Introduction
Acute myeloid leukemia (AML) is a type of bone
marrow leukocyte (white leukocyte) caused by abnormal proliferation
of leukemia (1). AML is characterized
by the rapid proliferation of abnormal cells in the bone marrow
that affects the generation of normal blood cells (2). Clinical analyses indicate that AML may
be induced by a variety of factors, including virus, ionizing
radiation, chemical substances and genetic factors (3,4). Fever,
infirmity and other complications are the characteristics of
patients with AML (4). A previous
study analyzed therapy and efficacy post-remission of AML (5). Mechanism analyses have suggested that
cellular molecular signaling pathways are involved in the
progression of AML in patients (6,7).
Therefore, understanding the potential mechanisms of AML is
essential for the development of therapy for AML.
The ecotropic viral integration site 1 (Evi1) is a
zinc finger transcription factor, which is highly expressed in
numerous human tumors (8,9). A report indicated that the
overexpression of the EVI1 oncogene is associated typically with
aggressive myeloid leukemia (10).
Jazaeri et al (11) reported
that Evi1 and EVI1s (Delta324) may be regarded as potential
therapeutic targets in ovarian cancer. In addition, the
transcription factor Evi1 may regulate cellular proliferation,
differentiation and apoptosis, and its overexpression may
contribute to an aggressive course of disease via transcriptional
repression of membrane-spanning-4-domains subfamily-A member-3
(MS4A3) in AML and other malignancies (12). Furthermore, the results indicated that
Evi1 is a transcriptional suppressor of the microRNA (miRNA)-143
gene, and miRNA-143 mediates its action via the K-Ras axis in human
colon cancer (13). Additionally,
Evi1 targets DeltaNp63 and upregulates the cyclin-dependent kinase
inhibitor p21, independent of p53, which may delay cell cycle
progression and cell proliferation in colon cancer cells (14). However, the potential underlying
mechanism mediated by Evi1 in AML requires further
investigation.
In the present study, Evi1 as a target of miR-431
was analyzed and a low expression of miR-431 was detected in AML
cells. It was previously reported by the present authors that Evi1
is a potential molecular target for leukemia therapy via the
TGFβ-induced EMT signaling pathway. Therefore, the present study
focused on the molecular mechanism of AML regulation by Evi1 via
miR-431, which further mediate the TGFβ-induced EMT signaling
pathway in leukemia. It was indicated that miR-431 may suppress the
invasion and proliferation of AML cells via the Evi1-mediated
MS4A3/TGFβ/EMT signaling pathway.
Materials and methods
Cell culture
HuT-78, HTL-90 (http://www.cnki.com.cn/Article/CJFDTotal-HNLG6S1.025.htm)
and normal T lymphocyte cells (15)
were obtained from Department of Anatomy, Southern Medical
University (Guangzhou, China) and cultured in Dulbecco's modified
Eagle's medium (DMEM; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) with 5 µg/m blasticidin and supplemented with 10% fetal bovine
serum (FBS, Thermo Fisher Scientific, Inc., Waltham, MA, USA). All
cells were cultured in a 37°C humidified atmosphere of 5%
CO2.
Cell proliferation assay
All miR-431 mimics were synthesized by Invitrogen
(Thermo Fisher Scientific, Inc.), including pmiR-431 pvector
(control). The proliferation of pvector or pmiR-431-transfected
HuT-78 cells was detected using Cell Counting Kit-8 (CCK-8) kit
according to the manufacturer's protocols. Briefly, HuT-78 cells
were cultured in 48-well plates at the density of 1×104
cells/well and then cultured for 24 h. Finally, 10 µl CCK-8
solution was added to each well and incubated for 2 h at 37°C. The
results were measured using a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) at 570 nm.
Methylation assay
gDNA was extracted using phenol-chloroform. A total
of 2 µg DNA in HuT-78 cells was treated with bisulfite using the
EpiTect Bisulfite kit (Qiagen, Inc., Valencia, CA, USA), and
polymerase chain reaction (PCR) was performed using PCR cloning kit
(Zero Blunt™ PCR Cloning Kit, Invitrogen™; Thermo Sisher
Scientific, Inc.) according to the manufacturer's protocol. Primer
sequences were as follows: Forwards,
5′-AGGTTTTAGAGTAGGATTGGAAATGT-3′ and reverse,
5′-ACCCCCTCTCCCAAAACTA-3′. The PCR conditions were set as
following: an initial denaturation at 95°C for 60 sec, followed by
45 cycles of 95°C for 15 sec, 58.5°C for 60 sec, 72°C for 1 min.
Pyrosequencing was conducted according to the manufacturer's
protocols of the PyroMark PCR kit (Qiagen, Inc.).
Reverse transcription-quantitative
(RT-q)PCR
HuT-78, HTL-90 and normal T lymphocyte cells were
cultured, and total RNA was isolated from HuT-78, HTL-90 and normal
T lymphocytes using TRIzol reagent (Life Technologies; Thermo
Fisher Scientific, Inc.) and transcribed into cDNA using Super
Script VILO cDNA Synthesis kit (Life Technologies; Thermo Fisher
Scientific, Inc.). The forward and reverse primers were synthesized
by Invitrogen (Thermo Fisher Scientific, Inc.). The sequences of
the primers were as follows: Evi1 forward,
5′-CACGGATCCGAGGCGCCATGTCAGAAC-3′ and reverse,
5′-CTGACTCGAGGGATTAGGGCTTCCTCTTGG-3′; β-actin forward,
5′-CGGAGTCAACGGATTTGGTC-3′ and reverse, 5′-AGCCTTCTCCATGGTCGTGA-3′.
The changes in relative mRNA expression were calculated using the
2−ΔΔCq method (16). The
qPCR thermocycling conditions were as follows: 95°C for 5 min, then
35 cycles of 95°C for 20 sec, 56°C for 20 sec and 72°C for 20 sec,
and a final extension at 72°C for 5 min. The results were expressed
as the n-fold way compared with the housekeeping gene
(β-actin).
Boyden chamber migration and invasion
assays
Serum-free HuT-78 cells were seeded
(1×105 cells/well) in 500 µl DMEM medium. For the
invasion assay, HuT-78 cells were suspended at a density of
1×105 in 500 µl serum-free DMEM. The cells were seeded
in the upper Matrigel invasion chamber (BD Biosciences, Franklin
Lakes, NY, USA) according to the manufacturer's protocols. For the
migration assay, the cells were subjected to a control insert (BD
Biosciences) instead of a Matrigel invasion chamber. The number of
tumor cells that migrated and invaded was counted in ≥3 randomly
stained fields for every membrane under a light microscope at ×40
magnification.
Transfection of miR-431
HuT-78 cells (1×106) were transfected
with 100 pmol pmiR-431 (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with pvector as control (Applied Biosystems;
Thermo Fisher Scientific, Inc.) by using a Cell Line Nucleofector
kit L (Lonza Group, Ltd., Basel, Switzerland). After 72 h
transfection, cells were used for further experiments. HuT-78 cells
transfected with miR-431 and then were treated by TGFβ (2 mg/ml)
for 12 h at 37°C for further analysis.
Construction of lentivirus for MS4A3
overexpression
S4A3 was cloned into a lentivirus plasmid using the
Lentivirus vector system (System Biosciences, LLC, Pallo Alto, CA,
USA) with vector as control. Plasmids were named pMS4A3 and pvector
(control), respectively. All DNA sequences were synthesized by
Invitrogen (Thermo Fisher Scientific, Inc.). pvector or pMS4A3
plasmid was transfected into HuT-78 cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). The cells that were transfected with pvector or
pMS4A3 were used for further analysis.
Western blotting
HuT-78 cells were collected and lysed in
radioimmunoprecipitation assay buffer (M-PER reagent for the cells
and T-PER reagent for the tissues, Thermo Fisher Scientific, Inc.)
followed by homogenization at 4°C for 10 min. Protein concentration
was measured by a BCA protein assay kit (Thermo Scientific, Inc.).
A total of (20 µg) protein was electrophoresed on 12.5%
polyacrylamide gradient gels and then transferred to nitrocellulose
membranes. Rabbit anti-human antibodies used in the immunoblotting
assays were: Evi1 (1:500; catalog no. ab28457; Abcam, Cambridge,
UK), MS4A3 (1:500; catalog no. ab173761; Abcam), TGFβ (1:200;
catalog no. ab92486; Abcam), fibronectin (FIB; 1:500; catalog no.
ab6328; Abcam), α-smooth muscle actin (α-SMA; 1:500; catalog no.
ab7817; Abcam), vimentin (VIM; 1:500; catalog no. ab92547; Abcam)
and β-actin (1:500; catalog no. ab8226; Abcam). Horseradish
peroxidase-conjugated anti-rabbit immunoglobulin G antibodies (cat.
no. 166-2408-MSDS, Bio-Rad Laboratories, Inc.) were used at a
1:5,000 dilution and detected using a Western Blotting Luminol
reagent (Roche Diagnostics, Basel, Switzerland). Densitometric
quantification of the protein expression was performed using
Quantity-One software (version 1.02; Bio-Rad Laboratories,
Inc.).
Luciferase reporter assay
Evi1 3′untranslated region (UTR) construct in a PIS2
vector (Invitrogen; Thermo Fisher Scientific, Inc.) was used for
luciferase reporter assay as described previously (17). Site-directed mutagenesis of the
miR-431 binding site in Evi1 3′UTR was analyzed using PCR
amplification with the construction of a mutated 3′-UTR as control.
HuT-78 and HTL-90 cells were transfected with Evi1 3′UTR
(cytomegalovirus-driven β-gal reporter system, BD Biosciences) and
miR-431 mimics or scrambled miR control using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 24 h at 37°C with untransfected cells as
controls. The cell pellets were analyzed using the luciferase
reporter assay according to the manufacturer's protocols (Promega
Corporation, Madison, WI, USA). The β-gal reporter system was used
as an internal control. The miR-431 mimics used in the present
study were annealed by the miR-431 mimic
5′-UAAUUAGUGUCAUGUAGUUAGG-3′ and 5′-AGACUACAUGAAGCUACCUAAU-3′, and
the control group was annealed by 5′-GUCACGGAUCGCGGCACAUTT-3′ and
5′-AAUUGCCACGCGUUGAAGATT-3′ (MD Bio, Inc. Gaithersburg, MD, USA).
The luciferase activities of the miR-431-transfected HuT-78 and
HTL-90 cell lines were quantified and subjected to statistical
analyses. Results were normalized against the cells transfected
with scrambled miR control.
Statistical analysis
All data were expressed as the mean ± standard
deviation of triplicate dependent experiments and analyzed by using
paired Student t-tests or one-way analysis of variance followed by
Tukey's post hoc test. All data were analyzed using SPSS Statistics
(version 19.0; SPSS, Inc., Chicago, IL, USA) and GraphPad Prism
(version 5.0; GraphPad Software, Inc., La Jolla, CA, USA).
Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) was also
used. *P<0.05 was considered to indicate a statistically
significant difference.
Results
Analysis of Evi1 and miR-431
expression in AML cells
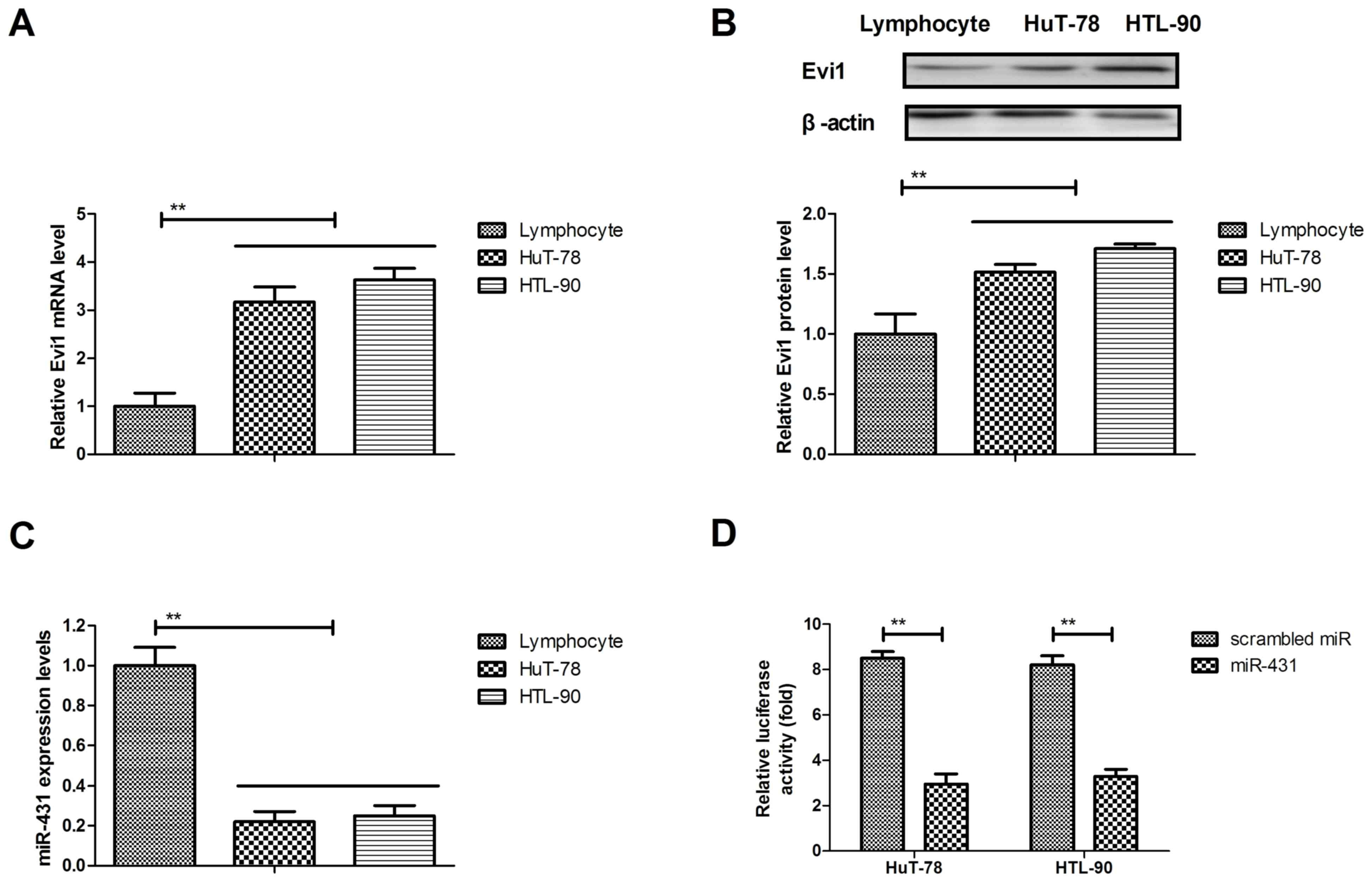
The expression levels of Evi1 and miR-431 were
analyzed in AML cells. The mRNA and protein expression levels of
Evi1 were upregulated in HuT-78 and HTL-90 cells compared with
(Fig. 1A and B). The results
demonstrated that miR-431 expression levels were significantly
downregulated in HuT-78 and HTL-90 compared with normal T
lymphocytes (Fig. 1C). The
dual-luciferase reporter assay indicated that Evi1 may be the
target gene of miR-431 in HuT-78 and HTL-90 cells (Fig. 1D). These results suggested that Evi1
may be upregulated and miR-431 expression may be downregulated in
HuT-78 and HTL-90 cells.
Transfection of miR-431 inhibits the
proliferation and invasion of AML cells
HuT-78 cell line is a typical and is the most
representative AML cells (18).
Therefore the role of miR-431 in AML cells was analyzed in the
present study. In order to investigate the role of miR-431 in
HuT-78 cells, the effects of miR-431 on proliferation and invasion
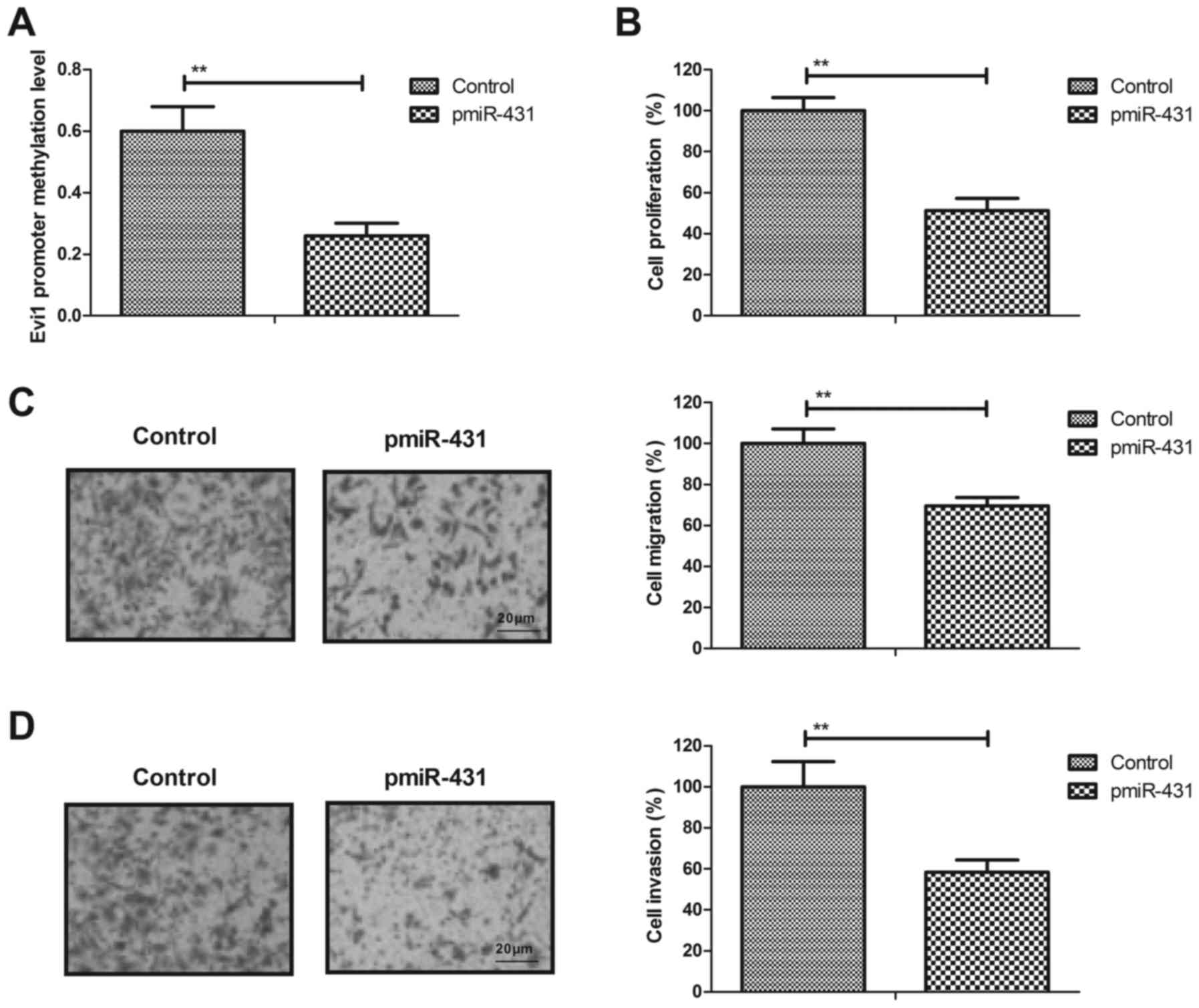
were analyzed in HuT-78 cells. The transfection of miR-431
decreased the promoter methylation levels of the Evi1 gene in
HuT-78 cells compared with the control (Fig. 2A). The results revealed that the
transfection of miR-431 increased miR-431 expression (data not
shown) and inhibited the proliferation of HuT-78 cells compared
with the control (Fig. 2B).
Furthermore, it was observed that the transfection of miR-431
suppressed the migration and invasion of HuT-78 cells (Fig. 2C and D). These results suggested that
the transfection of miR-431 may inhibit the proliferation and
invasion of AML cells.
Transfection of miR-431 inhibits MS4A3
and TGFβ/EMT signaling pathway in AML cells
To identify the potential mechanism mediated by
miR-431, the effects of miR-431 on the TGFβ/EMT signaling pathway
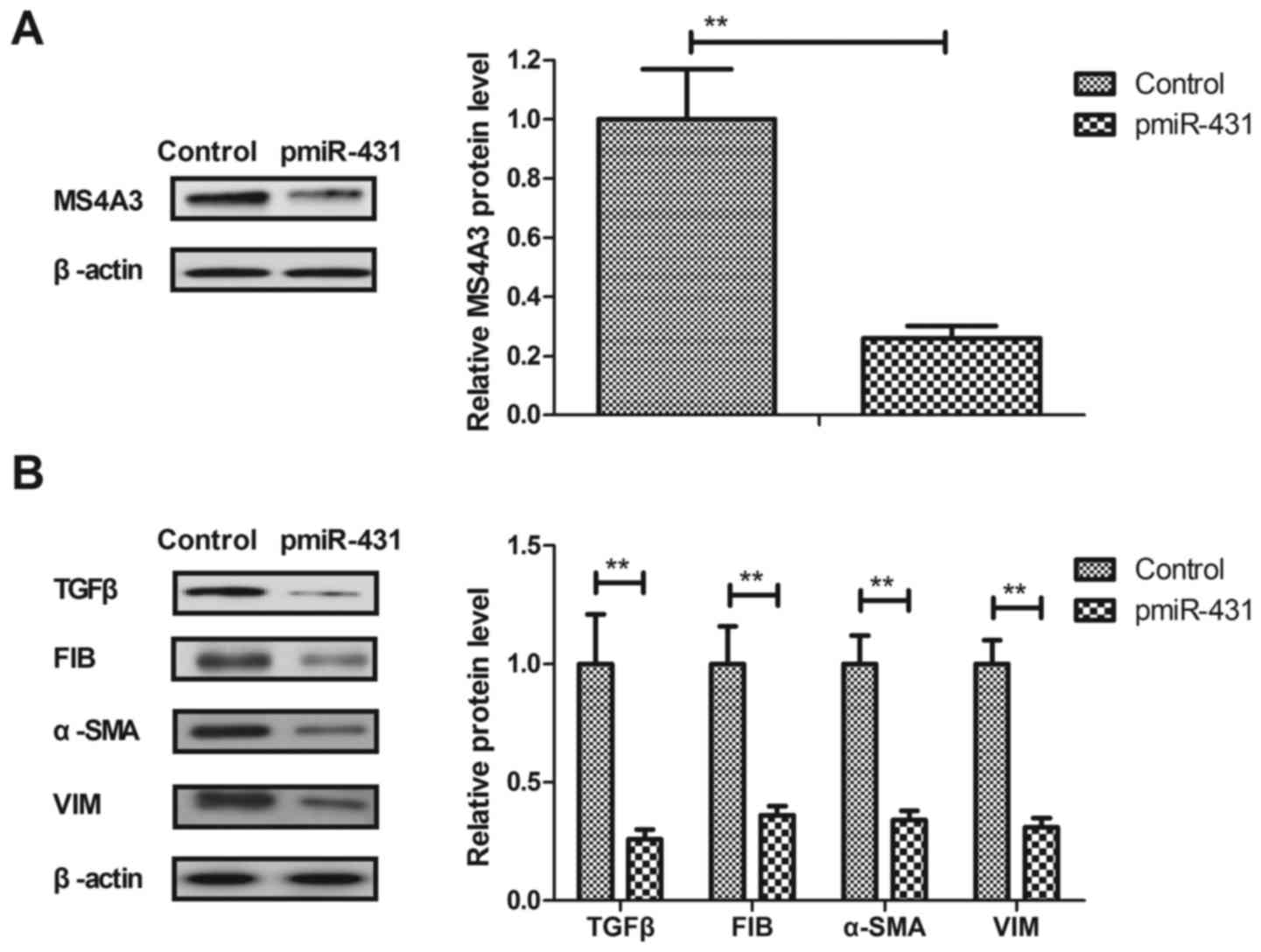
in HuT-78 cells were investigated in the present study. As
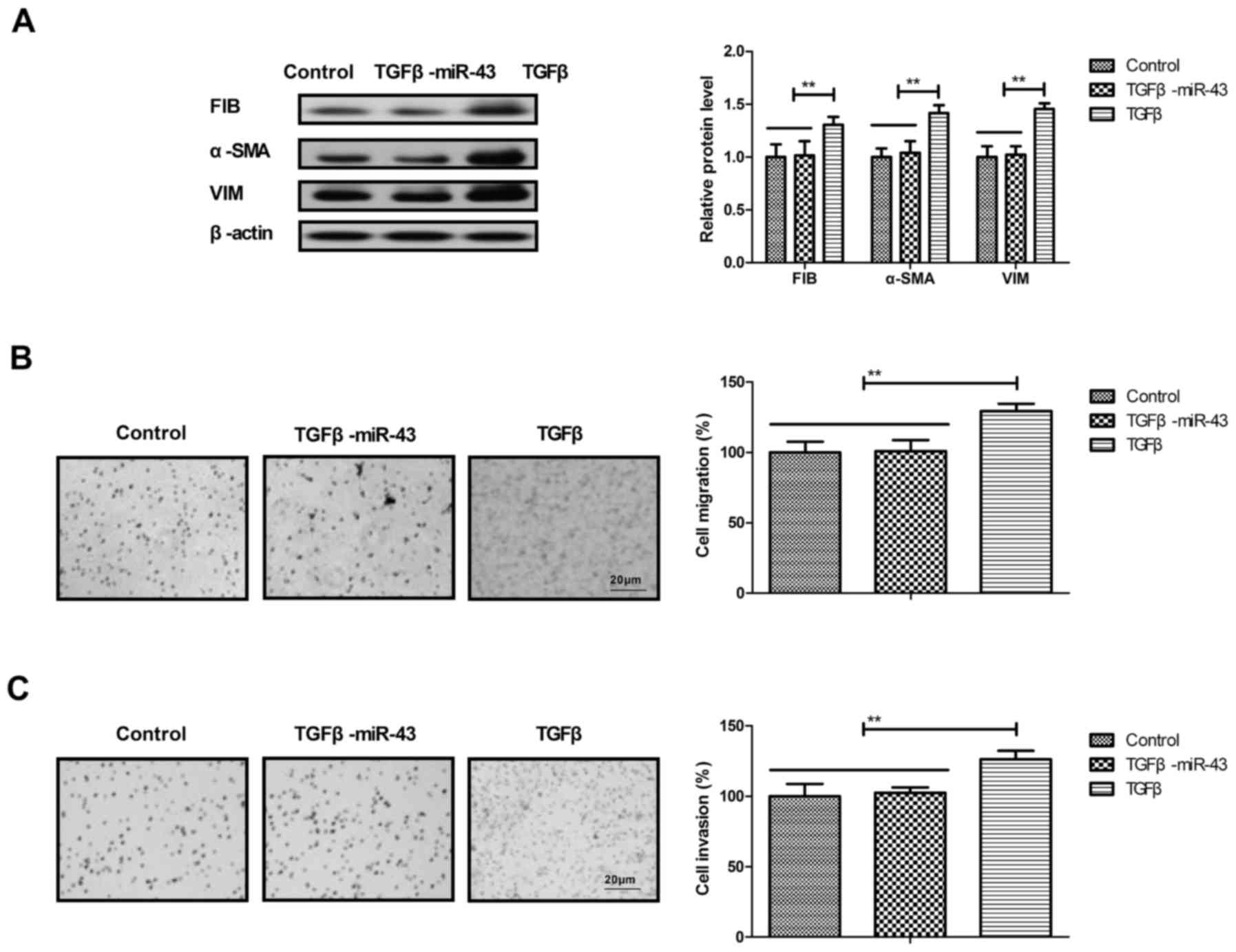
presented in Fig. 3A, the
transfection of miR-431 suppressed the expression levels of MS4A3
in HuT-78 cells. It was demonstrated that the transfection of
miR-431 decreased the expression levels of TGFβ and EMT markers
(FIB, α-SMA and VIM) in HuT-78 cells (Fig. 3B). These results indicated that the
transfection of miR-431 may inhibit MS4A3 and TGFβ/EMT signaling
pathways in AML cells.
Overexpression of MS4A3 inhibits
miR-431 mediated-inhibition on the expression levels of TGFβ and
EMT markers
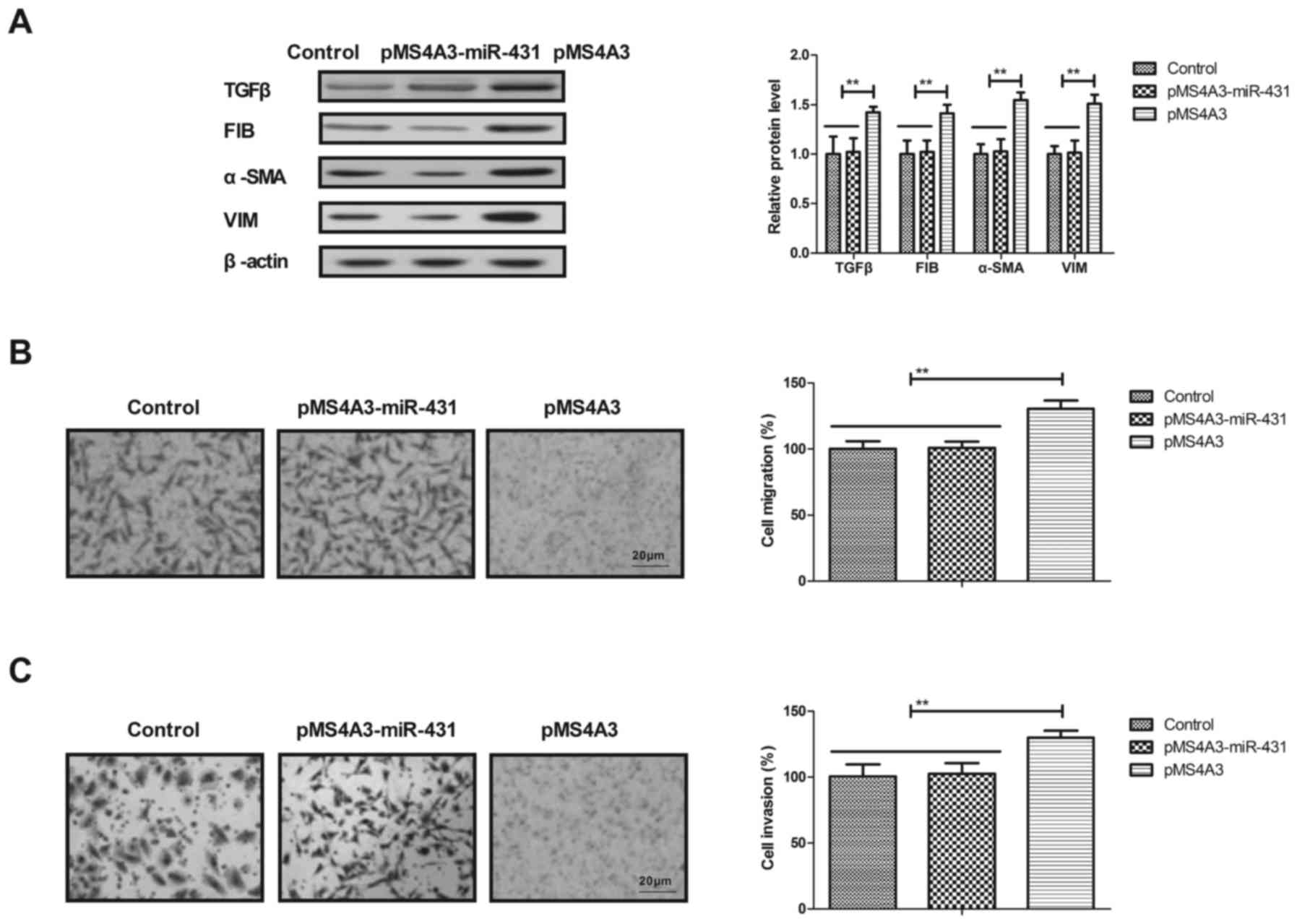
To investigate the potential molecular mechanism of
miR-431 in the TGFβ/EMT signaling pathway, the effects of MS4A3 on
the expression levels of TGFβ and EMT markers in HuT-78 cells were
analyzed in the present study. It was demonstrated that the
overexpression of MS4A3 inhibited the expression levels of TGFβ and
EMT markers in AML cells (Fig. 4A).
The transfection of miR-431 mimic suppressed the migration and
invasion of AML cells, and this effect was also reversed by MS4A3
overexpression in HuT-78 cells (Fig. 4B
and C). These results suggested that the overexpression of
MS4A3 inhibited miR-431 mediated-inhibition on the expression
levels of TGFβ and EMT markers.
Evi1 regulates the progression of AML
via MS4A3-mediated TGFβ/EMT signaling pathway
The TGFβ/EMT signaling pathway serves an important
role in the progression of AML (19,20).
Therefore, the role of Evi1 on MS4A3-mediated TGFβ/EMT signaling
pathway in HuT-78 cells was analyzed in the present study. It was
demonstrated that the addition of TGFβ eliminated the
downregulation of EMT markers that is mediated by miR-431 mimics in
AML cells (Fig. 5A). The addition of
TGFβ promoted the migration and invasion of AML cells compared with
the control and reversed the inhibition on migration and invasion
of AML cells that is mediated by miR-431 mimic (Fig. 5B and C). These results suggested that
Evi1 may regulate the progression of AML via MS4A3-mediated
TGFβ/EMT signaling.
Discussion
The overexpression of the Evi1 oncogene gene is
associated with typically aggressive myeloid leukemia (21,22). In
the present study, a putative target of Evi1 in the progression of
AML was analyzed. A previous study reported that Evi1
overexpression was predominantly detected within subtypes of
pediatric AML in distinct subtypes of pediatric AML (23). The results of the present study
indicated that Evi1 was upregulated and miR-431 expression was
downregulated in HuT-78 and HTL-90 cells compared with normal T
lymphocytes. The findings also indicated that the transfection of
miR-431 inhibited the proliferation and invasion of AML cells,
which further suppressed the MS4A3/TGFβ/EMT signaling pathway in
AML cells. Notably, the results of the present study indicated that
Evi1 may regulate the proliferation and invasion of AML via
MS4A3-mediated TGFβ/EMT signaling.
The Evi1 gene may serve as an alternative minimal
residual disease marker in AML on the basis of increasing
expression levels of MDS1-EVI/EVI 29, 36 and 93 days prior to
hematologic manifestation, particularly in samples where other
specific markers are lacking (24).
Previous studies have reported that high expression of Evi1 may
predict the outcome of younger adult patients (<22 years old)
with AML, which was associated with distinct cytogenetic
abnormalities (23,25). In the present study, it was observed
that the inhibition of Evi1 expression mediated by miR-431
transfection significantly inhibited the proliferation and invasion
of AML cells. A previous study reported that EVI1 may bind to and
downregulate Serpin family B member 2, which is involved in the
Janus kinase-signal transducers and activators of transcription
signaling pathway, suggesting that Evi1 is a target gene in the
treatment of AML (26). The results
of the present study reported that miR-431 may regulate
MS4A3-mediated TGFβ and EMT signaling pathway in AML cells.
MS4A3 is a member of a family of four-transmembrane
proteins, which acts as a cell surface signaling molecule and an
intracellular adapter protein (27).
Heller et al (12) indicated
that Evi1 may promote tumor growth via transcriptional repression
of MS4A3. The results of the present study confirmed previous
results and reported that Evi1 regulated MS4A3 via regulation of
miR-431 levels. Pan et al (28) analyzed the association between the
downregulation of miR-431 expression and clinicopathological
characteristics in hepatocellular carcinoma tissues. In the present
study, the results revealed that miR-431 transfection inhibited the
invasion of AML cells, which was abolished by the overexpression of
MS4A3.
The TGFβ/EMT signaling pathway serves a crucial role
in mediating tumor suppressive effects in human cutaneous melanoma
(20). TGFβ-mediated formation of
retinoblastoma protein-E2F complexes has been demonstrated to be
involved in the proliferation and invasion of human myeloid
leukemia cells (29). The results of
the present study indicated that the overexpression of MS4A3
inhibited the expression TGFβ and EMT markers, which is mediated by
miR-431 mimic in AML cells. Additionally, the transfection of
miR-431 mimic suppressed the migration and invasion of AML cells,
and this effect was also reversed by MS4A3 overexpression in HuT-78
cells. Notably, the addition of TGFβ abolished the downregulation
of EMT markers, which is mediated by transfection of miR-431 mimic.
This finding suggests that Evi1 may regulate the progression of AML
via the miR-431/EVI1/MS4A3-mediated TGFβ/EMT signaling pathway.
In conclusion, the silencing of Evi1 by miR-431
transfection exerted inhibitory effects on MS4A3-mediated TGFβ/EMT
signaling pathway in AML cells. The findings of the present study
demonstrated that the oncogenic agent miR-431 may contribute to the
inhibition of Evi1-positive AML cells. In addition, the present
study revealed that the targeting of Evi1 by miR-431 transfection
reversed the downregulation of EMT markers (FIB, α-SMA and VIM) in
HuT-78 cells, which attenuated the proliferation and invasion of
AML cells. These findings suggested that Evi1 may be a novel
therapeutic agent for the treatment of AML.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Xiamen
Science and Technology Plan Project (grant no. 3502Z20164066) and
The Fujian Province medical innovation project (grant no.
2017-CXB-19). National Natural Science Foundation of China (grant
no. 61427807) and National Key R&D Program of China (grant no.
2017YFC1103403).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MJ performed the experiments and wrote the paper. XZ
contributes to the data analysis and WH is the designer for this
study.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
No applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Varkaris A, Gunturu K, Kewalramani T and
Tretter C: Acute myeloid leukemia after radium-223 therapy: Case
report. Clin Genitourin Cancer. 15:e723–726. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang L, Wang SA, DiNardo C, Li S, Hu S,
Xu J, Zhou W, Goswami M, Medeiros LJ and Tang G:
Tetraploidy/near-tetraploidy acute myeloid leukemia. Leuk Res.
53:20–27. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yamaguchi H: Gene mutations in acute
myeloid leukemia. Rinsho Ketsueki. 57:2535–2542. 2016.(In
Japanese). PubMed/NCBI
|
|
4
|
Uchida T, Hagihara M, Hua J and Inoue M:
The effects of azacitidine on the response and prognosis of
myelodysplastic syndrome and acute myeloid leukemia involving a
bone marrow erythroblast frequency of >50. Leuk Res. 53:35–38.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao SY, Tuerxun N, Chen R and Hao JP:
Analysis of therapy and efficacy after remission of acute myeloid
leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 24:1721–1724.
2016.(In Chinese). PubMed/NCBI
|
|
6
|
Liu C, Zhao L, Zhao J, Xu Q, Song Y and
Wang H: Reduced ADAMTS-13 level negatively correlates with
inflammation factors in plasma of acute myeloid leukemia patients.
Leuk Res. 53:57–64. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Montewis A and Eveillard M: Acute myeloid
leukemia with erythrophagocytosis indicative of KAT6A
rearrangement. Blood. 128:3142016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pradhan AK, Halder A and Chakraborty S:
Physical and functional interaction of the proto-oncogene EVI1 and
tumor suppressor gene HIC1 deregulates Bcl-xL mediated block in
apoptosis. Int J Biochem Cell Biol. 53:320–328. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Louz D, van den Broek M, Verbakel S,
Vankan Y, van Lom K, Joosten M, Meijer D, Löwenberg B and Delwel R:
Erythroid defects and increased retrovirally-induced tumor
formation in Evi1 transgenic mice. Leukemia. 14:1876–1884. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nanjundan M, Nakayama Y, Cheng KW, Lahad
J, Liu J, Lu K, Kuo WL, Smith-McCune K, Fishman D, Gray JW and
Mills GB: Amplification of MDS1/EVI1 and EVI1, located in the
3q26.2 amplicon, is associated with favorable patient prognosis in
ovarian cancer. Cancer Res. 67:3074–3084. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jazaeri AA, Ferriss JS, Bryant JL, Dalton
MS and Dutta A: Evaluation of EVI1 and EVI1s (Delta324) as
potential therapeutic targets in ovarian cancer. Gynecol Oncol.
118:189–195. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Heller G, Rommer A, Steinleitner K, Etzler
J, Hackl H, Heffeter P, Tomasich E, Filipits M, Steinmetz B,
Topakian T, et al: EVI1 promotes tumor growth via transcriptional
repression of MS4A3. J Hematol Oncol. 8:282015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao JS, Zhang Y, Tang X, Tucker LD,
Tarwater PM, Quesenberry PJ, Rigoutsos I and Ramratnam B: The Evi1,
microRNA-143, K-Ras axis in colon cancer. FEBS Lett. 585:693–699.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nayak KB, Kuila N, Das Mohapatra A, Panda
AK and Chakraborty S: EVI1 targets ΔNp63 and upregulates the cyclin
dependent kinase inhibitor p21 independent of p53 to delay cell
cycle progression and cell proliferation in colon cancer cells. Int
J Biochem Cell Biol. 45:1568–1576. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang M, Xiao X, Liang Y, Peng M, Jiang Y,
Xu Y and Gong G: Effect of methylation inhibitor on demethylation
pattern of the PD-1 gene in promoter region and PD-1 expression in
human T lymphocyte cell line. Zhong Nan Da Xue Xue Bao. Yi Xue Ban.
36:1163–1169. 2011.(In Chinese).
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Banerjee S, Yang S and Foster CB: A
luciferase reporter assay to investigate the differential
selenium-dependent stability of selenoprotein mRNAs. J Nutr
Biochem. 23:1294–1301. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pham MH, Kondapalli N, Reckord CL and
Foglesong PD: Interleukin-2 induces the activities of DNA
topoisomerase I and DNA topoisomerase II in HuT 78 cells. Biochem
Biophys Res Commun. 390:577–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yue X, Zhao Y, Zhang C, Li J, Liu Z, Liu J
and Hu W: Leukemia inhibitory factor promotes EMT through
STAT3-dependent miR-21 induction. Oncotarget. 7:3777–3790. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Humbert L, Ghozlan M, Canaff L, Tian J and
Lebrun JJ: The leukemia inhibitory factor (LIF) and p21 mediate the
TGFβ tumor suppressive effects in human cutaneous melanoma. BMC
Cancer. 15:2002015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Noordermeer SM, Monteferrario D, Sanders
MA, Bullinger L, Jansen JH and van der Reijden BA: Improved
classification of MLL-AF9-positive acute myeloid leukemia patients
based on BRE and EVI1 expression. Blood. 119:4335–4337. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vázquez I, Maicas M, Cervera J, Agirre X,
Marin-Béjar O, Marcotegui N, Vicente C, Lahortiga I, Gomez-Benito
M, Carranza C, et al: Down-regulation of EVI1 is associated with
epigenetic alterations and good prognosis in patients with acute
myeloid leukemia. Haematologica. 96:1448–1456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Balgobind BV, Lugthart S, Hollink IH,
Arentsen-Peters ST, van Wering ER, de Graaf SS, Reinhardt D,
Creutzig U, Kaspers GJ, de Bont ES, et al: EVI1 overexpression in
distinct subtypes of pediatric acute myeloid leukemia. Leukemia.
24:942–949. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weisser M, Haferlach C, Haferlach T and
Schnittger S: Feasibility of using the combined MDS-EVI1/EVI1 gene
expression as an alternative molecular marker in acute myeloid
leukemia: A report of four cases. Cancer Genet Cytogenet.
177:64–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gröschel S, Lugthart S, Schlenk RF, Valk
PJ, Eiwen K, Goudswaard C, van Putten WJ, Kayser S, Verdonck LF,
Lübbert M, et al: High EVI1 expression predicts outcome in younger
adult patients with acute myeloid leukemia and is associated with
distinct cytogenetic abnormalities. J Clin Oncol. 28:2101–2107.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Glass C, Wuertzer C, Cui X, Bi Y, Davuluri
R, Xiao YY, Wilson M, Owens K, Zhang Y and Perkins A: Global
identification of EVI1 target genes in acute myeloid leukemia. PloS
One. 8:e671342013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kutok JL, Yang X, Folkerth R and Adra CN:
Characterization of the expression of HTm4 (MS4A3), a cell cycle
regulator, in human peripheral blood cells and normal and malignant
tissues. J Cell Mol Med. 15:86–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan L, Ren F, Rong M, Dang Y, Luo Y, Luo D
and Chen G: Correlation between down-expression of miR-431 and
clinicopathological significance in HCC tissues. Clin Transl Oncol.
17:557–563. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu XT: TGFbeta-mediated formation of
pRb-E2F complexes in human myeloid leukemia cells. Biochem Biophys
Res Commun. 369:277–280. 2008. View Article : Google Scholar : PubMed/NCBI
|