Introduction
The severity and incurability of cancer are
attributed to cancer metastasis, the initiation of which requires
enhanced migratory and invasive capabilities (1). Tumor cells undergo
epithelial-to-mesenchymal transition (EMT) by inducing the
expression of mesenchymal markers and inhibiting the expression of
epithelial markers during tumor progression, and EMT enables cancer
cells to invade and metastasize (2).
EMT is a process characterized by the decrease of E-cadherin
expression and the increase of the expression of mesenchymal
markers, such as vimentin, β-catenin and N-cadherin (3). Thus, E-cadherin suppresses metastasis
and its inhibition promotes the development of malignant epithelial
cancers (4). In addition, EMT may
render cancer cells resistant to various chemotherapeutic/targeted
agents via reducing apoptotic sensitivity (5). EMT attenuates the activation of
caspase-8 via DR4/DR5 accompanied by E-cadherin inhibition.
E-cadherin interacts with DR4/DR5, thereby promoting caspase-8
activation and apoptosis (6). Thus,
E-cadherin has potentially significant biological implications in
the cross-regulation between EMT and apoptosis (7).
Several transcription factors, such as ZEB and Slug,
can repress E-cadherin (8,9). In addition, the expression of these
transcription factors is controlled by a complex network of
signaling molecules, including mitogen-activated protein kinases
(MAPKs), glycogen synthase kinase-3β (GSK-3β), phosphatidylinositol
3-kinase (PI3K) and nuclear factor κB (NF-κB) (10). MAPKs (ERK, JNK and p38) can promote
EMT and tumor cell metastasis (11,12). p38
activation is required for EMT, accompanied by E-cadherin
downregulation (13). Furthermore,
p38 plays a dual role in chemotherapeutic agent-induced apoptosis
(14). Several chemotherapeutic
agents require p38 to induce apoptosis (15). However, p38 can also mediate
resistance to apoptosis, as p38 activation results in induction of
cyclooxygenase-2 (COX-2) overexpression, which triggers resistance
to apoptosis in cancer cells (16,17).
Aspirin is a non-selective COX inhibitor and has
been used as an anti-inflammatory drug for >100 years (18). Regular use of aspirin is effective in
preventing several common cancers, including colon, breast, liver
and lung cancer (19–22). Aspirin acts by targeting several tumor
cell functions, including migration, and has been reported to
reduce the risk of cancer initiation and progression (1,23). Aspirin
modulates matrix metalloproteinase-2 (MMP-2) and E-cadherin
production and, therefore, possesses anti-metastatic properties
(24). Erlotinib, an epidermal growth
factor receptor tyrosine kinase inhibitor (EGFR-TKI), blocks the
autophosphorylation of EGFR and suppresses cell proliferation,
along with induction of apoptosis and anti-angiogenic effects,
inhibiting invasion and metastasis (25,26). In
the present study, we hypothesized that the combination of aspirin
with erlotinib may exert synergistic antitumor effects by
inhibiting cancer cell proliferation and metastasis. First, our
data demonstrated that aspirin combined with erlotinib exerted a
synergistic anti-proliferative effect and promoted apoptosis in
multiple human cancer cells. Furthermore, we also found that the
combination of aspirin and erlotinib was significantly more
effective in inhibiting cancer cell migration and invasion, which
are crucial for cancer metastasis. In addition, the synergistic
anti-angiogenic effects of aspirin plus erlotinib were confirmed
in vitro and in vivo. Finally, the synergistic
anti-proliferative and anti-metastatic effects of the combination
of aspirin with erlotinib were further validated in an A549
xenograft model in vivo. These findings suggested that
aspirin plus erlotinib may be an efficient combination regimen for
patients with metastatic cancer.
Materials and methods
Materials
Aspirin was purchased from Sigma-Aldrich (St. Louis,
MO, USA) and erlotinib was obtained from LC Laboratories (Woburn,
MA, USA). SB-203580, a p38 inhibitor, was obtained from Selleck
Chemicals (Houston, TX, USA).
Cell culture
Human lung carcinoma cell lines (NCI-H1299 and
A549), ovarian carcinoma cell line (HO-8910), colon carcinoma cell
line (HCT-116) and gastric carcinoma cell line (SGC-7901) were
obtained from Shanghai institute of biochemistry and cell biology.
A549 was maintained in Ham's F12 medium + 10% fetal bovine serum
(FBS). SGC-7901 was maintained in RPMI-1640 medium + 10% FBS.
HO-8910, NCI-H1299 and HCT-116 were maintained in DMEM + 10%
FBS.
Sulphorhodamine (SRB) cytotoxicity
assay
The cytotoxic activity was measured by the SRB
method, as previously described (27).
Colony-forming assay
Cancer cells (500–1,000 cells/dish) were plated into
six-well plates, treated with drugs every 3–4 days for 2 weeks, and
then stained by crystal violet (28).
Analysis of apoptosis and
determination of mitochondrial membrane depolarization
Cancer cells (3×105/well) were exposed to
the drugs, harvested and washed with PBS. Then, propidium iodide
(PI) staining was used to detect apoptosis, and the mitochondrial
membrane depolarization was determined by
5,5′,6,6′tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide (JC-1) staining as described previously (27).
Protein preparation from tissue and
cell samples and western blot analysis
The western blotting was performed as described
previously (29). The mouse
antibodies used for western blotting were obtained from different
resources: Anti-β-actin monoclonal antibody (Ab) (from BD
Biosciences, Franklin Lakes, NJ, USA) and anti-XIAP monoclonal Ab
(Santa Cruz Biotechnology, Dallas, TX, USA). The rabbit antibodies
used for western blotting were purchased from different resources:
anti-PARP polyclonal Ab, anti-procaspase-3 polyclonal Ab,
anti-Mcl-1 polyclonal Ab and anti-p38 polyclonal Ab (Santa Cruz
Biotechnology), anti-phospho-p38 (Thr-180/Tyr-182) polyclonal Ab
and anti-E-cadherin polyclonal Ab (Cell Signaling Technology,
Danvers, MA, USA).
Wound-healing assay
Cells were seeded in 24-well plates and cultured
until they reached confluence. Confluent monolayer cells were
gently scratched with a sterile pipette tip and then washed three
times with PBS to clear cell debris and suspended cells. Fresh
serum-free medium was added, and the cells were allowed to close
the wound for 48 h under normal conditions. Images of the wound in
the same relative position were captured with a computer-assisted
microscope (Olympus, Tokyo, Japan).
Cell invasion assay
Cell invasion experiments were performed using
24-well modified Boyden chambers (Costar, NY, USA) containing a
polycarbonate membrane with 8.0-µm pores according to the
manufacturer's instructions. First, the membranes were coated with
25 µg Matrigel (BD Biosciences). Cells were seeded at a density of
1×106 cells/well in the upper chamber with culture
medium (200 µl) alone, while the bottom of the plate was filled
with culture medium (500 µl) supplemented with 20% FBS. Cells that
invaded the underside of the membrane were fixed in 1% methanol and
stained by crystal violet.
Chick embryo chorioallantoic membrane
(CAM) assay
Inhibition of angiogenesis was determined using the
CAM assay. Fertilized chicken eggs were incubated at 37°C in a 50%
humidified atmosphere. On day 7, the eggshell was cracked and
gently opened into the plate to avoid any unnecessary physical
stress. It was made sure that the yolk sac membrane remained intact
and that the embryo was viable. Then, a sterile filter paper square
saturated with aspirin, erlotinib or their combination was placed
in areas between vessels, but not onto any large vessels. After a
48-h incubation, the membranes were examined by microscopy and
photographic documentation. Angiogenesis was quantified by counting
blood vessel branches; at least 10 viable embryos were tested for
each treatment.
Plasmids transfection
Cells (3×104) were seeded in 6-well
plates. E-cadherin (RG220731; Origene Technologies, Rockville, MD,
USA) and empty vector plasmid were transfected into cells using
Lipofectamine 3000 (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturers instructions.
siRNA transfection
Cells (5×104) were seeded in 6-well
plates. P38 siRNA and control siRNA (Genepharma, Shanghai, China)
were transfected into cells using Oligofectamine reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Sense p38 siRNA sequence was
5′-GCAUAAUGGCCGAGCUGUUTT-3′.
Antitumor activity in vivo and
histopathological evaluation of tumor metastasis
A549 xenografts were performed as previously
described (29). BALB/c (nu/nu) mice
were maintained under sterile conditions using an individually
ventilated cage system, randomized to 4 groups and then treated
with vehicle, aspirin (100 mg/kg, i.g. administration) daily and/or
erlotinib (20 mg/kg, i.p. administration) twice per week for 29
days (n=6). Finally, the mice were sacrificed at the end of the
treatment. The liver were fixed in 10% buffered formalin and
embedded in paraffin, 5-µm tissue sections were stained with
hematoxylin and eosin (H&E). All animal handling was performed
in accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals, and approved by the Zhejiang
University City College Animal Care and Use Committee (Hangzhou,
Zhejiang, China).
TUNEL staining
TUNEL assay was done using TUNEL apoptosis assay
kits (Beyotime Institute of Biotechnology, Shanghai, China) as
recommended by the manufacturer.
Statistical analyses
One-way ANOVA followed by Tukey's post hoc test and
Two-tailed student's t-tests were used to examine the significance
of differences among groups. Data points in graphs represent the
mean ± SD (*P<0.05; **P<0.01). For SRB assay, combination
index (CI) values were calculated using Calcusyn (Biosoft, Great
Shelford, Cambridge, UK) and the mean CI values were chosen for
presentation. A CI value <0.9 indicated synergism; 0.9 to 1.10,
additive; and >1.10, antagonism.
Results
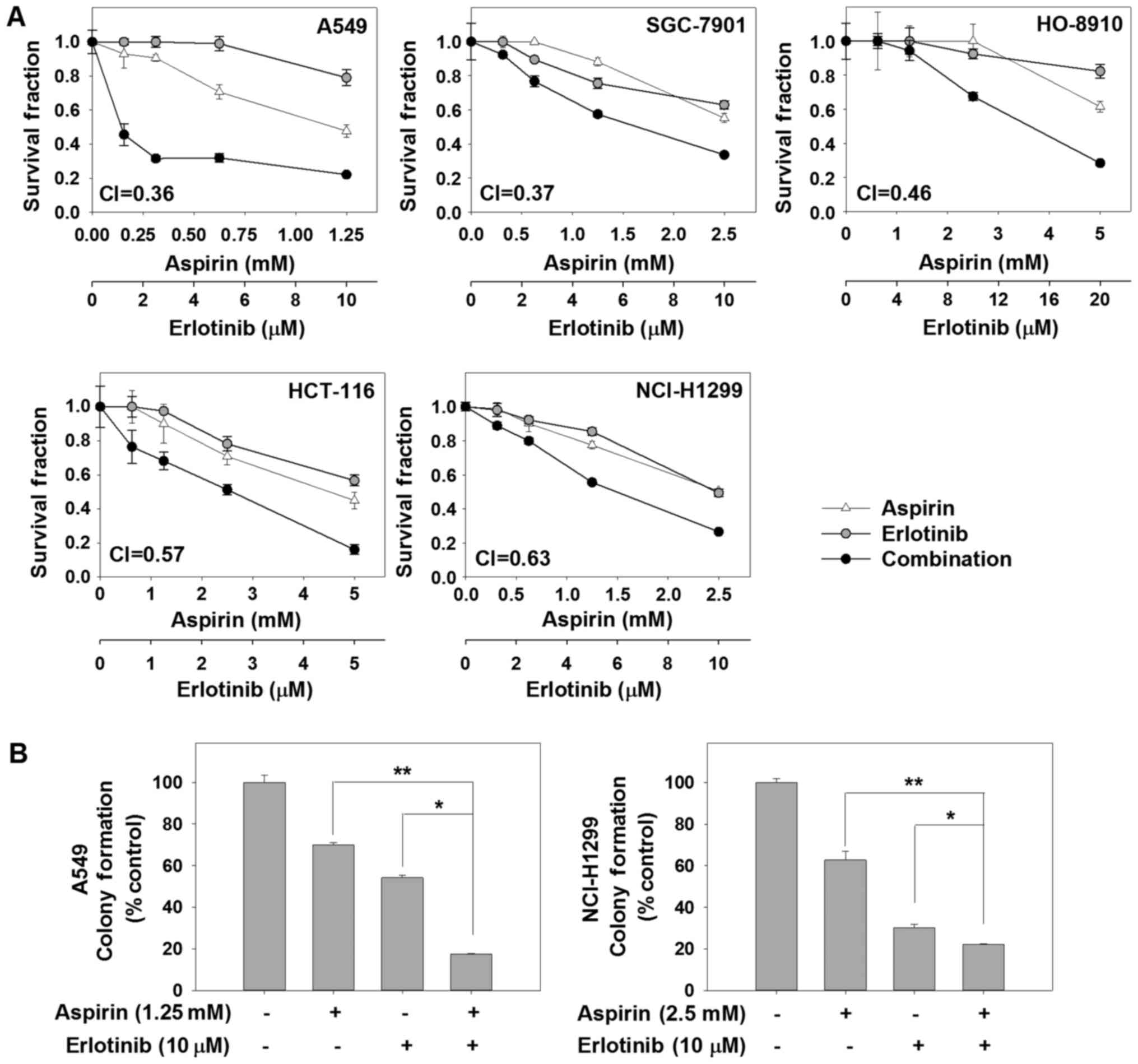
Cytotoxicity of aspirin plus erlotinib
in human carcinoma cell lines
First, we assessed the anticancer activity of the
combination of aspirin and erlotinib by SRB assay in 5 human
carcinoma cell lines. The survival curves for aspirin and/or
erlotinib are shown in Fig. 1A. We
found that aspirin plus erlotinib significantly reduced the
survival fraction in human cancer cells compared with each agent
alone. To verify the synergistic anticancer effect of aspirin and
erlotinib, CI values were calculated. Aspirin plus erlotinib
exerted synergistic cytotoxic effects on 5 human carcinoma cell
lines (CI values <0.7). In a long-term colony-forming assay, the
combination of aspirin and erlotinib resulted in significant
inhibition on the proliferation of A549 and NCI-H1299 cells, while
monotherapy induced a moderate inhibition (P<0.01, ANOVA)
(Fig. 1B). Taken together, these
finding indicate that the combination of aspirin and erlotinib was
more effective in limiting colony formation and cell growth of
human cancer cells in vitro compared with either agent
alone.
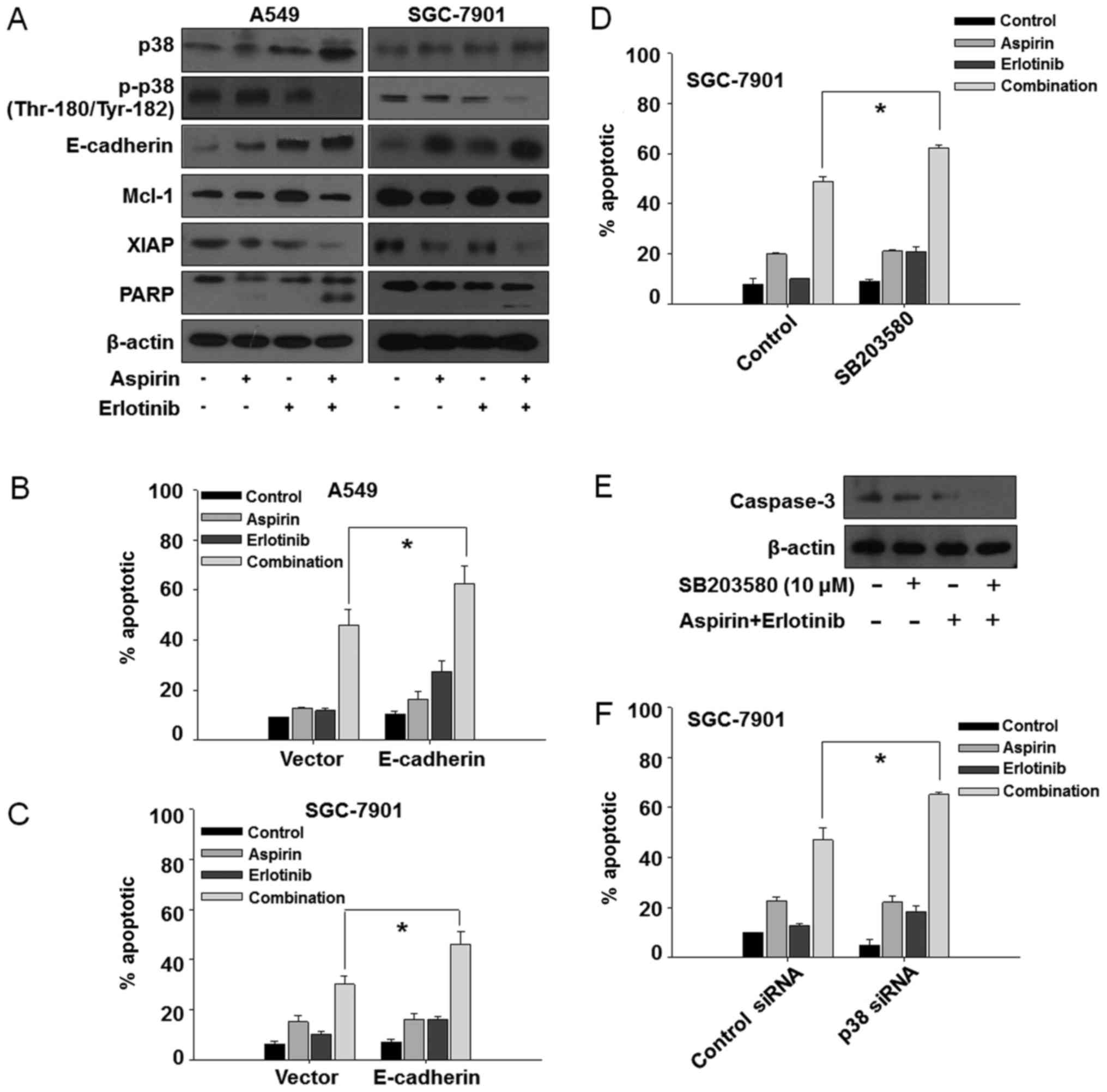
Aspirin plus erlotinb induces
mitochondrial mediated apoptosis via p38/E-cadherin pathway
We first investigated whether the synergistic
anticancer effects of aspirin plus erlotinib were related to the
induction of apoptosis. The percentage of apoptotic A549 cells was
6.95% in the control group, 28.96% with aspirin treatment, 18.67%
with erlotinib treatment, and 62.23% in the aspirin plus erlotinib
group (Fig. 2A, top panel). Aspirin
plus erlotinib significantly enhanced apoptosis in both A549 and
SGC-7901 cells compared with either drug alone (P<0.01, ANOVA;
Fig. 2B and C). Next, we investigated
whether aspirin plus erlotinib affected the mitochondrial membrane
potential. As shown in Fig. 2A
(bottom panel), D and E, aspirin plus erlotinib increased the
percentage of mitochondrial membrane depolarized carcinoma cells
compared with either drug alone (P<0.01, ANOVA). Thus, our data
suggested that the synergistic effects of aspirin plus erlotinib
are mediated via the mitochondrial apoptotic pathway. In addition,
aspirin plus erlotinib markedly induced PARP cleavage and XIAP
suppression in two of the cancer cell lines (Fig. 3A). The induction of EMT and activation
of p38 are associated with resistance to erlotinib, and blockade of
p38 was found to be able to suppress EMT in erlotinib-resistant
cancer cells (30). Furthermore, the
upregulation of E-cadherin increased the sensitivity of several
TKIs, such as gefitinib and erlotinib (31,32). Thus,
we were interested in examining the involvement of p38 and
E-cadherin in the aspirin and erlotinib combination treatment.
Interestingly, as shown in Fig. 3A,
the enhanced apoptosis induced by aspirin plus erlotinib was
accompanied by p38 inhibition and overexpression of E-cadherin in
A549 and SGC-7901 cells, indicating that aspirin may reverse
erlotinib resistance via the p38/E-cadherin pathway. To further
investigate the involvement of the p38/E-cadherin pathway in the
synergistic effects of aspirin and erlotinib, we first performed
E-cadherin overexpression experiments by transfecting cancer cells
with an E-cadherin plasmid. As shown in Fig. 3B and C, overexpression of E-cadherin
increased the apoptosis induced by aspirin plus erlotinib in A549
and SGC-7901 cells. Next, a p38 inhibitor (SB-203580) was found to
increase the apoptosis induced by aspirin plus erlotinib in
SGC-7901 cells (Fig. 3D and E).
Moreover, p38 depletion by siRNA also enhanced aspirin plus
erlotinib-induced apoptosis (Fig.
3F). Therefore, these data indicated that the p38/E-cadherin
pathway may be involved in the enhanced apoptosis induced by
aspirin plus erlotinib treatment.
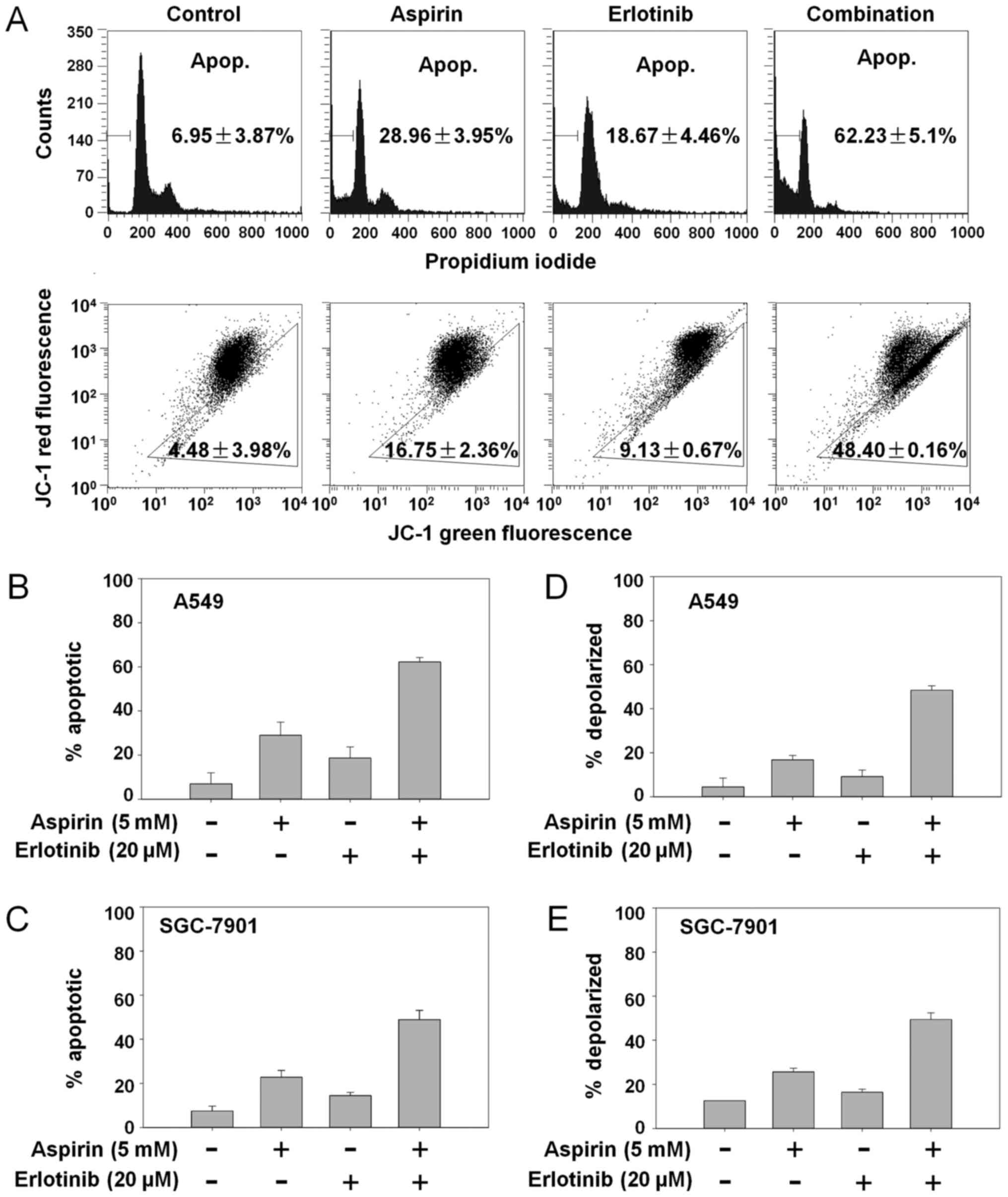 | Figure 2.Aspirin plus erlotinib induced
apoptosis via mitochondrial pathway. (A) A549 cells were incubated
with aspirin (5 mM), erlotinib (20 µM) or the combination for 48 h,
and then cells were stained with PI (top panel)/JC-1 (bottom panel)
followed by flow cytometry detection. (B and C) Cancer cells in
6-well plates were treated with drugs for 48 h and detected by flow
cytometry after PI staining. (D and E) Cancer cells were treated
with drugs for 48 h and detected by flow cytometry after JC-1
staining. PI, propidium iodide; JC-1,
5,5′,6,6′tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide. |
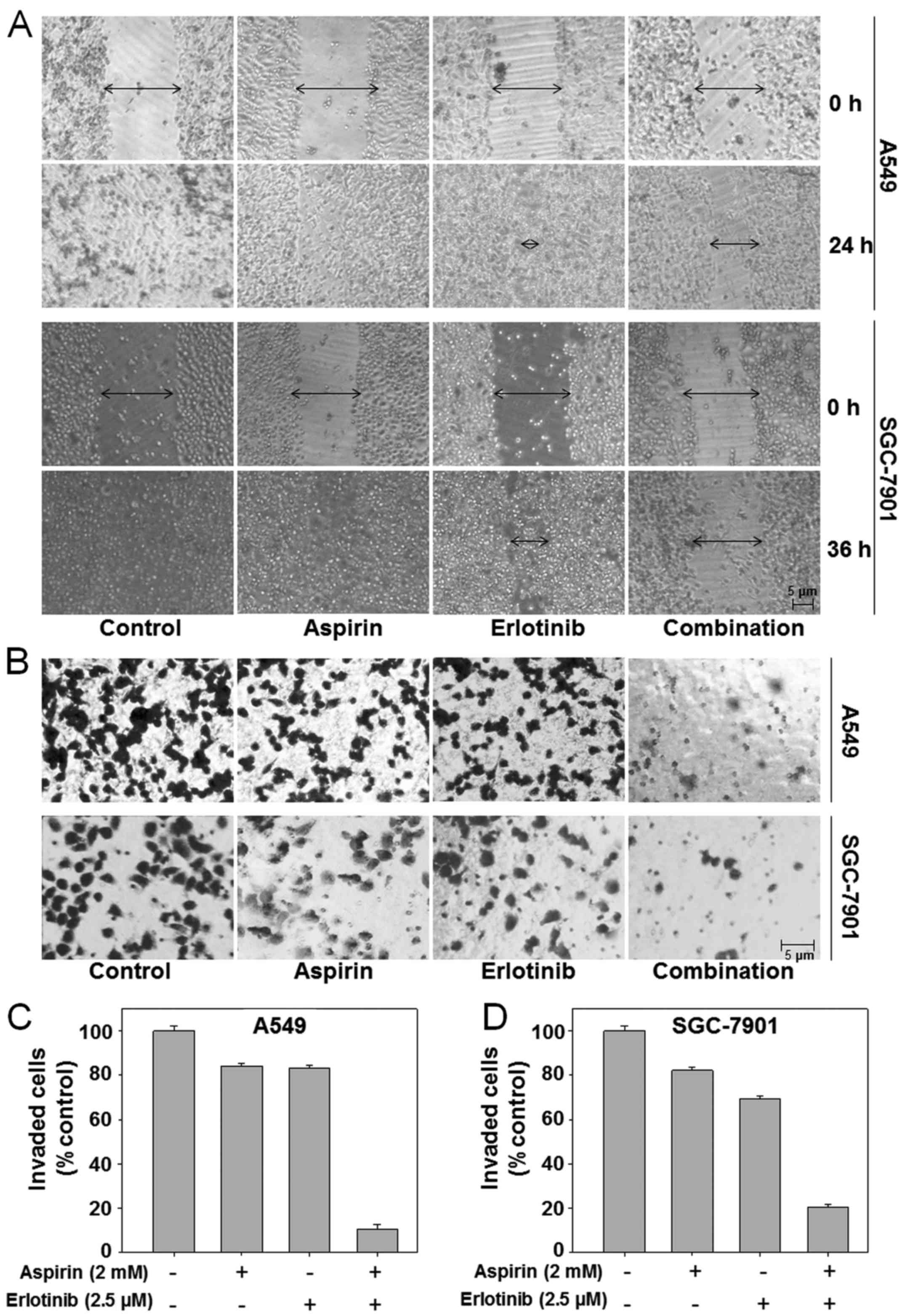
Aspirin plus erlotinib inhibits
invasion and migration of human carcinoma cells
E-cadherin inhibition is an important step in EMT
and a hallmark of metastatic cells (33). Our data already demonstrated that
aspirin plus erlotinib could increase the expression of E-cadherin;
thus, we hypothesized that aspirin plus erlotinib may inhibit EMT
and cancer metastasis. EMT enables cancer cell migration and
invasion, and a scratch assay was conducted on A549 and SGC-7901
cells to assess cell migration, which is a defining feature of the
mesenchymal phenotype. As shown in Fig.
4A (top panel), untreated A549 cells migrated within 24 h after
wounding, whereas there was moderate inhibition of cell migration
when the A549 cells were treated with erlotinib at 2.5 µM. However,
the suppressive effect on migration was most prominent in A549
cells treated with aspirin plus erlotinib. Similar results were
observed in SGC-7901 cells (Fig. 4A,
bottom panel). In addition, the effects of aspirin plus erlotinib
on cancer cell invasion were evaluated by Matrigel Transwell
invasion assays. Our data demonstrated that aspirin or erlotinib
achieved moderate inhibition of cell invasion in the A549 and
SGC-7901 cell lines. However, aspirin plus erlotinib led to a
significant reduction in the number of invading cancer cells
compared with the effects of either aspirin or erlotinib alone
(P<0.01, ANOVA) (Fig. 4B-D). Our
results indicated that the combination of aspirin and erlotinib
enhanced the inhibition of cancer cell migration and invasion
compared with either drug alone.
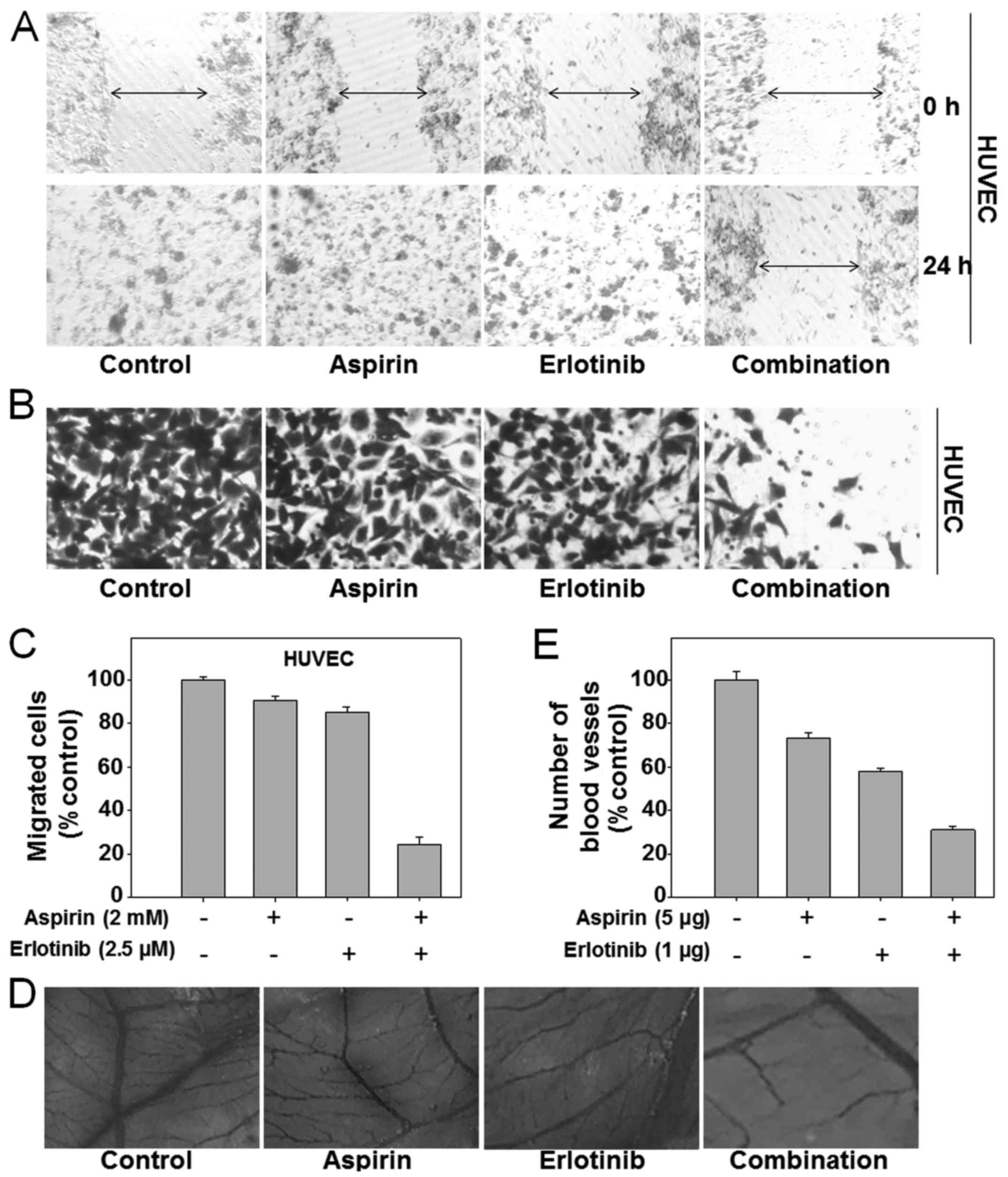
Synergistic inhibition of angiogenesis
by aspirin plus erlotinib
Endothelial cells can invade the surrounding
basement membrane, and then migrate into the stroma during tumor
angiogenesis. Finally, endothelial cells organize to form new
capillaries, which are crucial for tumor metastasis (34). To evaluate the anti-angiogenic
function of the aspirin plus erlotinib combination, we first
detected its effects on migration and invasion of human umbilical
vein endothelial cells (HUVECs). There was no inhibition of cell
migration when HUVECs were treated with aspirin or erlotinib alone.
However, when the two drugs were combined, the inhibitory effect on
HUVEC migration was enhanced in the scratch assay (Fig. 5A). Similarly, aspirin plus erlotinib
treatment induced a substantial decrease in the number of invading
HUVECs compared with aspirin or erlotinib alone (P<0.01; ANOVA)
(Fig. 5B and C). These observations
suggested that aspirin plus erlotinib may inhibit angiogenesis via
reducing invasion and migration of endothelial cells. Next, the CAM
assay was performed to further investigate the anti-angiogenic
effect of aspirin plus erlotinib. As shown in Fig. 5D, aspirin plus erlotinib blocked
angiogenesis in the CAM assay. Quantitative analysis revealed that
aspirin, erlotinib, and aspirin plus erlotinib caused a 26.7, 42.2
and 68.9% reduction in the number of blood vessels, respectively
(P<0.01; ANOVA) (Fig. 5E). Our
data obtained from the three models were sufficient to confirm the
synergistic anti-angiogenic activity of aspirin plus erlotinib.
Synergistic antitumor activity of
aspirin and erlotinib in vivo
An A549 xenograft model was constructed to verify
the anticancer and anti-metastatic efficacy of aspirin plus
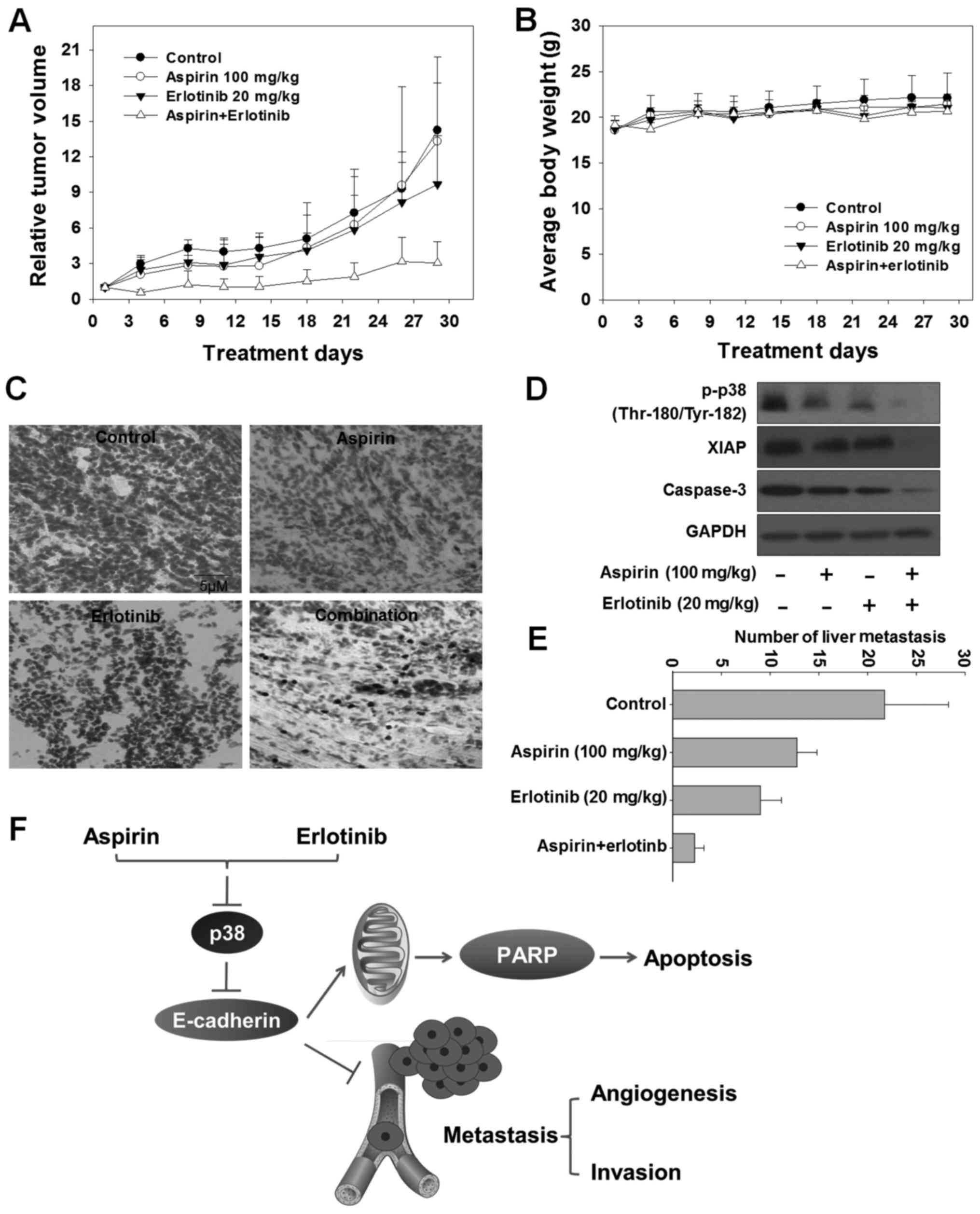
erlotinib. As demonstrated in Fig. 6A
and Table I, aspirin exerted no
significant anticancer effect [mean relative tumor volume
(RTV)aspirin: 13.3 vs. mean RTVcontrol: 14.2;
P>0.05, t-test]. Erlotinib exerted a moderate anticancer effect
(mean RTVerlotinib: 9.7 vs. mean RTVcontrol:
14.2, P<0.05, t-test). Aspirin plus erlotinib induced
significant tumor growth inhibition (mean
RTVcombination: 3.1 vs. mean RTVcontrol:
14.2, P<0.01, t-test), which was markedly higher compared with
that of either aspirin or erlotinib alone (mean
RTVcombination: 3.1 vs. mean RTVaspirin:
13.3, P<0.05, t-test and mean RTVcombination: 3.1 vs.
mean RTVerlotinib: 9.7, P<0.05, t-test). In addition,
the loss of body weight did not differ significantly on day 29
compared with day 0 in the aspirin plus erlotinib group (P>0.05;
ANOVA) (Fig. 6B). The TUNEL assay was
performed to detect the apoptosis induced by aspirin plus erlotinib
in the A549 xenograft model. The number of TUNEL-positive cells
increased in the tumor tissues of mice receiving the combination
treatment (Fig. 6C). Furthermore,
caspase activation and XIAP inhibition were observed in the tumor
tissues of mice receiving combination treatment, highlighting that
apoptosis was involved in the tumor growth inhibitory effects
induced by aspirin and erlotinib in vivo (Fig. 6D). In addition, the number of liver
metastases was significantly reduced in the combination-treated
group compared with the aspirin or erlotinib alone group in the
A549 xenograft model (P<0.01; ANOVA) (Fig. 6E). In conclusion, the synergistic
antitumor effects of aspirin plus erlotinib were confirmed in
vivo.
 | Table I.In vivo efficacy of aspirin in
combination with erlotinib against A549 xenografts. |
Table I.
In vivo efficacy of aspirin in
combination with erlotinib against A549 xenografts.
|
| No. of animals | Body weight
(g) |
|
|
|
|---|
|
|
|
|
|
|
|
|---|
| Group | Start | End | Start | End | Inhibition rate
(%) | Mean RTV | T/C (100%) |
|---|
| Control | 6 | 6 | 18.7±1.5 | 22.1±2.7 | – | 14.2 | – |
| Aspirin (100
mg/kg) | 6 | 6 | 18.6±1.0 | 21.5±1.1 | 0 | 13.3 | 93.7 |
| Erlotinib (20
mg/kg) | 6 | 6 | 18.7±1.0 | 21.1±0.8 | 19.2 | 9.7a | 68.3 |
| Combination | 6 | 6 | 19.2±0.5 | 22.6±0.8 | 55.8 | 3.1b–d | 21.8 |
Discussion
Cancer cells are sensitive to aspirin in patients
with PIK3CA mutation, but not in those with PIK3CA wild-type tumors
(35,36). Our data indicated that aspirin plus
erlotinib exerted synergistic anti-proliferative effects on PIK3CA
wild-type cancer cell lines (A549, NCI-H1299 and HO-8910) and
PIK3CA mutant cell lines (HCT-116 and SGC-7901), indicating that
PIK3CA mutation was not associated with the synergistic
anti-proliferative effect exerted by aspirin plus erlotinib on
human cancer cells. Thus, aspirin plus erlotinib may improve the
antitumor efficacy both in aspirin-sensitive and aspirin-resistant
cancer cells. The co-administration of aspirin and erlotinib
markedly improved the antitumor efficacy without increasing the
toxicity in a A549 xenograft model in vivo.
EGFR-TKIs, such as afatinib, gefitinib and
erlotinib, have been proven to be clinically effective for patients
with metastatic or locally advanced non-small cell lung cancer
(NSCLC) (37). However, treatment
with EGFR-TKIs eventually fails due to the development of acquired
drug resistance within 9–14 months of treatment (38). EGFR-TKI-resistant NSCLC cells undergo
EMT by repressing E-cadherin and upregulating p38 expression
(39). The loss of E-cadherin
expression and upregulation of p38 activate multiple pathways that
inhibit apoptosis, induce tumor metastasis and erlotinib
resistance, and eventually lead to failure of erlotinib treatment
(30,40). Thus, overexpression of E-cadherin and
inhibition of p38 may be effective in overcoming EMT induction and
apoptosis evasion-mediated erlotinib resistance. Our data indicated
that aspirin plus erlotinib significantly induced
mitochondrial-mediated apoptosis, and significantly induced the
activation of E-cadherin and suppression of p38 both in A549 and
SGC-7901 cells. Moreover, elevated expression of E-cadherin
increased apoptosis induced by aspirin plus erlotinib; in addition,
the inhibition of p38 by SB-203580 or siRNA also enhanced the
apoptosis induced by aspirin plus erlotinib. These data indicated
that the p38/E-cadherin signaling pathway was implicated in the
enhanced apoptosis induced by aspirin plus erlotinib, and aspirin
may reverse erlotinib resistance via the p38/E-cadherin
pathway.
As p38 and E-cadherin also play a key role in EMT
and tumor metastasis, we hypothesized that aspirin plus erlotinib
may significantly inhibit EMT and the metastatic process. EMT has
been shown to play a crucial role in the invasion and metastasis of
epithelial tumors, as it enables cancer cell migration and invasion
(41). Our data indicated that
combined treatment with aspirin and erlotinib enhanced the
inhibition on cancer cell migration and invasion. Metastasis and
tumor growth depend on the development of a neovasculature around
and in the tumor (42). Angiogenesis
is a neovascularization process that involves critical steps,
including endothelial cell invasion, migration and proliferation
(43). Our results determined that
aspirin plus erlotinib inhibited angiogenesis by suppressing
migration and invasion of endothelial cells. Furthermore, the
synergistic anti-angiogenic effects of aspirin plus erlotinib were
confirmed by the CAM assay. Most importantly, aspirin plus
erlotinib significantly decreased the number of liver metastases
in vivo. Overall, these findings demonstrated that aspirin
plus erlotinib significantly inhibited tumor metastasis via
inhibiting EMT and angiogenesis; thus, aspirin plus erlotinib may
be an efficient combination regimen for patients with metastatic
cancer.
In summary, to the best of our knowledge, our data
are the first to demonstrate that combining aspirin with erlotinib
could significantly inhibit the proliferation and induce apoptosis
of human cancer cells. In addition, our data indicated that aspirin
and erlotinib inhibited EMT and angiogenesis, consequently
suppressing tumor metastasis. Furthermore, the p38/E-cadherin
signaling pathway was involved in the synergistic anticancer
activity of aspirin plus erlotinib (Fig.
6F). Therefore, aspirin appears to be a pertinent sensitizer to
erlotinib for treating patients with metastatic cancer.
Acknowledgements
Not applicable.
Funding
The present study was funded by National Natural
Science Foundation of China (grant no. 1702887), Hangzhou Major
Science and Technology Project (grant no. 20172016A01), Fund of
Hangzhou Medical Key Discipline Construction (grant no.
2017-51-07), Zhejiang Provincial Foundation of Natural Science
(grant no. LQ16H310004), Public-service Technology Research Plan of
Zhejiang Province (grant nos. 2015C33269 and 2016C33210),
High-level Talents Coming Back from Abroad Innovation and
Entrepreneurship Program in Hangzhou (grant no. 2051), Zhejiang
Provincial Program for the cultivation of high-level innovative
health talents (grant no. 2010-190-4), Scientific and Technological
Developing Scheme of Hangzhou City (grant nos. 20150733Q14 and
20140633B03).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
CZ and NML were responsible for the conception and
design of the study. XH and XW were responsible for collecting the
data. LWW analyzed and interpreted the data. CZ drafted this
manuscript. NML revised it critically for important intellectual
content. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal handling was performed in accordance with
the National Institutes of Health Guide for the Care and Use of
Laboratory Animals, and approved by the Zhejiang University City
College Animal Care and Use Committee.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Glossary
Abbreviations
Abbreviations:
|
CI
|
combination index
|
|
PI
|
propidium iodide
|
|
Ab
|
antibody
|
|
EMT
|
epithelial to mesenchymal
transition
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
EGFR-TKI
|
epidermal growth factor receptor
tyrosine kinase inhibitor
|
|
V
|
tumor volume
|
|
RTV
|
relative tumor volume
|
|
HUVEC
|
human umbilical vein endothelial
cells
|
|
CAM
|
chick embryo chorioallantoic
membrane
|
|
GSK-3β
|
glycogen synthase kinase-3β
|
|
NF-κB
|
nuclear factor-κB.
|
References
|
1
|
Khan P, Manna A, Saha S, Mohanty S,
Mukherjee S, Mazumdar M, Guha D and Das T: Aspirin inhibits
epithelial-to-mesenchymal transition and migration of oncogenic
K-ras-expressing non-small cell lung carcinoma cells by
down-regulating E-cadherin repressor Slug. BMC Cancer. 16:392016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kurimoto R, Iwasawa S, Ebata T, Ishiwata
T, Sekine I, Tada Y, Tatsumi K, Koide S, Iwama A and Takiguchi Y:
Drug resistance originating from a TGF-β/FGF-2-driven
epithelial-to-mesenchymal transition and its reversion in human
lung adenocarcinoma cell lines harboring an EGFR mutation. Int J
Oncol. 48:1825–1836. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsuda Y, Miura K, Yamane J, Shima H,
Fujibuchi W, Ishida K, Fujishima F, Ohnuma S, Sasaki H, Nagao M, et
al: SERPINI1 regulates epithelial-mesenchymal transition in an
orthotopic implantation model of colorectal cancer. Cancer Sci.
107:619–628. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh A and Settleman J: EMT, cancer stem
cells and drug resistance: An emerging axis of evil in the war on
cancer. Oncogene. 29:4741–4751. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu M, Marsters S, Ye X, Luis E, Gonzalez L
and Ashkenazi A: E-cadherin couples death receptors to the
cytoskeleton to regulate apoptosis. Mol Cell. 54:987–998. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McConkey DJ, Choi W, Marquis L, Martin F,
Williams MB, Shah J, Svatek R, Das A, Adam L, Kamat A, et al: Role
of epithelial-to-mesenchymal transition (EMT) in drug sensitivity
and metastasis in bladder cancer. Cancer Metastasis Rev.
28:335–344. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bolos V, Peinado H, Perez-Moreno MA, Fraga
MF, Esteller M and Cano A: The transcription factor Slug represses
E-cadherin expression and induces epithelial to mesenchymal
transitions: A comparison with Snail and E47 repressors. J Cell
Sci. 116:499–511. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peinado H, Olmeda D and Cano A: Snail, Zeb
and bHLH factors in tumour progression: An alliance against the
epithelial phenotype? Nat rev Cancer. 7:415–428. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang H, Fang R, Wang XF, Zhang F, Chen DY,
Zhou B, Wang HS, Cai SH and Du J: Stabilization of Snail through
AKT/GSK-3β signaling pathway is required for TNF-α-induced
epithelial-mesenchymal transition in prostate cancer PC3 cells. Eur
J Pharmacol. 714:48–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zohn IE, Li Y, Skolnik EY, Anderson KV,
Han J and Niswander L: p38 and a p38-interacting protein are
critical for downregulation of E-cadherin during mouse
gastrulation. Cell. 125:957–969. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen R, Yang Q and Lee JD: BMK1 kinase
suppresses epithelial-mesenchymal transition through the Akt/GSK3β
signaling pathway. Cancer Res. 72:1579–1587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cheng JC, Klausen C and Leung PC: Hydrogen
peroxide mediates EGF-induced down-regulation of E-cadherin
expression via p38 MAPK and snail in human ovarian cancer cells.
Mol Endocrinol. 24:1569–1580. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sui X, Kong N, Ye L, Han W, Zhou J, Zhang
Q, He C and Pan H: p38 and JNK MAPK pathways control the balance of
apoptosis and autophagy in response to chemotherapeutic agents.
Cancer Lett. 344:174–179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olson JM and Hallahan AR: p38 MAP kinase:
A convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Limami Y, Pinon A, Leger DY, Pinault E,
Delage C, Beneytout JL, Simon A and Liagre B: The
P2Y2/Src/p38/COX-2 pathway is involved in the resistance to ursolic
acid-induced apoptosis in colorectal and prostate cancer cells.
Biochimie. 94:1754–1763. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Salim H, Akbar NS, Zong D, Vaculova AH,
Lewensohn R, Moshfegh A, Viktorsson K and Zhivotovsky B: miRNA-214
modulates radiotherapy response of non-small cell lung cancer cells
through regulation of p38MAPK, apoptosis and senescence. Br J
Cancer. 107:1361–1373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan KH, Yao CJ, Chang HY, Lai GM, Cheng AL
and Chuang SE: The synergistic anticancer effect of troglitazone
combined with aspirin causes cell cycle arrest and apoptosis in
human lung cancer cells. Mol Carcinog. 49:235–246. 2010.PubMed/NCBI
|
|
19
|
Chan AT, Ogino S and Fuchs CS: Aspirin and
the risk of colorectal cancer in relation to the expression of
COX-2. N Engl J Med. 356:2131–2142. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bardia A, Keenan TE, Ebbert JO, Lazovich
D, Wang AH, Vierkant RA, Olson JE, Vachon CM, Limburg PJ, Anderson
KE and Cerhan JR: Personalizing aspirin use for targeted breast
cancer chemoprevention in postmenopausal women. Mayo Clin Proc.
91:71–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Petrick JL, Sahasrabuddhe VV, Chan AT,
Alavanja MC, Beane-Freeman LE, Buring JE, Chen J, Chong DQ,
Freedman ND, Fuchs CS, et al: NSAID Use and risk of hepatocellular
carcinoma and intrahepatic cholangiocarcinoma: The liver cancer
pooling project. Cancer Prev Res. 8:1156–1162. 2015. View Article : Google Scholar
|
|
22
|
Brasky TM, Baik CS, Slatore CG, Potter JD
and White E: Non-steroidal anti-inflammatory drugs and small cell
lung cancer risk in the VITAL study. Lung Cancer. 77:260–264. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rothwell PM, Wilson M, Price JF, Belch JF,
Meade TW and Mehta Z: Effect of daily aspirin on risk of cancer
metastasis: A study of incident cancers during randomised
controlled trials. Lancet. 379:1591–1601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Guillem-Llobat P, Dovizio M, Bruno A,
Ricciotti E, Cufino V, Sacco A, Grande R, Alberti S, Arena V,
Cirillo M, et al: Aspirin prevents colorectal cancer metastasis in
mice by splitting the crosstalk between platelets and tumor cells.
Oncotarget. 7:32462–32477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ito K, Semba T, Uenaka T, Wakabayashi T,
Asada M and Funahashi Y: Enhanced anti-angiogenic effect of E7820
in combination with erlotinib in epidermal growth factor
receptor-tyrosine kinase inhibitor-resistant non-small-cell lung
cancer xenograft models. Cancer Sci. 105:1023–1031. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hirai F, Edagawa M, Shimamatsu S, Toyozawa
R, Toyokawa G, Nosaki K, Yamaguchi M, Seto T, Takenoyama M and
Ichinose Y: Evaluation of erlotinib for the treatment of patients
with non-small cell lung cancer with epidermal growth factor
receptor wild type. Oncol Lett. 14:306–312. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li YL, Sun J, Hu X, Pan YN, Yan W, Li QY,
Wang F, Lin NM and Zhang C: Epothilone B induces apoptosis and
enhances apoptotic effects of ABT-737 on human cancer cells via
PI3K/AKT/mTOR pathway. J Cancer Res Clin Oncol. 142:2281–2289.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang C, Shi J, Mao SY, Xu YS, Zhang D,
Feng LY, Zhang B, Yan YY, Wang SC, Pan JP, et al: Role of p38 MAPK
in enhanced human cancer cells killing by the combination of
aspirin and ABT-737. J Cell Mol Med. 19:408–417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang C, Cai TY, Zhu H, Yang LQ, Jiang H,
Dong XW, Hu YZ, Lin NM, He QJ and Yang B: Synergistic antitumor
activity of gemcitabine and ABT-737 in vitro and in vivo through
disrupting the interaction of USP9X and Mcl-1. Mol Cancer Ther.
10:1264–1275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fernando RI, Hamilton DH, Dominguez C,
David JM, McCampbell KK and Palena C: IL-8 signaling is involved in
resistance of lung carcinoma cells to erlotinib. Oncotarget.
7:42031–42044. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Witta SE, Gemmill RM, Hirsch FR, Coldren
CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita
M, et al: Restoring E-cadherin expression increases sensitivity to
epidermal growth factor receptor inhibitors in lung cancer cell
lines. Cancer Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yauch RL, Januario T, Eberhard DA, Cavet
G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al:
Epithelial versus mesenchymal phenotype determines in vitro
sensitivity and predicts clinical activity of erlotinib in lung
cancer patients. Clin Cancer Res. 11:8686–8698. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heerboth S, Housman G, Leary M, Longacre
M, Byler S, Lapinska K, Willbanks A and Sarkar S: EMT and tumor
metastasis. Clin Transl Med. 4:62015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bhat TA, Nambiar D, Tailor D, Pal A,
Agarwal R and Singh RP: Acacetin inhibits in vitro and in vivo
angiogenesis and downregulates Stat signaling and VEGF expression.
Cancer Prev Res. 6:1128–1139. 2013. View Article : Google Scholar
|
|
35
|
Liao X, Lochhead P, Nishihara R, Morikawa
T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, et
al: Aspirin use, tumor PIK3CA mutation and colorectal-cancer
survival. N Engl J Med. 367:1596–1606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Turturro SB, Najor MS, Ruby CE, Cobleigh
MA and Abukhdeir AM: Mutations in PIK3CA sensitize breast cancer
cells to physiologic levels of aspirin. Breast Cancer Res Treat.
156:33–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Burotto M, Manasanch EE, Wilkerson J and
Fojo T: Gefitinib and erlotinib in metastatic non-small cell lung
cancer: A meta-analysis of toxicity and efficacy of randomized
clinical trials. Oncologist. 20:400–410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang B, Jiao J, Liu Y, Guo LX, Zhou B, Li
GQ, Yao ZJ and Zhou GB: Gefitinib analogue induces apoptosis of
T790M EGFR-harboring lung cancer cells by up-regulation of the BH-3
only protein Noxa. PLoS One. 7:e487482012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rastogi I, Rajanna S, Webb A, Chhabra G,
Foster B, Webb B and Puri N: Mechanism of c-Met and EGFR tyrosine
kinase inhibitor resistance through epithelial mesenchymal
transition in non-small cell lung cancer. Biochem Biophys Res
Commun. 477:937–944. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang Y, Sheng Q, Spillman MA, Behbakht K
and Gu H: Gab2 regulates the migratory behaviors and E-cadherin
expression via activation of the PI3K pathway in ovarian cancer
cells. Oncogene. 31:2512–2520. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jain RK: Normalization of tumor
vasculature: An emerging concept in antiangiogenic therapy.
Science. 307:58–62. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lamalice L, Le Boeuf F and Huot J:
Endothelial cell migration during angiogenesis. Circ Res.
100:782–794. 2007. View Article : Google Scholar : PubMed/NCBI
|