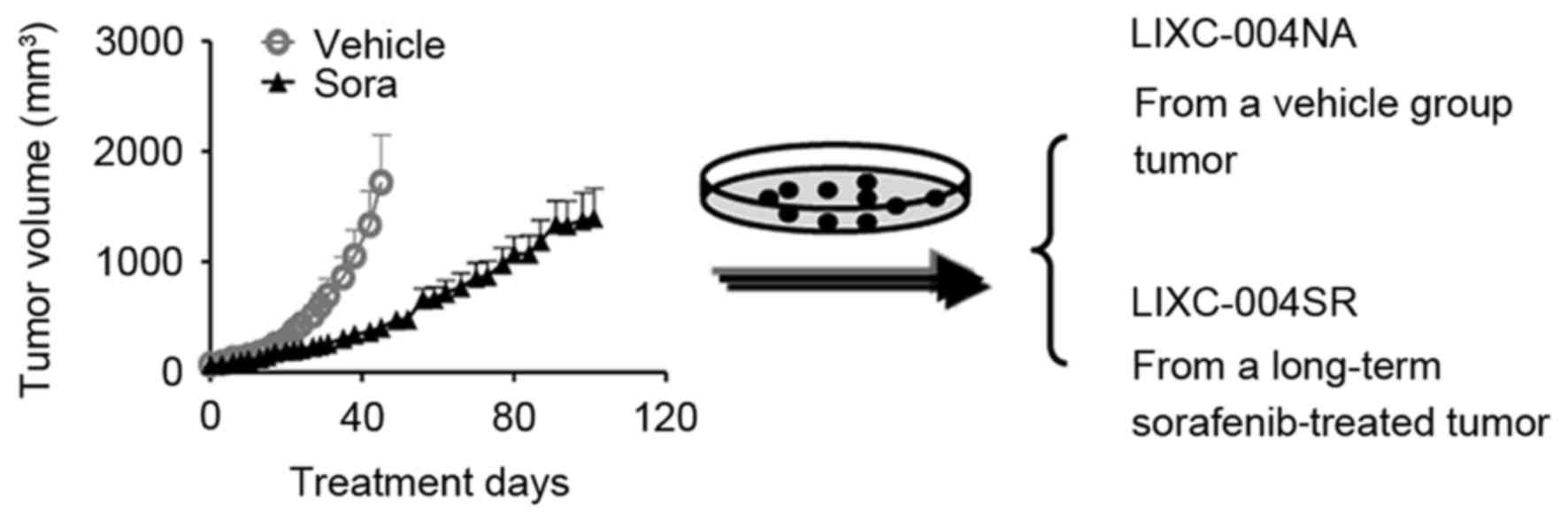
Introduction
Liver cancer is responsible for the second highest
number of cancer-associated fatalities in men worldwide, following
lung cancer. Hepatocellular carcinoma (HCC) accounts for 70–85% of
all liver cancer types (1). Sorafenib
is a multi-kinase inhibitor that inhibits angiogenesis and tumor
cell proliferation (2). Sorafenib
treatment has demonstrated survival benefits and good tolerability
in clinical trials (3), and has
become the standard treatment option for advanced HCC. Despite its
success, sorafenib treatment extended patient overall survival by
only 2–3 months in previous studies (3,4). Even
among those who initially responded well to sorafenib treatment,
the majority of patients developed drug resistance during treatment
(3,5).
Currently, there is no effective systemic therapy available for
patients following failure of sorafenib therapy (6,7).
Therefore, preventing or treating sorafenib resistance has become
an urgent unmet medical requirement.
Acquired drug resistance develops in the context of
tumor-host interactions. In order to understand the key mechanisms
of sorafenib resistance, one such model was developed from an HCC
patient-derived xenograft (PDX) model following long-term
sorafenib-treatment. A PDX model was selected as these models
preserve numerous clinical characteristics and their drug response
is markedly associated with clinical drug efficacy (8–10). To
facilitate mechanistic studies, cell lines were established from
the vehicle-treated and sorafenib-treated xenografts. Through
comparing the two cell lines and respective xenografts, the present
study demonstrated that the in vivo sorafenib resistance of
the LIXC-004SR cell line appears to be partially mediated by
alternative angiogenesis pathways.
Materials and methods
Patient samples
Surgically removed human HCC samples were obtained
from Nantong Cancer Hospital (Nantong, Jiangsu, China) and written
informed consent was obtained according to protocols approved by
the ChemPartner Institutional Ethical Committee.
Animals
A total of 5 female 6-8-week-old severe combined
immune deficiency (SCID) mice and ~150 female 6-8-week-old Nu/Nu
mice (16–20 g) were purchased from Beijing Vital River Laboratory
Animal Technology Co. Ltd. (Beijing, China) and used for different
experiments, including the PDX model establishment, sorafenib
resistant model generation, tumorigenicity of cell lines, and in
vivo efficacy study of these cell lines. Animals were housed
under specific pathogen-free conditions at ChemPartner. Animals for
at least three days for acclimation prior to the study. Animals
were provided pelleted food and water ad libitum and kept in a room
conditioned at 20–25°C, 40–70% relative humidity with a 12 h
light/dark cycle according to guidelines provided by the
Association for Assessment and Accreditation of Laboratory Animal
Care. The experiment was performed according to the protocol
approved by the ChemPartner Institutional Animal Care and Use
Committee.
Reagents
Sorafenib was purchased from LC Laboratories
(Woburn, MA, USA) and AZD4547 [a selective inhibitor for fibroblast
growth factor (FGF) receptor 1] was purchased from Shanghai
Biochempartner Co., Ltd. (Shanghai, China). For in vivo
studies, sorafenib was dissolved in cremophor (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany), ethanol and distilled water
(12.5:12.5:75, respectively), and administered orally each day at a
dosage of 40 mg/kg (2,11). AZD4547 was dissolved in 1% Tween-80
(Sigma-Aldrich; Merck KGaA) in distilled water, sonicated for 30
min and mixed thoroughly and administered orally each day at a
dosage of 12.5 mg/kg (dosage volume, 10 ml/kg) (12).
Establishment of cell lines from
PDX
A PDX model, LIX004, was established by implantation
of primary HCC tumor fragments from a 39-year-old female patient
subcutaneously into the right flank of SCID mice with Matrigel (BD
Biosciences, Franklin Lakes, NJ, USA) and subsequent passaging in
Nu/Nu mice. The primary tumor cells isolated from a tumor obtained
from vehicle-treated (10 ml/kg) or sorafenib-treated (40 mg/kg,
orally once daily, for ~100 days) mice were used for HCC cell line
establishment (Fig. 1) (13). All tissue culture media and
supplements were purchased from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). Tumor cells were suspended in serum-free medium
and were incubated continuously in a 37°C and 5% CO2
incubator (Thermo Fisher Scientific, Inc.). Half of the medium was
changed every 3–4 days and the cells were subcultured at 70–80%
confluence. When the cancer cell lines grew stably, the culture
medium was gradually changed to RPMI-1640 medium supplemented with
10% fetal bovine serum and10 µg/ml human insulin. Cultures were
tested negative for Mycoplasma contamination using polymerase chain
reaction analysis (MycoScan™ Mycoplasma Detection Kit; HD
Biosciences, Shanghai, China), according to the manufacturer's
protocol. Cells in the exponential growth phase were used in
subsequent experiments following passage 20.
Cells
HUVECs were obtained from All Cells-Shanghai
(Shanghai, China) and were used in studies within 7 passages.
Chromosome analysis
Metaphase cells were arrested using colchicines
(Sigma-Aldrich; Merck KGaA) and fixed in methanol/acetic acid
solution (3:1) prior to being stained with Giemsa solution (Thermo
Fisher Scientific, Inc.) at room temperature for 20 min. The slides
were mounted and the number of chromosomes was determined and
observed using an Eclipse microscope (magnification, ×1,000; Nikon
Instruments Inc., Melville, NY, USA). The frequency was analyzed
using Origin software (version 7.0; OriginLab, Northampton, MA,
USA).
Short-tandem repeat (STR) assay
Cell line purity was verified by microsatellite/STR
analysis. The genomic DNA was extracted and the STR amplicons were
analyzed using an AmpF/STR® Identifiler® PCR
Amplification kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols.
In vivo xenograft studies
In total, ~150 female Nu/Nu nude mice aged 6–8 weeks
were used in various in vivo studies. Cultured tumor cells
were subcutaneously injected into the right flank of the mice
(5.0×106 cells/mouse) with 50% Matrigel. Animals with a
tumor volume of 100–150 mm3 (~60%) were selected and
randomized for daily treatment with either the vehicle or sorafenib
(n=8 or 10 per group). Un-enrolled animals (~40%) were euthanized
with CO2 anesthesia. Treated animals were monitored
daily with tumor volume and body weight was recorded ≥2 times per
week. If animals exhibited signs of distress, weight loss >20%
of initial body weight and/or tumor volume >2,000
mm3, they were sacrificed.
Immunohistochemical (IHC)
analysis
Cells were cultured as a monolayer on sterile
chamber slides prior to being fixed with 4% formaldehyde at 4°C
overnight. Tumor tissues were 4% formaldehyde-fixed at 4°C for 48 h
and paraffin-embedded and 5 µm slides were prepared. The slides
were rehydrated in Xylene and a descending alcohol series. Antigen
heat retrieval (at 100°C) was performed in EDTA buffer (pH 9.0) or
citrate buffer (pH 6.0, all from Fuzhou Maixin Biotech. Co., Ltd.,
Fuzhou, Fujian, China) and washed in running water and then the IHC
wash buffer (Fuzhou Maixin Biotech Co., Ltd.). The slides were
blocked in 4% goat serum (Thermo Fisher Scientific, Inc.) in PBS at
room temperature for 30 min, and then incubated with different
primary antibodies at 4°C overnight, including anti-cytokeratin
antibody (cat no. 130–080–101; diluted at 1:50; Miltenyi Biotec
GmbH, Bergisch, Gladbach, Germany), anti-α1-fetoprotein
(AFP) antibody (cat no. 7723-1; diluted at 1:200; Epitomics; Abcam,
Cambridge, MA, USA), anti-vimentin antibody (cat no. 2707-1;
diluted at 1:200; Epitomics; Abcam), and anti-CD34 antibody (cat
no. 2150-1; diluted at 1:200; Epitomics; Abcam), which were diluted
in the 4% goat serum blocking buffer. Slides were washed with the
wash buffer and then incubated with secondary horseradish
peroxidase-conjugated secondary antibody (cat no. K400311, Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA) at room
temperature for 30 min, and then slides were developed using the
3,3′-diaminobenzidine method (Fuzhou Maixin Biotech, Co., Ltd) at
room temperature for 4 min. Slides were then mounted and scanned
using a ScanScope XT slide scanner (Aperio Group LLC, Sausalito,
CA, USA) and CD34+ areas were quantified using Image
Scope software (vision 11.0, Aperio Group LLC).
Conditioned medium (CM)
collection
LIXC-004NA or LIXC-004SR cells were seeded at a
density of 2×106 cells per 75-cm2 flask.
After 24 h, cells were washed twice with sterile PBS and medium was
changed to serum-free medium (RPMI-1640) supplemented with 0.2%
bovine serum albumin (purchased from Thermo Fisher Scientific,
Inc.) and incubated at 37°C for a further 48 h. Culture supernatant
was collect and passed through a 0.45-µm filter.
HUVEC proliferation and western blot
analysis
A HUVEC proliferation assay was performed with the
cells inoculated into the poly-D-lysine-coated 96-well plates (BD
Biosciences) and incubated at 37°C overnight (2,000 cells/well).
Culture medium was changed to 50% LIXC-004NA or LIXC-004SR CM and
incubated at 37°C for another 48 h. Cells were fixed in 4%
formaldehyde at 4°C overnight and treated with 0.1% Triton X-100 at
room temperature for 10 min. Cell numbers were then quantified
using Acumen eX3 (TTP Labtech Ltd., Melbourn, UK) following
staining with 1.5 µM propidium iodide (Thermo Fisher Scientific,
Inc.) at room temperature for 15 min in the dark.
HUVECs were seeded onto 6-well plates and incubated
at 37°C overnight, then serum-starved for 3 h, followed by compound
treatment for 1 h prior to being stimulated with LIXC-004NA or
LIXC-004SR CM for 10 min. Cells were washed with PBS twice, and
western blot analysis was performed on the total protein.
Cell or tumor samples were lysed in cell lysis
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA)
containing a Halt protease inhibitor (Thermo Fisher Scientific,
Inc.) and a phosphatase inhibitor cocktail (Sigma-Aldrich; Merck
KGaA). The protein concentration was determined using a
bicinchoninic acid method (Beyotime Institute of Biotechnology,
Haimen, China) and a total of 20 µg protein per sample was loaded
onto NuPAGE™ 4–12% Bis-Tris Protein Gels. Proteins were transferred
onto nitrocellulose membranes (all from Thermo Fisher Scientific,
Inc.). Membranes were incubated with blocking buffer (LI-COR
Biosciences, Lincoln, NE, USA) at room temperature for 1 h and
incubated with primary antibodies at 4°C overnight, including
anti-extracellular-signal-regulated kinases (ERK) 1/2 antibody (cat
no. 4695), phosphorylated (pho)-ERK1/2 (Thr202/Tyr204) antibody
(cat no. 4370), protein kinase B (Akt) (cat no, 9272) antibody and
pho-Akt (Ser437) antibody (Cat. No. 4060, all primary antibodies
were diluted at 1:1,000, and purchased from Cell Signaling
Technology, Inc.), followed by the Alexa Fluor 680-conjugated
secondary antibody incubation (cat no. A21076; diluted at 1:10,000;
Thermo Fisher Scientific, Inc.) at room temperature for 60 min.
Blots were scanned using Odyssey systems (LI-COR Biosciences).
Anti-GAPDH antibody (cat no. 5632-1; diluted at 1:2,000; Epitomics;
Abcam) was used as a loading control.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from each cell line using an
RNeasy kit (Qiagen, Inc., Valencia, CA, USA) and complementary DNA
(cDNA) was produced from 1 µg RNA using a RevertAid First Strand
cDNA Synthesis kit (Thermo Fisher Scientific, Inc.). TaqMan primers
for matrix metalloproteinase-1 (MMP-1; Hs00899658_m1), fibroblast
growth factor 5 (FGF5; Hs00738132_m1), vascular endothelial growth
factor A (VEGFA; Hs00899658_m1) and GAPDH (Hs02758991_g1) were used
for RT-qPCR on a ViiA 7 System (Thermo Fisher Scientific, Inc.).
All TaqMan primers were purchased from Thermo Fisher Scientific,
Inc., and a TaqMan gene expression master mix kit (Thermo Fisher
Scientific, Inc.) was used according to the manufacturer's
protocol. Each sample was assayed in duplicate and the ∆∆Cq method
was used to determine relative gene expression levels (14).
Statistical analysis
Data are presented as the mean ± standard deviation
and the data of the in vivo efficacy studies are expressed
as the mean ± standard error of the mean. Statistical differences
between groups were analyzed using Student's t-test, one-way
analysis of variance (ANOVA) or two-way ANOVA with Bonferroni post
hoc test using Prism (version 5; GraphPad Software, La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
LIXC-004SR cells are generated from
sorafenib-treated HCC PDX tumors and exhibit signs of multi-drug
resistance
To reproduce the acquired sorafenib resistance,
Nu/Nu mice bearing LIX004 PDX tumors were treated with 40 mg/kg
sorafenib daily. Despite retardation of tumor progression, the
animals were not cured and, following ~100 days of sorafenib
treatment, all animals developed tumors, with certain tumors
reaching a volume of nearly 2,000 mm3 (Fig. 1).
To determine the mechanisms of sorafenib resistance
in the tumor, primary tumor cell lines were established. The cell
line generated from the tumor, which had developed following
long-term sorafenib treatment, was LIXC-004SR, and the cell line
derived from a vehicle-treated tumor was LIXC-004NA (Fig. 1). LIXC-004SR cells exhibited a
slightly shorter in vitro doubling time when compared with
that of the LIXC-004NA cells (24.67 and 32.37 h, respectively).
LIXC-004SR displayed less cytokeratin expression and each cell line
expressed vimentin (Fig. 2A). STR
analyses revealed that the two cell lines matched perfectly with
the original PDX model (Fig. 2B) and
thus that the two cell lines and the PDX had been derived from the
same individual.
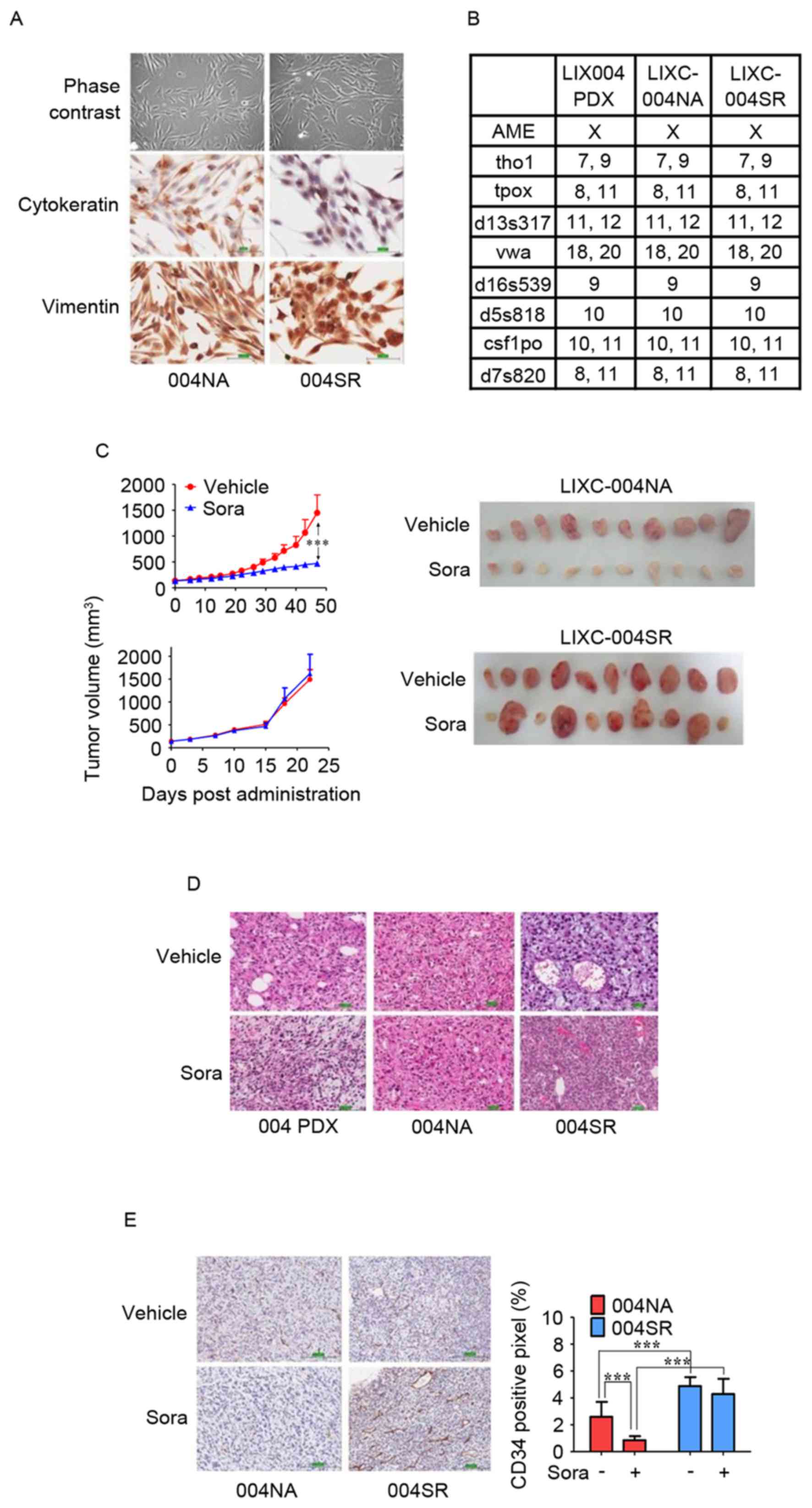 | Figure 2.In vitro and in vivo
characterization of LIXC-004NA and LIXC-004SR cell lines. (A) IHC
staining of the cultured cells under phase contrast microscopy
(upper panels), cytokeratin (middle panels) and vimentin (lower
panels; magnification, ×200). (B) Short-tandem repeat analyses of
the two cell lines and the original PDX tumor. (C) Tumor growth
curves (left panels) and images (right panels) of the tumors in
response to treatment with vehicle or sorafenib. (D) Hematoxylin
and eosin staining of original 004 PDX tumor (left panels), 004NA
tumor (middle panels) and 004SR tumor (right panels; magnification,
×100). (E) IHC using anti-CD34 antibody (left panels;
magnification, ×100), and blood vessel density by quantification of
the percentage of CD34+ pixels (right panel).
***P<0.001 by two-way analysis of variance. IHC,
immunohistochemistry; 004 PDX, LIX-004 PDX; 004NA, LIXC-004NA;
004SR, LIXC-004SR; Sora, sorafenib; CD, cluster of
differentiation. |
Furthermore, the two cell lines exhibited similar
sensitivity to sorafenib, fluorouracil and docetexal, while
LIXC-004SR displayed a decreased sensitivity to doxorubicin,
vinblastine and erlotinib (Table I),
despite the fact that the model had never been exposed to these
drugs previously.
 | Table I.In vitro inhibition of
hepatocellular carcinoma tumor cell growth by different drug
therapies. |
Table I.
In vitro inhibition of
hepatocellular carcinoma tumor cell growth by different drug
therapies.
|
| IC50,
µM |
|---|
|
|
|
|---|
| Cell line | Sorafenib | Docetaxel | Doxorubicin | Vinblastine | 5-FU | Erlotinib |
|---|
| LIXC-004NA | 13.295 | >20 | 0.675 | <0.032 | >200 | 13.998 |
| LIXC-004SR | 10.110 | >20 | >10 | 0.306 | >200 | >200 |
Sorafenib resistance of the LIXC-004SR
xenograft in vivo
The two cell lines were tested for tumorigenicity
in vivo and were able to produce xenograft tumors (Fig. 2C). When reviewed by a pathologist, the
two xenografts were diagnosed as poorly differentiated HCC, as was
the original PDX model (Fig. 2D).
When the average tumor size reached 100–150
mm3, the tumor-bearing animals were randomized and
treated with vehicle or 40 mg/kg sorafenib daily. The xenograft
derived from the LIXC-004NA cell line responded to sorafenib
treatment. Smaller, paler tumors were observed in the
sorafenib-treated animals (Fig. 2C),
suggesting that angiogenesis and tumor growth had been successfully
inhibited. In contrast, when LIXC-004SR tumor-bearing animals were
treated with sorafenib, 6/10 mice displayed tumors of a similar
size when compared with those displayed by the vehicle group, while
the other 4 animals developed tumors that were larger than any of
those displayed by the vehicle control group (Fig. 2C), suggesting that the LIXC-004SR
tumors were resistant to sorafenib in a number of the animals. The
studies were repeated twice and similar tumor growth and drug
response profiles were observed. Notably, the LIXC-004SR tumors
grew faster than the LIXC-004NA tumors, using approximately half of
the time to reach a tumor volume of ~1,500 mm3 (Fig. 2C). In light of this, LIXC-004SR
represents one of the few acquired sorafenib-resistant tumor models
established from a PDX model.
Activation of the sorafenib-resistant
angiogenic pathway in the LIXC-004SR xenograft
IHC was performed to quantify the state of
angiogenesis in the two tumor models (in vivo with or without
sorafenib treatment). LIXC-004SR tumors that were not treated with
sorafenib exhibited increased blood vessel density compared with
the LIXC-004NA tumors (Fig. 2E),
which may contribute to the faster tumor growth observed in
vivo. Notably, in accordance with the tumor appearance,
sorafenib significantly reduced blood vessel density in LIXC-004NA
tumors, but not in LIXC-004SR tumors (Fig. 2E), indicating that an alternative
angiogenic pathway may contribute to the sorafenib resistance in
LIXC-004SR tumors.
Western blot analyses of the tumors indicated that
sorafenib treatment induced similar changes in MEK, Akt and ERK
phosphorylation in LIXC-004NA and LIXC-004SR tumors (data not
shown), suggesting that the direct tumor cell inhibition or death
may not contribute to the differences in sorafenib responses
between these tumors. Microarray analyses were performed on the
LIXC-004SR and LIXC-004NA cells. Among the differentially expressed
genes, a group of genes involved in angiogenesis, including MMP-1,
FGF5 and VEGFA, were upregulated in the LIXC-004SR cells when
compared with expression in the LIXC-004NA cells (data not shown).
This over-expression was confirmed by RT-qPCR analyses (Fig. 3A).
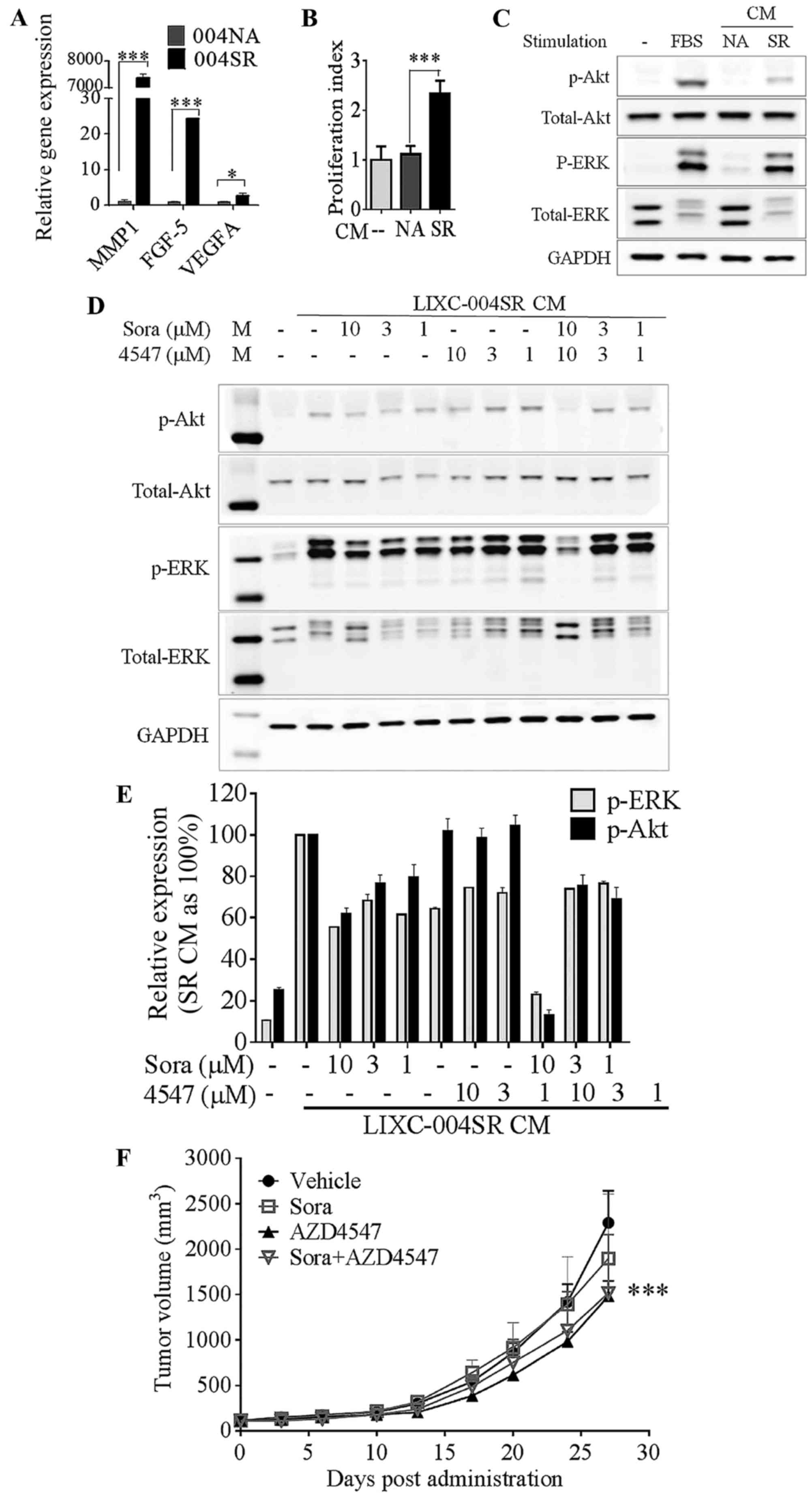 | Figure 3.LIXC-004SR activates an alternative
angiogenic pathway. (A) Reverse transcription-quantitative
polymerase chain reaction analyses of selected differentially
expressed genes; MMP-1, FGF-5 and VEGF-A. *P<0.05,
***P<0.0001; two-way ANOVA with Bonferroni post tests. (B) HUVEC
proliferation. ***P<0.001, one-way ANOVA. (C) HUVEC Akt and ERK
phosphorylation determination by western blot analysis. (D) Western
blot analyses of 004SR supernatant-induced HUVEC Akt and ERK
phosphorylation in the presence of sorafenib and AZD4547 (upper
panel) and quantification of the western blot analyses (lower
panel, data represent two separate experiments). (E) 004SR
tumor-bearing mice were treated with vehicle, sorafenib, AZD4547,
or the combination of AZD4547 and sorafenib. (F) AZD4547 inhibited
tumor progression (***P<0.001 vs. the vehicle control group on
day 27; two-way ANOVA with Bonferroni post hoc tests). The study
was repeated once with similar results. 004SR, LIXC-004SR; MMP-1,
matrix metalloproteinase 1; FGF-5, fibroblast growth factor 5;
VEGF-A, vascular endothelial growth factor A; ANOVA, analysis of
variance; HUVEC, human umbilical vein endothelial cells; ERK,
extracellular-signal-regulated kinase; CM, conditioned medium. |
LIXC-004SR and LIXC-004NA culture supernatant was
used in further analyses, as soluble factors mediate tumor-host
communication in vivo. LIXC-004SR, but not LIXC-004NA,
culture supernatant was able to significantly induce HUVEC
proliferation (Fig. 3B), and induce
Akt and ERK phosphorylation in HUVECs (Fig. 3C), suggesting that pro-angiogenic
factors were present in the LIXC-004SR culture supernatant. This
HUVEC Akt and ERK phosphorylation was only inhibited by the
combination of sorafenib and an FGFR1 selective inhibitor, AZD4547
almost completely (Fig. 3D). These
data suggest that LIXC-004SR cells were able to induce endothelial
cell signaling and proliferation that was insensitive to sorafenib
treatment. The FGF pathway may be one of the mechanisms
involved.
To examine whether or not the FGF pathway serves a
role in tumor growth in vivo, LIXC-004SR tumor-bearing mice
were treated with vehicle, sorafenib, AZD4547, or the combination
of sorafenib and AZD4547. The LIXC-004SR tumors were resistant to
sorafenib treatment. The tumor growth was inhibited by AZD4547 and
was not further inhibited by the addition of sorafenib (Fig. 3E). The results suggest that the FGF
pathway contributed towards LIXC-004SR tumor growth in
vivo.
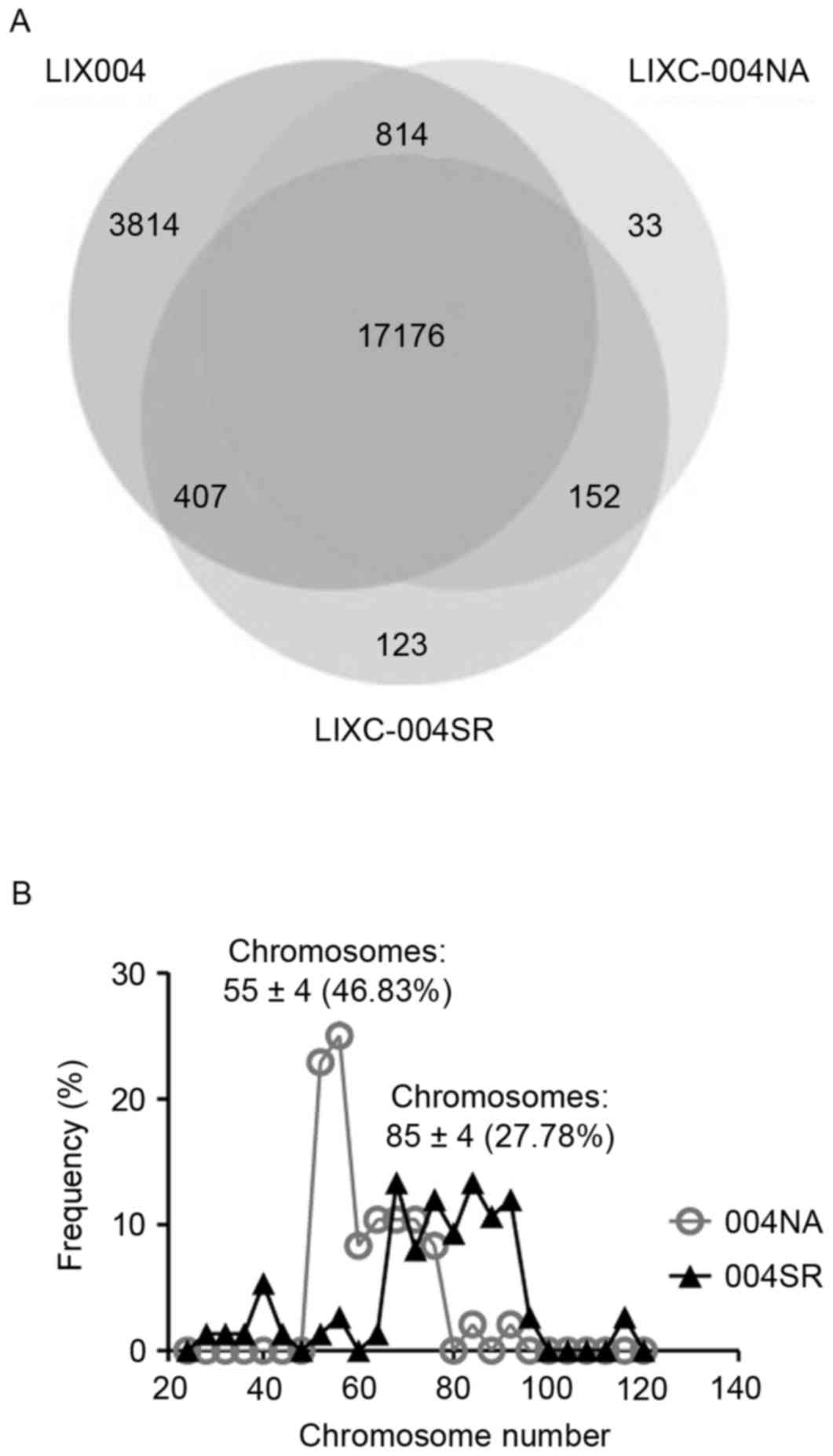
LIXC-004SR cells display genome
instability
Whole exome sequencing and RNA sequencing analyses
of the cell lines were performed and compared with the whole genome
sequencing data from the LIX-004 PDX tumor. It was revealed that
>98.5% of the single nucleotide polymorphisms (SNPs) observed in
the two cell lines were present in the LIX-004 PDX tumor (Fig. 4A), demonstrating that the cell lines
effectively preserved the genetic mutations of the PDX model.
Notably, 123 SNPs were revealed to be unique to LIX-004SR cells
compared with the 33 unique SNPs observed in the LIX-004NA
cells.
To investigate the potential source of this acquired
drug resistance, chromosomal analyses were performed, as genome
instability was suggested to be an enabling Hallmark of Cancer
(15). LIXC-004NA cells exhibited a
peak of 55±4 chromosomes with ~50% of the cells exhibiting a
chromosome number within this range (Fig.
4B). This number is close to the normal human chromosomal
number of 46. LIXC-004SR cells, on the other hand, presented with a
more diverse distribution of chromosomal numbers with a peak at
85±4 chromosomes and with only ~25% of the cells within the range
(Fig. 4B). These data suggest that
genome instability may be associated with the tumor size increase
observed following long-term sorafenib treatment.
Discussion
At present, sorafenib is the only approved targeted
therapy for liver cancer (5).
However, acquired sorafenib resistance has prevented patients from
experiencing the long-term benefits of this drug. A number of drug
resistance studies have focused on tumor cell lines either
naturally resistant or induced to become resistant in tissue
culture. Such ‘tumor-centric’ approaches have led to
‘tumor-centric’ mechanisms of drug resistance (16). However, these types of approaches do
not take into account the role served by tumor-host interaction. In
previous studies, tumor microenvironment/tumor stroma has been
revealed to contribute toward tumor drug resistance (17,18). The
present study used in vivo PDX models to recapitulate the
process of drug resistance in vivo, since PDX models are
able to preserve a number of the characteristics of clinical tumor
samples and the drug response is more strongly associated with
clinical drug efficacy when compared with conventional cell
line-derived xenograft models (19).
One of the drawbacks of the model used in the present study is that
it requires the use of immune-compromised animals, namely Nu/Nu
mice. These animals lack functional T cells and thus, do not
account for the mechanisms that these cells are involved in.
However, these animals have a number of the components of the tumor
microenvironment, including endothelial cells, fibroblasts,
pericytes, myeloid cells, B cells and the extracellular matrix
(19). In addition, certain key
factors in tumor-host interactions, including VEGF and
granulocyte-colony stimulating factor, may induce similar signals
through receptors in mice. Therefore, despite the lack of T cells,
PDX models possess several aspects of relevant tumor-host
interactions.
Another drawback of the system used in the present
study is that the tumor was implanted subcutaneously, instead of
orthotopically in the liver. This may lead to loss of the tumor
organ-microenvironment. By contrast, one of the key advantages of
PDX models is that the tumor is maintained in vivo and it
carries sufficient structural information to ensure its normal
growth behavior (19). In the present
study, adenocarcinomas formed gland-like structures despite having
been implanted subcutaneously and even did so following several
rounds of in vivo passaging (20). Furthermore, the pathological diagnosis
of these tumors was poorly differentiated HCC, as was the original
clinical diagnosis, indicating the preservation of at least some of
the clinical characteristics of the human tumor sample. In the
present study, the resistance of LIXC-004SR to sorafenib developed
in the presence of tumor-host interaction, making it a more
relevant model for drug resistance research.
In the present study, the FGF pathway is suggested
to be involved in acquired sorafenib resistance. This finding is
infrequently reported in ‘tumor-centric’ sorafenib-resistance
studies (21–24). However, this novel finding appears to
be reasonable, as the FGF pathway has been reported to be one of
the most dysregulated pathways in patients with HCC (6,25). FGF5
has been reported to be upregulated in pancreatic cancer patient
samples and to contribute to the malignant phenotype (16,26–28). A
more recent study revealed an association between FGF5 and a more
aggressive HCC phenotype, and suggested FGF5 as an oncogene in HCC,
and thus its potential as a novel therapeutic target for HCC
(29). Tovar et al (30) also revealed that FGF signaling
contributed toward sorafenib resistance in cell line-derived HCC
xenografts following sorafenib treatment in vivo.
In the present study, LIXC-004SR cells also
demonstrated signs of genome instability when compared with the
LIXC-004NA cells. As genome instability is an enabling Hallmark of
Cancer (15), it may contribute to
the acquired in vivo sorafenib resistance. Given the
heterogeneity of human cancer, particularly liver cancer, and the
chromosome instability observed in the present study, this type of
study is unlikely to cover all the potential mechanisms of acquired
drug resistance. The fact that the FGFR1 inhibitor only resulted in
a certain degree of inhibition of LIXC-004SR xenograft tumor growth
in vivo suggests that additional mechanism(s) exist in this
model.
Overall, the approach of the present study has
produced a relevant animal model of acquired sorafenib resistance
from a PDX model. From this in vivo model, the alternative
angiogenic pathways and genome instability, which were previously
overlooked in drug resistance studies focusing only on tumor cells
in tissue culture, were rediscovered. The model also exhibited
characteristics of multi-drug resistance. The generation of a
sister cell line, with an identical genetic background and only
differing in drug sensitivity, makes this pair of cell lines and
associated xenograft and PDX models valuable tools in the study of
drug resistance. The understanding and further utilization of this
model system may facilitate research and drug development regarding
the prevention and treatment of drug resistance, and will hopefully
contribute toward sustained therapeutic efficacy and long-term
survival in cancer patients.
Acknowledgements
The present study was supported by the Shanghai
Municipal Commission of Science and Technology (grant no.
14DZ2252000), the National Natural Science Foundation of China
(grant no. 81274146), the 333 High Level Project of Jiangsu
Province (grant no. BRA2014245) and Funding for Priority Academic
Program Development of Jiangsu Higher Education Institution (PAPD).
The authors would also like to thank Dr Charles Yang (Department of
Biology, Shanghai ChemPartner Co. Ltd.) for providing editorial
assistance and advice.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
PDX
|
patient-derived xenograft
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
research. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al: Sorafenib in advanced hepatocellular carcinoma. N Engl J
Med. 359:378–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Abou-Alfa GK, Schwartz L, Ricci S, Amadori
D, Santoro A, Figer A, De Greve J, Douillard JY, Lathia C, Schwartz
B, et al: Phase II study of sorafenib in patients with advanced
hepatocellular carcinoma. J Clin Oncol. 24:4293–4300. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, et al: Efficacy and safety of
sorafenib in patients in the Asia-Pacific region with advanced
hepatocellular carcinoma: A phase III randomized, double-blind,
placebo-controlled trial. Lancet. 10:25–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moeini A, Cornellà H and Villanueva A:
Emerging signaling pathways in hepatocellular carcinoma. Liver
Cancer. 1:83–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Santoro A, Rimassa L, Borbath I, Daniele
B, Salvagni S, Van Laethem JL, Van Vlierberghe H, Trojan J, Kolligs
FT, Weiss A, et al: Tivantinib for second-line treatment of
advanced hepatocellular carcinoma: A randomised, placebo-controlled
phase 2 study. Lancet Oncol. 14:55–63. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villarroel MC, Rajeshkumar NV,
Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, Hruban RH,
Eshleman JR, Klein A, Laheru D, et al: Personalizing cancer
treatment in the age of global genomic analyses: PALB2 gene
mutations and the response to DNA damaging agents in pancreatic
cancer. Mol Cancer Ther. 10:3–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hidalgo M, Bruckheimer E, Rajeshkumar NV,
Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B, Strawn S, Wick
MJ, Martell J and Sidransky D: A pilot clinical study of treatment
guided by personalized tumorgrafts in patients with advanced
cancer. Mol Cancer Ther. 10:1311–1316. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Das Thakur M, Salangsang F, Landman AS,
Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M and Stuart
DD: Modelling vemurafenib resistance in melanoma reveals a strategy
to forestall drug resistance. Nature. 494:251–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wilhelm SM, Carter C, Tang L, Wilkie D,
McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al:
BAY 43–9006 exhibits broad spectrum oral antitumor activity and
targets the RAF/MEK/ERK pathway and receptor tyrosine kinases
involved in tumor progression and angiogenesis. Cancer Res.
64:7099–7109. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gavine PR, Mooney L, Kilgour E, Thomas AP,
Al-Kadhimi K, Beck S, Rooney C, Coleman T, Baker D, Mellor MJ, et
al: AZD4547: An orally bioavailable, potent, and selective
inhibitor of the fibroblast growth factor receptor tyrosine kinase
family. Cancer Res. 72:2045–2056. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xin H, Wang K, Hu G, Xie F, Ouyang K, Tang
X, Wang M, Wen D, Zhu Y and Qin X: Establishment and
characterization of 7 novel hepatocellular carcinoma cell lines
from patient-derived tumor xenografts. PLoS One. 9:e853082014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhai B and Sun XY: Mechanisms of
resistance to sorafenib and the corresponding strategies in
hepatocellular carcinoma. World J Hepatol. 5:345–352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Olive KP, Jacobetz MA, Davidson CJ,
Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA,
Caldwell ME, Allard D, et al: Inhibition of Hedgehog signaling
enhances delivery of chemotherapy in a mouse model of pancreatic
cancer. Science. 324:1457–1461. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sebens S and Schafer H: The tumor stroma
as mediator of drug resistance-a potential target to improve cancer
therapy? Curr Pharm Biotechnol. 13:2259–2272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sausville EA and Burger AM: Contributions
of human tumor xenografts to anticancer drug development. Cancer
Res. 66:3351–3354. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu G, Li F, Ouyang K, Xie F, Tang X, Wang
K, Han S, Jiang Z, Zhu M, Wen D, et al: Intrinsic gemcitabine
resistance in a novel pancreatic cancer cell line is associated
with cancer stem cell-like phenotype. Int J Oncol. 40:798–806.
2012.PubMed/NCBI
|
|
21
|
Chen KF, Chen HL, Tai WT, Feng WC, Hsu CH,
Chen PJ and Cheng AL: Activation of phosphatidylinositol
3-kinase/Akt signaling pathway mediates acquired resistance to
sorafenib in hepatocellular carcinoma cells. J Pharmacol Exp Ther.
337:155–161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Serova M, de Gramont A, Tijeras-Raballand
A, Dos Santos C, Riveiro ME, Slimane K, Faivre S and Raymond E:
Benchmarking effects of mTOR, PI3K, and dual PI3K/mTOR inhibitors
in hepatocellular and renal cell carcinoma models developing
resistance to sunitinib and sorafenib. Cancer Chemother Pharmacol.
71:1297–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tai WT, Cheng AL, Shiau CW, Liu CY, Ko CH,
Lin MW, Chen PJ and Chen KF: Dovitinib induces apoptosis and
overcomes sorafenib resistance in hepatocellular carcinoma through
SHP-1-mediated inhibition of STAT3. Mol Cancer Ther. 11:452–463.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van Malenstein H, Dekervel J, Verslype C,
Van Cutsem E, Windmolders P, Nevens F and van Pelt J: Long-term
exposure to sorafenib of liver cancer cells induces resistance with
epithelial-to-mesenchymal transition, increased invasion and risk
of rebound growth. Cancer Lett. 329:74–83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sawey ET, Chanrion M, Cai C, Wu G, Zhang
J, Zender L, Zhao A, Busuttil RW, Yee H, Stein L, et al:
Identification of a therapeutic strategy targeting amplified FGF19
in liver cancer by Oncogenomic screening. Cancer Cell. 19:347–358.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kornmann M, Ishiwata T, Beger HG and Korc
M: Fibroblast growth factor-5 stimulates mitogenic signaling and is
overexpressed in human pancreatic cancer: Evidence for autocrine
and paracrine actions. Oncogene. 15:1417–1424. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian X, Chen G, Zhou S, Henne-Bruns D,
Bachem M and Kornmann M: Interactions of pancreatic cancer and
stellate cells are mediated by FGFR1-III isoform expression.
Hepatogastroenterology. 59:1604–1608. 2012.PubMed/NCBI
|
|
28
|
Allerstorfer S, Sonvilla G, Fischer H,
Spiegl-Kreinecker S, Gauglhofer C, Setinek U, Czech T, Marosi C,
Buchroithner J, Pichler J, et al: FGF5 as an oncogenic factor in
human glioblastoma multiforme: Autocrine and paracrine activities.
Oncogene. 27:4180–4190. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fang F, Chang RM, Yu L, Lei X, Xiao S,
Yang H and Yang LY: MicroRNA-188-5p suppresses tumor cell
proliferation and metastasis by directly targeting FGF5 in
hepatocellular carcinoma. J Hepatol. 63:874–885. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tovar V, Cornella H, Moeini A, Vidal S,
Hoshida Y, Sia D, Peix J, Cabellos L, Alsinet C, Torrecilla S, et
al: Tumour initiating cells and IGF/FGF signalling contribute to
sorafenib resistance in hepatocellular carcinoma. Gut. 66:530–540.
2017. View Article : Google Scholar : PubMed/NCBI
|