptosis repeat containing 5 | 37 |
| BUB1B | BUB1 mitotic
checkpoint serine/threonine kinase B | 36 |
| PCNA | Proliferating cell
nuclear antigen | 35 |
| CENPA | Centromere protein
A | 34 |
| MAD2L1 | MAD2 mitotic arrest
deficient-like 1 | 33 |
Pathway crosstalk
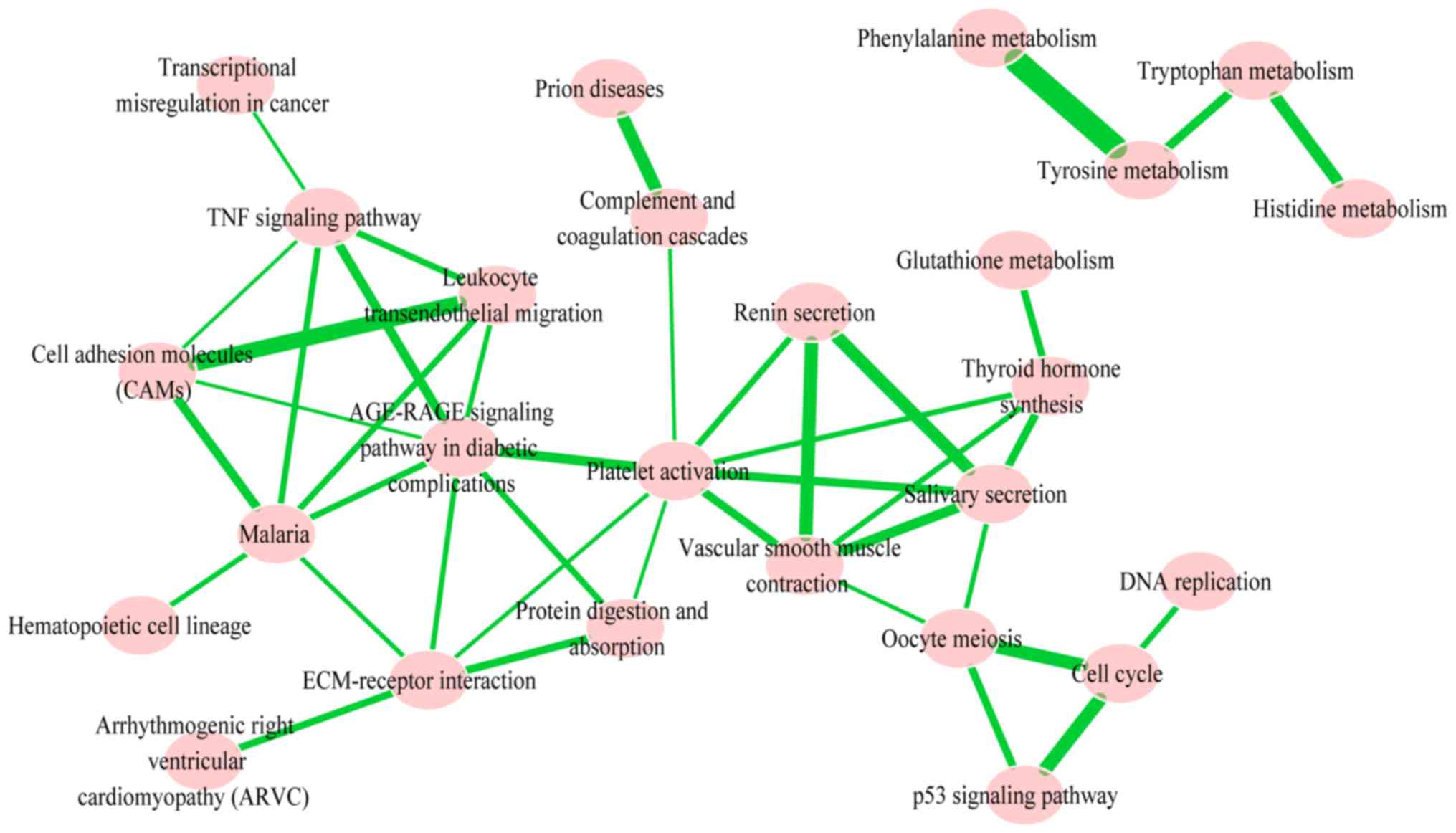
In order to identify the significantly-enriched
pathways and to understand the interaction between them, pathway
crosstalk analysis among the 26 significant pathways was performed;
in total, these pathways contained 41 edges. Based on the crosstalk
analysis, the pathways were divided into two major groups, and each
of the included pathways shared more crosstalk events than those
outside of the pathways identified in the crosstalk analysis and
may be associated with similar biological processes (Fig. 4). One group primarily included
metabolism of amino acids tyrosine, phenylalanine, tryptophan and
histidine. The other group primarily contained tumor necrosis
signaling pathways: CAM; tumor necrosis factor signaling pathway;
leukocyte transendothelial migration; malaria and stress reaction
(renin secretion, salivary secretion, platelet activation, vascular
smooth muscle contraction, extracellular matrix (ECM)-receptor
interaction and oocyte meiosis). These pathways had >4 degrees
and high rank-values with other pathways in this crosstalk.
Survival analysis
To validate the 9 hub genes identified, the KMplot
was used to analyze the survival potential of patients with
upregulated hub genes. Following the gene upload, 8 genes were
available in KMplot database, and there were 1,926 patients as
candidates. All the hub genes were upregulated genes, 7 of which
were a significantly associated with low survival rates (P<0.05;
Fig. 5). However, PCNA was
upregulated, but did not exhibit a significant association.
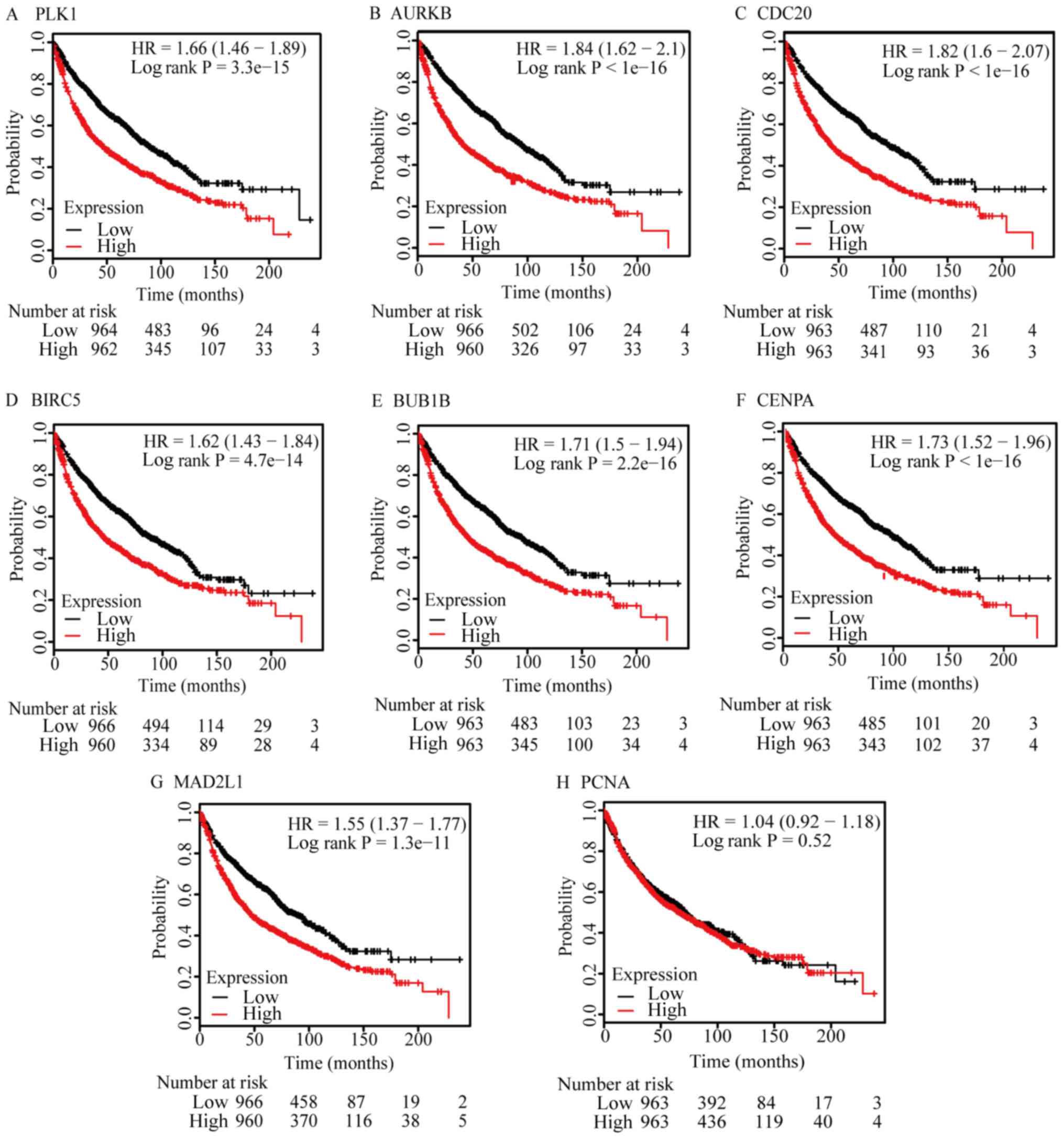 | Figure 5.Survival analysis of hub genes. (A)
PLK, (B) AURKB, (C) CDC20, (D) BIRC5, (E) BUB1B, (F) CENPA and (G)
MAD2L1 expression indicated significantly lower survival rates,
compared with low expression samples. (H) PCNA expression did not
exhibit a significantly different survival rates. PLK, polo-like
kinase 1; AURKB, aurora kinase B; CDC20, cell division cycle 20;
BIRC5, baculoviral initiator of apoptosis repeat containing 5;
BUB1B, BUB1 mitotic checkpoint serine/threonine kinase B; CENPA,
centromere protein A; MAD2L1, MAD2 mitotic arrest deficient-like 1;
PCNA, proliferating cell nuclear antigen. |
Discussion
NSCLC accounts for ~80% of all types of diagnosed
lung cancer (19). The cause of NSCLC
is complex with various factors, including smoking, air pollution
and radon exposure (20); therefore,
understanding the biological mechanisms of NSCLC is important for
clinical diagnosis and treatment. As microarrays have a wide range
of applications in oncology, including identification of
disease-associated biomarkers, alternative splicing and gene
function prediction, microarray data were extracted from GSE33532,
and 729 upregulated and 1,066 downregulated DEGs between NSCLC and
normal samples were identified using bioinformatics analysis. In
order to obtain additional analysis of these DEGs, GO and KEGG
analyses were performed using DAVID software.
The GO analysis results indicated that the
upregulated DEGs were primarily associated with ‘cell cycle phase’,
‘mitotic cell cycle’, ‘cell cycle process’, ‘M phase of mitotic
cell cycle’ and ‘nuclear division’, while the downregulated DEGs
were primarily associated with ‘response to wounding’, ‘anatomical
structure morphogenesis’, ‘response to stimulus’, ‘response to
chemical stimulus’ and ‘single-multi cellular organism process’.
These results are in agreement with those of previously published
studies suggesting that irregular and abnormal cell cycles or cell
proliferation are closely associated with tumor proliferation and
apoptosis (21–24), and that there is an association
between repetitive wounding/stimuli and lung cancer (25).
The KEGG pathway analysis result revealed that
upregulated DEGs were involved in ‘Cell cycle’, ‘DNA replication’,
‘p53 signaling pathway’, ‘ECM-receptor interaction’ and ‘protein
digestion and absorption’. Previous studies have indicated that the
p53-independent structure-activity associations of mesogenic
compounds are associated with cytotoxic effects (26,27), and
that disturbances in the p53 signaling pathway is associated with
NSCLC (28). According to previous
studies, ECM-receptor interactions were involved in cell adhesion
(29), and it has been revealed that
the ECM molecule hyaluronan induced focal adhesion, to signal the
cytoskeletal changes required for the elevated cell motility
observed in the processes of tumor cell progression, metastasis and
invasion (30).
Notably, the downregulated DEGs were enriched in
disease malaria during the KEGG enrichment analysis, which may
suggest that anti-malaria compounds, artemisinin,
dihydroartemisinin and artesunate, also have anticancer potential.
The antimalarial drug, artemisinin, was previously used in the
treatment of lung cancer, and Tong et al (31) identified that artemisinin inhibited
tumor metastasis through Wnt/β-catenin signaling. In addition, a
previous study by Ashton et al (32) suggested that a commonly used
anti-malarial drug, atovaquone, effectively increased the oxygen
content inside cancer cells, therefore improving the efficiency of
radiation treatment. Atovaquone rapidly decreased the oxygen
consumption rate by >80% in a range of cancer cell lines at
pharmacological concentrations. In additional experiments,
atovaquone killed 90% of cancer cells in an in vitro lung
cancer tumor model involving radiotherapy (32).
By constructing a PPI network with DEGs, the present
study identified the top degree hub genes: CDK1, PLK1, AURKB,
CDC20, BIRC5, BUB1B, PCNA, CENPA and MAD2L1. CDK1 exhibited the
highest degree of connectivity among these hub genes. CDK1, a
protein-coding gene, serves a key role in the control of the
eukaryotic cell cycle by modulating the centrosome cycle and
mitotic onset, promoting G2-M transition and regulating G1 progress
and G1-S transition via association with multiple interphase
cyclins (33). CDK1 is an adverse
prognostic biomarker of lung adenocarcinoma (LUAD), and increased
expression of CDK1 was revealed to be associated with a higher risk
of cancer recurrence and poor survival, compared with the normal
expression of CDK1 in patients with LUAD (34). Danilov et al (35) also demonstrated that using dinaciclib
to inhibit the expression of CDK1 induced anaphase catastrophe in
lung cance (35). The second hub
gene, PLK1, performs several important functions throughout the M
phase of the cell cycle, including the regulation of centrosome
maturation and spindle assembly, the removal of cohesins from
chromosome arms, the inactivation of anaphase-promoting
complex/cyclosome (APC/C) inhibitors and the regulation of mitotic
exit and cytokinesis (36). A
previous study demonstrated that PLK1 may promote tumor cell
survival by regulating Myc stabilization; inhibitors of PLK1
preferentially induced potent apoptosis level in MYCN-amplified
tumor cells from neuroblastoma and small cell lung cancer, and
synergistically potentiated the therapeutic efficacies of Bcl-2
antagonists (37). AURKB, the third
hub gene, encodes a member of the aurora kinase subfamily of
serine/threonine kinases, and participates in the regulation of
alignment and segregation of chromosomes during mitosis and meiosis
(38). A previous study has confirmed
the antitumor and radiosensitizing activities of daurinol in human
lung cancer cells through the inhibition of AURKB (39). In addition, CDC20 serves as a
regulatory protein interacting with several other proteins at
multiple points in the cell cycle (40). A previous study suggested that CDC20
is a critical regulator in glioma tumors, initiating cell
proliferation and survival (41).
High levels of CDC20 expression are a key component of the spindle
assembly checkpoint; CDC20 has been identified in various
malignancies and serves a vital role in tumorigenesis and
progression (42–44). BIRC5, the member of the IAP gene
family, encodes negative regulatory proteins that prevent apoptotic
cell death. Han et al (45)
suggested that the upregulation of the anti-apoptosis gene BIRC5
will lead to the inhibition of p53 signaling in H1650GR cells.
BUB1B serves a role in the inhibition of the APC/C, delaying the
onset of anaphase and ensuring proper chromosome segregation
(46). Chen et al (47) also revealed that BUB1B may serve as
target gene in lung carcinoma as a result of PPI network analysis.
In addition, high levels of BUB1B expression are associated with
disease progression and poor survival in patients with lung
adenocarcinoma (47). PCNA acts as a
homotrimer and assists in increasing the processivity of leading
strand synthesis during DNA replication (48). Bodduluru et al (49) revealed that benzo[a]pyrene may induce
pulmonary carcinogenesis by modulating PCNA expression. CENPA
encodes a centromere protein that contains a histone H3-associated
histone fold domain required for targeting to the centromere
(50). This domain is one of the
basic components of the human active kinetochore, which serves an
important role in cell-cycle regulation, cell survival and genetic
stability (51). Toh et al
(52) identified that CENPA may be
considered as a prospective diagnostic and prognostic biomarker of
lung adenocarcinoma. MAD2L1 is a component of the mitotic spindle
assembly checkpoint that prevents the onset of anaphase until all
chromosomes are properly aligned at the metaphase plate (53). As aforementioned, MAD2L1 is closely
associated with the carcinogenesis of lung adenocarcinoma (34). Guo et al (54) performed a case-control analysis,
indicating an association between the concentration of MAD2L1
Leu84Met SNP gene product and the risk of lung cancer in an allele
dose-dependent manner, with the result demonstrating that the
expression level of MAD2L1 Leu84Met SNP was linearly associated
with the risk of lung cancer.
Considering the enrichment results of the top 5
modules from the PPI network genes in the present study, it was
demonstrated that NSCLC was associated with ‘cell cycle’,
‘chemokine signaling pathway’, ‘protein digestion and absorption’,
‘DNA replication’ and ‘malaria’.
In the chemokine signaling pathway (hsa04062; KEGG
database), chemokines are a type of small chemoattractant peptide,
which may provide directional cues for cell trafficking; this is
the key for the protective host response (55). In addition, chemokines regulate a
plethora of biological processes in hematopoietic cells that lead
to cellular activation, differentiation and survival (55,56). A
previous study has revealed that chemokines are vital in the
pathogenesis of NSCLC and NSCLC cells are rich in the secreted
protein CXCL12 (57). Another study
has suggested that the methylation of CXCL12 has a marked
correlation with NSCLC prognosis (58), and that CXCL12-mediated adhesion and
survival signals are associated with chemo-resistance in lung
cancer (59).
During the survival analysis of the present study,
KMplot was used to assess the effect of high expression levels of
the hub genes in patients with lung cancer. There were 8 genes
available in the database, with only CDK1 not matching HGU133A and
HGU133Aplus2 probe set IDs in the KMplot database. Notably, 7 of
the 8 genes indicated significantly low survival rates, compared
with low expression samples (P<0.05); the remaining gene, PCNA,
did not. In addition, a number of previous studies have analyzed
the association between the expression of PCNA and NSCLC
postoperative survival time and did not identify a significant
correlation (60–63). This may be due to the fact that the
prognosis of lung cancer may be affected by a variety of factors,
including pathological type and stage, differentiation, treatment,
complications, age, physical condition and the expression of PCNA
(60,64–67). It is
difficult to predict the prognosis of lung cancer by considering
the effecters of PCNA alone.
In conclusion, the results from the present study
provided a wider analysis of the DEGs associated with NSCLC, and
identified certain key pathways in the progress of NSCLC, which may
provide guidance for future studies. Nevertheless, a number of
biomarkers associated with NSCLC remain uncharacterized; additional
biological and bioinformatics analyses are required.
Acknowledgements
The authors would like to thank Dr Yanchao Mu
(Clinical Laboratory of Anyang Children's Hospital, Henan, China)
for their technical guidance.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
QT and HZ wrote the main part of the manuscript and
took part in the planning and execution of the experiments. MK and
XM took part in the development of the analysis code, planned and
carried out the main part of the experiments. XC provided language
guidance designed this experiment and revised the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spira A and Ettinger DS: Multidisciplinary
management of lung cancer. N Engl J Med. 350:379–392. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ettinger DS, Akerley W, Borghaei H, Chang
AC, Cheney RT, Chirieac LR, D'Amico TA, Demmy TL, Ganti AK,
Govindan R, et al: Non-small cell lung cancer. J Natl Compr Canc
Netw. 10:1236–1271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bejjani BA and Shaffer LG: Clinical
utility of contemporary molecular cytogenetics. Annu Rev Genomics
Hum Genet. 9:71–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang C, Li HR, Fan JB, Wang-Rodriguez J,
Downs T, Fu XD and Zhang MQ: Profiling alternatively spliced mRNA
isoforms for prostate cancer classification. BMC Bioinformatics.
7:2022006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu M, Deng X, Joshi T, Xu D, Stacey G and
Cheng J: Reconstructing differentially co-expressed gene modules
and regulatory networks of soybean cells. BMC Genomics. 13:4372012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Denoix PF: Enquete permanent dans les
centres anticancereaux. Bull Inst Nat Hyg. 1:70–75. 1946.(In
French).
|
|
8
|
Meister M, Belousov A, Xu EC, et al:
Intra-tumor heterogeneity of gene expression profiles in early
stage non-small cell lung cancer. J Bioinf Res Stud. 1:12014.
|
|
9
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Phipson B, Lee S, Majewski IJ, Alexander
WS and Smyth GK: Robust hyperparameter estimation protects against
hypervariable genes and improves power to detect differential
expression. Ann Appl Stat. 10:946–963. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Martucci D, Masseroli M and Pinciroli F:
Gene ontology application to genomic functional annotation,
statistical analysis and knowledge mining. Stud Health Technol
Inform. 102:108–131. 2004.PubMed/NCBI
|
|
12
|
Kanehisa M: The KEGG database. Novartis
Found Symp. 247:91–103, 119-128, 244–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Langley SR, Dwyer J, Drozdov I, Yin X and
Mayr M: Proteomics: From single molecules to biological pathways.
Cardiovasc Res. 97:612–622. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Burrell RA, McGranahan N, Bartek J and
Swanton C: The causes and consequences of genetic heterogeneity in
cancer evolution. Nature. 501:338–345. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu M, Fan R, Liu X, Cheng F and Wang J:
Pathways and networks-based analysis of candidate genes associated
with nicotine addiction. PLoS One. 10:e01274382015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Szasz AM, Lanczky A, Nagy A, Förster S,
Hark K, Green JE, Boussioutas A, Busuttil R, Szabó A and Győrffy B:
Cross-validation of survival associated biomarkers in gastric
cancer using transcriptomic data of 1,065 patients. Oncotarget.
7:49322–49333. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ramalingam SS, Owonikoko TK and Khuri FR:
Lung cancer: New biological insights and recent therapeutic
advances. CA Cancer J Clin. 61:91–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gridelli C, Rossi A, Carbone DP, Guarize
J, Karachaliou N, Mok T, Petrella F, Spaggiari L and Rosell R:
Non-small-cell lung cancer. Nat Rev Dis Primers. 1:150092015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
An Q, Han C, Zhou Y, Li F, Li D, Zhang X,
Yu Z, Duan Z and Kan Q: Matrine induces cell cycle arrest and
apoptosis with recovery of the expression of miR-126 in the A549
non-small cell lung cancer cell line. Mol Med Rep. 14:4042–4048.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jung CK, Jung JH, Lee KY, Kang CS, Kim M,
Ko YH and Oh CS: Centrosome abnormalities in non-small cell lung
cancer: correlations with DNA aneuploidy and expression of cell
cycle regulatory proteins. Pathol Res Pract. 203:839–847. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rao PC, Begum S, Jahromi MA, Jahromi ZH,
Sriram S and Sahai M: Cytotoxicity of withasteroids: Withametelin
induces cell cycle arrest at G2/M phase and mitochondria-mediated
apoptosis in non-small cell lung cancer A549 cells. Tumour Biol.
37:12579–12587. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Xu J, Zhao C, Zhao L and Feng B:
Antiproliferative, cell-cycle dysregulation effects of novel
asiatic acid derivatives on human non-small cell lung cancer cells.
Chem Pharm Bull (Tokyo). 61:1015–1023. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Haura EB: Is repetitive wounding and bone
marrow-derived stem cell mediated-repair an etiology of lung cancer
development and dissemination? Med Hypotheses. 67:951–956. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hai J, Sakashita S, Allo G, Ludkovski O,
Ng C, Shepherd FA and Tsao MS: Inhibiting MDM2-p53 interaction
suppresses tumor growth in patient-derived non-small cell lung
cancer xenograft models. J Thorac Oncol. 10:1172–1180. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
In JK, Kim JK, Oh JS and Seo DW:
5-Caffeoylquinic acid inhibits invasion of non-small cell lung
cancer cells through the inactivation of p70S6K and Akt activity:
Involvement of p53 in differential regulation of signaling
pathways. Int J Oncol. 48:1907–1912. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fukushi S, Yoshino H, Yoshizawa A and
Kashiwakura I: p53-independent structure-activity relationships of
3-ring mesogenic compounds' activity as cytotoxic effects against
human non-small cell lung cancer lines. BMC Cancer. 16:5212016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Albelda SM and Buck CA: Integrins and
other cell adhesion molecules. FASEB J. 4:2868–2880. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hall CL and Turley EA: Hyaluronan: RHAMM
mediated cell locomotion and signaling in tumorigenesis. J
Neurooncol. 26:221–229. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tong Y, Liu Y, Zheng H, Zheng L, Liu W, Wu
J, Ou R, Zhang G, Li F, Hu M, et al: Artemisinin and its
derivatives can significantly inhibit lung tumorigenesis and tumor
metastasis through Wnt/β-catenin signaling. Oncotarget.
7:31413–31428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ashton TM, Fokas E, Kunz-Schughart LA,
Folkes LK, Anbalagan S, Huether M, Kelly CJ, Pirovano G, Buffa FM,
Hammond EM, et al: The anti-malarial atovaquone increases
radiosensitivity by alleviating tumour hypoxia. Nature Commun.
7:123082016. View Article : Google Scholar
|
|
33
|
Castedo M, Perfettini JL, Roumier T and
Kroemer G: Cyclin-dependent kinase-1: Linking apoptosis to cell
cycle and mitotic catastrophe. Cell Death Differ. 9:1287–1293.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Shi YX, Zhu T, Zou T, Zhuo W, Chen YX,
Huang MS, Zheng W, Wang CJ, Li X, Mao XY, et al: Prognostic and
predictive values of CDK1 and MAD2L1 in lung adenocarcinoma.
Oncotarget. 7:85235–85243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Danilov AV, Hu S, Orr B, Godek K,
Mustachio LM, Sekula D, Liu X, Kawakami M, Johnson FM, Compton DA,
et al: Dinaciclib induces anaphase catastrophe in lung cancer cells
via inhibition of cyclin-dependent kinases 1 and 2. Mol Cancer
Ther. 15:2758–2766. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lane HA and Nigg EA: Antibody
microinjection reveals an essential role for human polo-like kinase
1 (Plk1) in the functional maturation of mitotic centrosomes. J
Cell Biol. 135:1701–1713. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xiao D, Yue M, Su H, Ren P, Jiang J, Li F,
Hu Y, Du H, Liu H and Qing G: Polo-like kinase-1 regulates Myc
stabilization and activates a feedforward circuit promoting tumor
cell survival. Mol Cell. 64:493–506. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Goldenson B and Crispino JD: The aurora
kinases in cell cycle and leukemia. Oncogene. 34:537–545. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Woo JK, Kang JH, Shin D, Park SH, Kang K,
Nho CW, Seong JK, Lee SJ and Oh SH: Daurinol enhances the efficacy
of radiotherapy in lung cancer via suppression of aurora kinase a/b
expression. Mol Cancer Ther. 14:1693–1704. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weinstein J, Jacobsen FW, Hsu-Chen J, Wu T
and Baum LG: A novel mammalian protein, p55CDC, present in dividing
cells is associated with protein kinase activity and has homology
to the Saccharomyces cerevisiae cell division cycle proteins Cdc20
and Cdc4. Mol Cell Biol. 14:3350–3363. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xie Q, Wu Q, Mack SC, Yang K, Kim L,
Hubert CG, Flavahan WA, Chu C, Bao S and Rich JN: CDC20 maintains
tumor initiating cells. Oncotarget. 6:13241–13254. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Choi JW, Kim Y, Lee JH and Kim YS: High
expression of spindle assembly checkpoint proteins CDC20 and MAD2
is associated with poor prognosis in urothelial bladder cancer.
Virchows Arch. 463:681–687. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang DZ, Ma Y, Ji B, Liu Y, Hwu P,
Abbruzzese JL, Logsdon C and Wang H: Increased CDC20 expression is
associated with pancreatic ductal adenocarcinoma differentiation
and progression. J Hematol Oncol. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kato T, Daigo Y, Aragaki M, Ishikawa K,
Sato M and Kaji M: Overexpression of CDC20 predicts poor prognosis
in primary non-small cell lung cancer patients. J Surg Oncol.
106:423–430. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Han X, Liu M, Wang S, Lv G, Ma L, Zeng C
and Shi Y: An integrative analysis of the putative
gefitinib-resistance related genes in a lung cancer cell line model
system. Curr Cancer Drug Targets. 15:423–434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Davenport JW, Fernandes ER, Harris LD,
Neale GA and Goorha R: The mouse mitotic checkpoint gene bub1b, a
novel bub1 family member, is expressed in a cell cycle-dependent
manner. Genomics. 55:113–117. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen H, Lee J, Kljavin NM, Haley B, Daemen
A, Johnson L and Liang Y: Requirement for BUB1B/BUBR1 in tumor
progression of lung adenocarcinoma. Genes Cancer. 6:106–118.
2015.PubMed/NCBI
|
|
48
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bodduluru LN, Kasala ER, Madhana RM, Barua
CC, Hussain MI, Haloi P and Borah P: Naringenin ameliorates
inflammation and cell proliferation in benzo(a)pyrene induced
pulmonary carcinogenesis by modulating CYP1A1, NFkappaB and PCNA
expression. Int Immunopharmacol. 30:102–110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chueh AC, Wong LH, Wong N and Choo KH:
Variable and hierarchical size distribution of
L1-retroelement-enriched CENP-A clusters within a functional human
neocentromere. Hum Mol Genet. 14:85–93. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Folco HD, Pidoux AL, Urano T and Allshire
RC: Heterochromatin and RNAi are required to establish CENP-A
chromatin at centromeres. Science. 319:94–97. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Toh SH, Prathipati P, Motakis E, Kwoh CK,
Yenamandra SP and Kuznetsov VA: A robust tool for discriminative
analysis and feature selection in paired samples impacts the
identification of the genes essential for reprogramming lung tissue
to adenocarcinoma. BMC Genomics. 12(Suppl 3): S242011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu L, Deng HX, Yang Y, Xia JH, Hung WY and
Siddque T: Assignment of mitotic arrest deficient protein 2
(MAD2L1) to human chromosome band 5q23.3 by in situ hybridization.
Cytogenet Cell Genet. 78:63–64. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo Y, Zhang X, Yang M, Miao X, Shi Y, Yao
J, Tan W, Sun T, Zhao D, Yu D, et al: Functional evaluation of
missense variations in the human MAD1L1 and MAD2L1 genes and their
impact on susceptibility to lung cancer. J Med Genet. 47:616–622.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zlotnik A, Burkhardt AM and Homey B:
Homeostatic chemokine receptors and organ-specific metastasis. Nat
Rev Immunol. 11:597–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zlotnik A and Yoshie O: The chemokine
superfamily revisited. Immunity. 36:705–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wald O, Shapira OM and Izhar U:
CXCR4/CXCL12 axis in non small cell lung cancer (NSCLC) pathologic
roles and therapeutic potential. Theranostics. 3:26–33. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Suzuki M, Mohamed S, Nakajima T, Kubo R,
Tian L, Fujiwara T, Suzuki H, Nagato K, Chiyo M, Motohashi S, et
al: Aberrant methylation of CXCL12 in non-small cell lung cancer is
associated with an unfavorable prognosis. Int J Oncol. 33:113–119.
2008.PubMed/NCBI
|
|
59
|
Hartmann TN, Burger JA, Glodek A, Fujii N
and Burger M: CXCR4 chemokine receptor and integrin signaling
co-operate in mediating adhesion and chemoresistance in small cell
lung cancer (SCLC) cells. Oncogene. 24:4462–4471. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ebina M, Steinberg SM, Mulshine JL and
Linnoila RI: Relationship of p53 overexpression and up-regulation
of proliferating cell nuclear antigen with the clinical course of
non-small cell lung cancer. Cancer Res. 54:2496–2503.
1994.PubMed/NCBI
|
|
61
|
Matturri L, Lavezzi AM, Grignani F,
Salomoni G and Roviaro GC: The prognostic value of cell
proliferation in non-small cell lung cancer assessed with tritiated
thymidine and anti-PCNA antibodies. Eur J Cancer. 30A:1397–1398.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Alemany Monraval P, Martorell Cebollada M,
Salvador Villalba I and Martínez Leandro E: Study of the expression
of proliferating cell nuclear antigen and p185 in non-small cell
lung carcinoma. Arch Bronconeumol. 32:(In Spanish):. 165–169. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Castellano VM, Sotelo T, Ballestin C,
Lopez-Encuentra A and Varela G: Analysis of proliferating cell
nuclear antigen (PCNA) expression in 24 cases of primary non-small
cell pulmonary carcinomas and correlation with survival. Arch
Bronconeumol. 32:(In Spanish):. 127–131. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guo X, Li D, Wu Y, Chen Y, Zhou X, Wang X,
Huang X, Li X, Yang H and Xing J: Genetic variants in genes of
tricarboxylic acid cycle key enzymes are associated with prognosis
of patients with non-small cell lung cancer. Lung Cancer.
87:162–168. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Di JZ, Peng JY and Wang ZG: Prevalence,
clinicopathological characteristics, treatment, and prognosis of
intestinal metastasis of primary lung cancer: A comprehensive
review. Surg Oncol. 23:72–80. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
North CM and Christiani DC: Women and lung
cancer: What is new? Semin Thorac Cardiovasc Surg. 25:87–94. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Lin J and Beer DG: Molecular predictors of
prognosis in lung cancer. Ann Surg Oncol. 19:669–676. 2012.
View Article : Google Scholar : PubMed/NCBI
|