Introduction
Pheochromocytoma, or paraganglioma (PPGL), is a rare
condition with an incidence of 1 per 100,000–300,000 (1,2). It
originates from chromaffin cells of the adrenal medulla and the
sympathetic and parasympathetic ganglia, and is observed in a wide
range of body parts from the skull base to the pelvic cavity.
Typical manifestations include paroxysmal hypertension, headaches,
palpitations and sweating induced by excess catecholamines.
Furthermore, PPGLs also secrete other hormones or cytokines,
including adrenocorticotropic hormone (ACTH), interleukin (IL)-6
and parathyroid hormone-related protein (PTHrp) (3,4). Patients
with PPGL could also manifest Cushing syndrome, fever and
hypercalcemia. However, certain patients have no clear symptoms. An
incidental adrenal tumor could be the first reason for seeing a
doctor.
ACTH-secreting pheochromocytoma is very rare and, to
date, ~40 cases have been reported (3,5–14). The majority of the tumors involved
unilateral adrenal glands, with the exception of two cases of
bilateral pheochromocytoma reported in 1988 and 2014 (5,13). The two
patients carried a germline RET gene mutation (5,13).
Surgery is the primary therapy in ectopic ACTH
syndrome. Medicines such as ketoconazole that could decrease
cortisol production are another option in ectopic ACTH syndrome,
particularly in the malignant form of the disease. Few drugs are
available for use in patients with ectopic ACTH syndrome in China;
therefore, it is necessary to find an available drug to treat the
condition.
The current study presents a third case of bilateral
ACTH-secreting pheochromocytoma, in which no germline mutation was
observed. The present study extensively examined the tumor in
vivo and in vitro in order to gain further insight into
the histopathological features, hormonal secretion pattern and
possible therapeutic medicine involved.
Patients and methods
Patient
A 54-year-old male who was unintentionally revealed
to have bilateral adrenal masses (upon physical examination for
potential coronary artery disease due to mild palpitations and
shortness of breath) was referred to Peking Union Medical College
Hospital (Peking Union Medical College, Chinese Academy of Medical
Sciences, Beijing, China). The patient had a history of
hypertension for 7 years, with a blood pressure that was stably
controlled at 140/90 mmHg by enalapril. The patient did not report
having experienced paroxysmal hypertension, headache, palpitation,
sweating, postural hypotension, constipation or weight change over
the preceding 7 years. The patient also did not report having
experienced thin skin, easy bruising or muscle weakness. Physical
examination revealed a body mass index of 28.6 kg/m2 and
a waist circumference of 97 cm. The patient presented with a mild
conjunctival edema but exhibited no signs of moon face,
supraclavicular fat, purple striae or facial plethora. Examination
did not reveal any café-au-lait spot, neurofibroma or Lisch
nodules. Laboratory tests revealed that ACTH-dependent
hypercortisolemia and normal urinary catecholamine excretion
(Table I). An enhanced computed
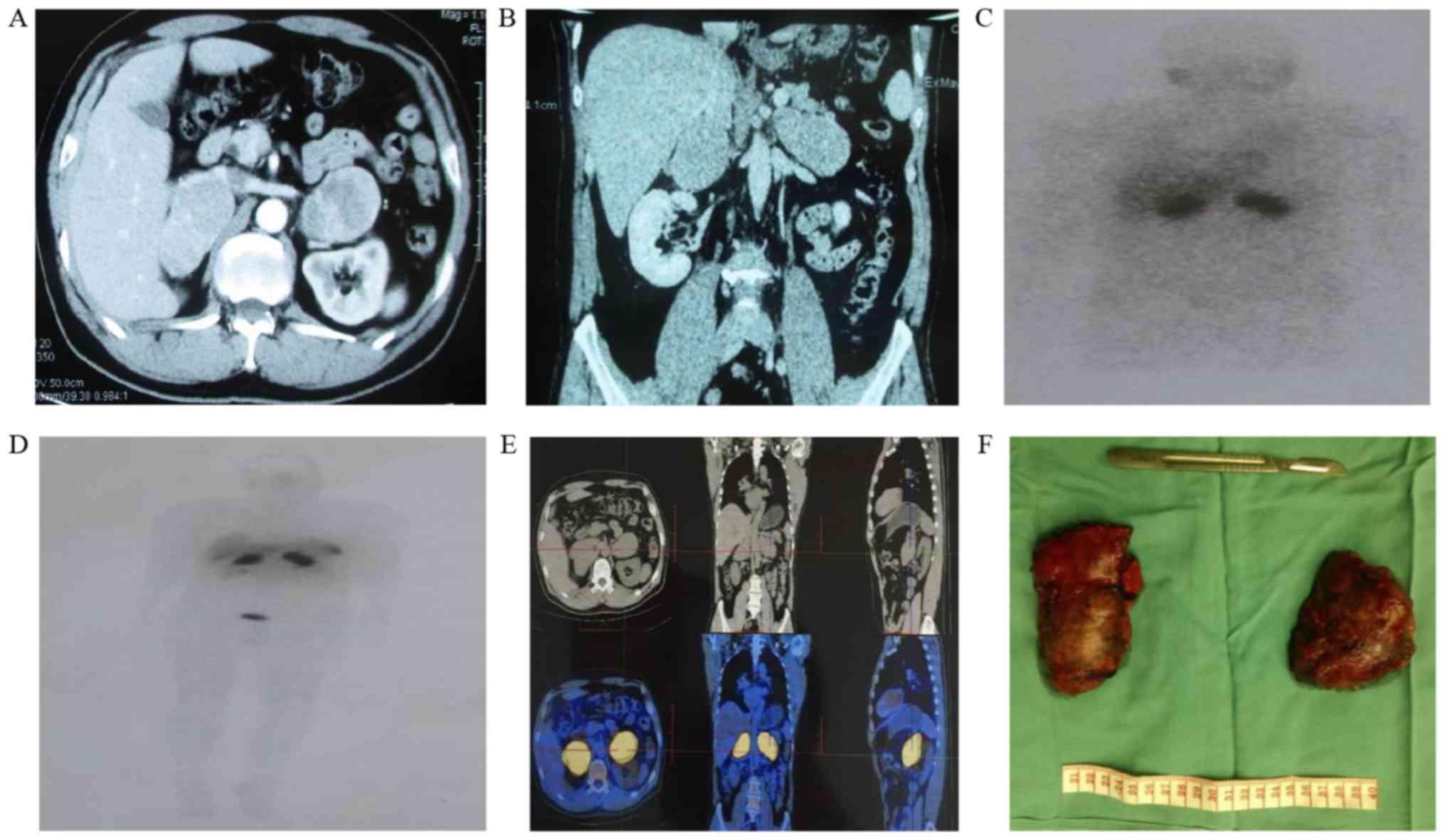
tomography (CT) scan revealed a 62×53 mm tumor in the left adrenal
gland and a 68×57 mm tumor in the right adrenal gland (Fig. 1A and B). The Hounsfield unit values in
pre-contrast, arterial and delayed phases were 41, 83 and 58,
respectively. 131I-metaiodobenzylguanidine scintigraphy
(Fig. 1C) and
99Tm-somatostatin receptor scintigraphy (Fig. 1D) exhibited intense focal uptake in
the bilateral adrenal tumors. An 18F-fluorodeoxyglucose
positron emission tomography scan revealed that the bilateral
tumors exhibited a maximum standardized uptake value of 32
(Fig. 1E), while no metastasis was
observed.
 | Table I.Clinical profiles. |
Table I.
Clinical profiles.
| Variables | On admission | 3 months after
surgery | Reference range |
|---|
| BP, mmHg | 140/90 | 130/80 |
|
| Antihypertension
drugs, mg | Enalapril 10 bid | Nifedipine 30 qd |
|
|
|
| Metaprolol 25
bid |
|
| Blood potassium,
mmol/l | 3.5 | 4.5 | 3.5–5.5 |
| FBG, mmol/l | 6.26 | 5.5 | 3.9–6.1 |
| HbA1C, % | 6.2 | ND | <6.5 |
| Cortisol, µg/dl | 24.57 | <0.5b | 4.0–22.3 |
| Overnight DST,
µg/dl | 21.29 | ND | 4.0–22.3 |
| ACTH, pg/ml | 17.1/23.8 | 96.9 | 0–46 |
| NE, µg/day | 25.26 | 40.24 | 16.69–40.65 |
| E, µg/day | 2.60 | 2.68 | 1.74–6.42 |
| DA, µg/day | 294.23 | 268.27 | 120.93–330.59 |
| UFC day ½,
µg/daya | 497.7/775.32 | ND | 12.3–103.5 |
| UFC day 4,
µg/daya | 788.97 | ND |
|
| UFC day 6,
µg/daya | 677.43 | ND |
|
Based on the aforementioned findings, the patient
was diagnosed with bilateral pheochromocytoma with ectopic ACTH
secretion. Measurements of serum calcitonin, parathyroid hormone,
calcium and phosphorus levels, as well as thyroid morphology, did
not support a diagnosis of multiple endocrine neoplasia type II.
Ocular fundus examination and abdominal CT did not support a
diagnosis of von Hippel-Lindau disease.
Upon receiving 4 mg doxazosin once a day for 1
month, the patient underwent bilateral resection of the adrenal
tumors and left adrenalectomy. Gross pathological examination
revealed two well-encapsulated, grey-red, oval-shaped solitary
lesions (Fig. 1F). The specimens were
pathologically confirmed to be bilateral pheochromocytomas.
The patient was administered with prednisone
immediately after surgery and, three months later, hormone profiles
were evaluated (Table I). The
patient's blood pressure remained abnormal, but was controlled at
130/80 mmHg by 30 mg nifedipine once a day and 25 mg metoprolol
once a day. Postoperatively, the patient did not notice any
difference in symptoms, but gained 2 kg in weight.
The present study was approved by the Ethics
Committee for Human Research of Peking Union Medical College
Hospital (IRB approval number: S-K222). Written informed consent
for publication was acquired from the patient and the control
cases.
Mutational analysis
Genomic DNA was extracted from peripheral blood
leukocytes using DNeasy Blood kits (Qiagen GmbH, Hilden, Germany)
and subjected to whole-exome sequencing by Novogene Corporation
(Chula Vista, CA, USA).
Immunohistochemistry
Tumor tissues were immersed in 4% paraformaldehyde
for 24 h and hydrated through a serial alcohol gradient, prior to
being embedded in paraffin wax blocks. Tissue sections (4-µm) were
dewaxed in xylene and rehydrated using decreasing concentrations of
ethanol. Antigens were unmasked by pressure cooker in citrate
buffer with pH 6.0. ACTH (dilution, 1:50; catalog no. M3501; Dako;
Agilent Technologies, Inc., Santa Clara, CA, USA), CYP11B1
(dilution, 1:200), Ki-67 (ready to use; catalog no. ZA-0525;
Zhongshanjinqiao, Beijing, China), chromogranin A (CgA; ready to
use; catalog no. ZA-0507; Zhongshanjinqiao), synaptophysin (Syn;
ready to use; catalog no. PA0299, Leica Microsystems GmbH, Wetzlar,
Germany), melan-A (dilution 1:50; catalog no. M7196; Dako; Agilent
Technologies, Inc.), anti-pan-cytokeratin (AE1/AE3) (dilution 1:50;
catalog no. M3515; Dako; Agilent Technologies, Inc.),
neuron-specific enolase (NSE; dilution 1:50, catalog no. M0873;
Dako; Agilent Technologies, Inc.) and S-100 (ready to use; catalog
no. GA504; Dako; Agilent Technologies, Inc.) were
immunohistochemically detected. Primary antibodies were incubated
at 37°C for 2 h. Endogenous peroxidase was blocked by 3% hydrogen
peroxide. The rat monoclonal antibody against CYP11B1 was provided
by Dr Gomez-Sanchez (Department of Medicine, University of
Mississippi Medical Center, Jackson, MS, USA). The immunostaining
sections were observed by light microscopy.
Electron microscopy
Approximately 1 mm3 of fresh resected
tumor tissue was fixed with 2.5% glutaraldehyde in 0.1 M phosphate
buffer at 4°C for >4 h, and post-fixed in 1% osmium tetroxide
for 1 h, dehydrated in graded alcohol and embedded into Epon 812
overnight at 37°C. The 50-nm ultrathin sections were stained with
uranyl acetate and lead citrate for 15 min, respectively.
Subsequently, they were examined under an electron microscope
JEM-IOIO, as previously described (15). The scale of electron microscope of
Fig. 3A-D was 2 µm, 500 nm, 1 and 1
µm respectively.
Tissue homogenization
A total of 25 mg tumor tissue was homogenized in 1
ml 0.9% sodium chloride (16). The
homogenate was collected for measurement of ACTH. Another three
patients from the same hospital served as control cases. The tumor
tissues from the control cases were preserved in liquid nitrogen
immediately after resection. These 3 patients were diagnosed with
pheochromocytomas without cortisol hypersecretion. The clinical
profiles are shown in Table II.
| Table II.Clinical and pathological profiles of
the 3 control cases. |
Table II.
Clinical and pathological profiles of
the 3 control cases.
| Case no. | Gender/age,
years | Manifestation | ACTH, pg/ml | UFC, µg | Location | Urine catecholamine,
µg |
131I-MIBG | Date of surgery | Pathology |
|---|
| 1 | M/44 | Paroxysmal
hypertension, pectoralgia and cold extremities | 20.7 | 19.2 | Right adrenal | Normal | Positive | July, 2016 | Pheochromocytoma |
| 2 | M/43 | Paroxysmal
hypertension and pectoralgia | 16.5 | 75.2 | Right adrenal | Normal | Positive | September, 2014 |
Pheochromocytoma |
| 3 | F/43 | Paroxysmal
headache, palpitation and hypertension | 23.8 | 41.3 | Right adrenal | Normal | Positive | November, 2016 |
Pheochromocytoma |
Primary cell culture of the tumor
Tumor tissue was placed in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) immediately after resection. The medium was collected for the
measurement of ACTH and catecholamines. Tumor tissue was digested
by 2% collagenase type I. Dispersed cells were centrifuged at 150 ×
g for 5 min at room temperature and subsequently re-suspended in
complete medium (DMEM containing 15% fetal bovine serum, 100 U/ml
penicillin and 100 µg/ml streptomycin) as previously described
(17). On day 4, the medium was
refreshed and the cells were treated with AZD8055 (1 mM; Selleck
Chemicals, Houston, TX, USA), or with the vehicle for 24 h. Culture
medium was harvested for the detection of ACTH and
catecholamines.
Statistical analysis
The data are expressed as the mean ± standard
deviation for the in vitro study. Student's t-test was
employed to compare the difference between two groups. P<0.05
was considered to indicate a statistically significant difference.
The statistical analyses were performed using the SPSS 21.0
software package (IBM Corp., Armonk, NY, USA).
Results
Mutational analysis
Whole-exome sequencing revealed that the following
19 pheochromocytoma-related genes: RET, VHL, NF1, SDHA, SDHB,
SDHC, SDHD, SDHAF2, HIF2A, FH, PHD1, PHD2, PHD3, HRAS, MDH2,
KIF1Bβ, MAX, TMEM127 and BAP1 were unmutated.
Pathological examination and
immunohistochemistry
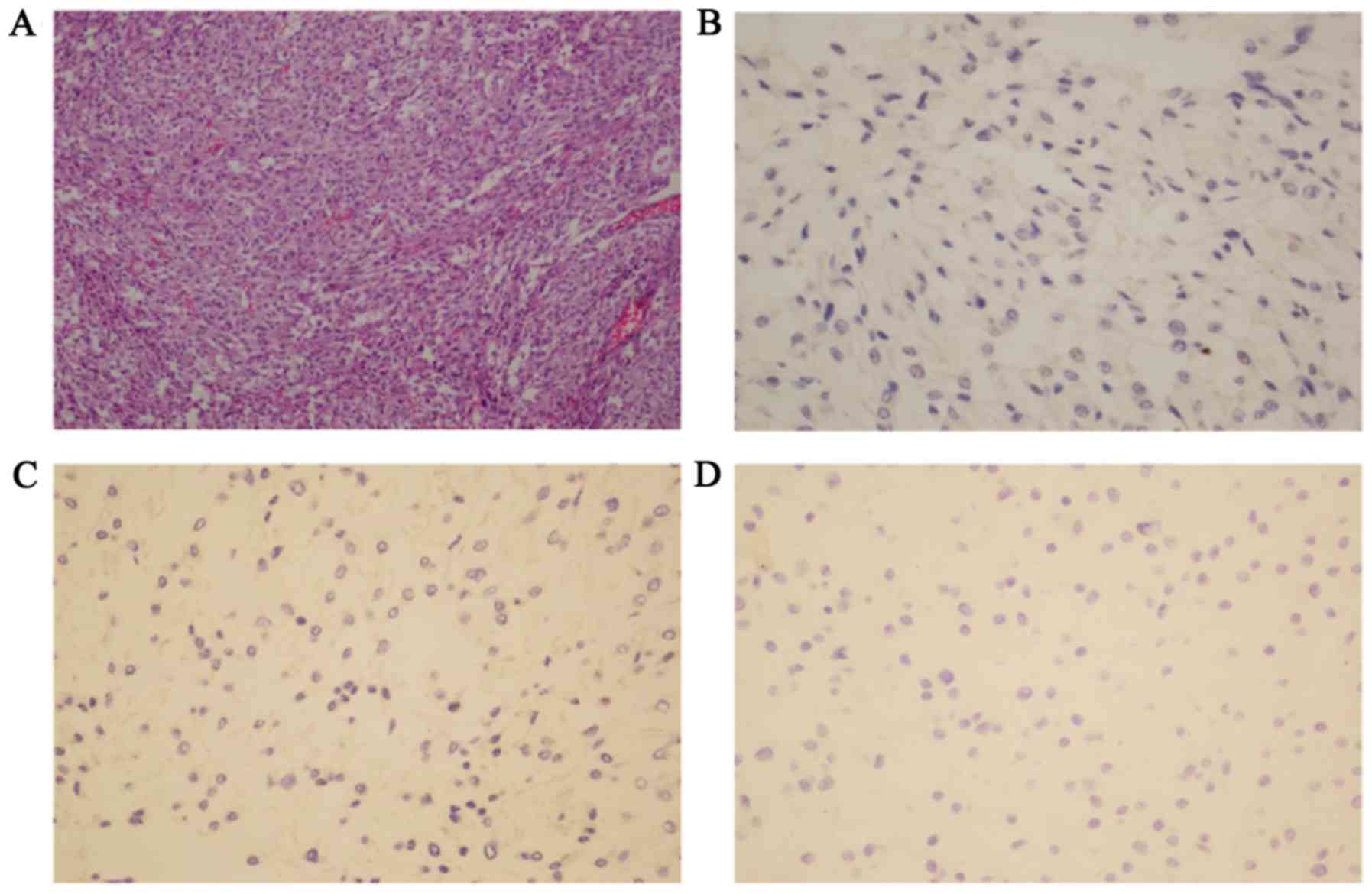
H&E staining (Fig.
2A) revealed that the tumor tissue was composed of round or
polygonal epithelioid cells arranged in a trabecular pattern. The
cells contained centrally-located nuclei with finely clumped
chromatin and a moderate amount of eosinophilic, granular
cytoplasm.
Immunohistochemically, the left and right side
pheochromocytomas were negatively stained for ACTH, CYP11B1,
Melan-A and S-100. The tumors were positively stained for NSE, CgA
and Syn, with scattered positivity for AE1/AE3. The overall
positive ratio for the Ki-67 index was <1% (Fig. 2B-D).
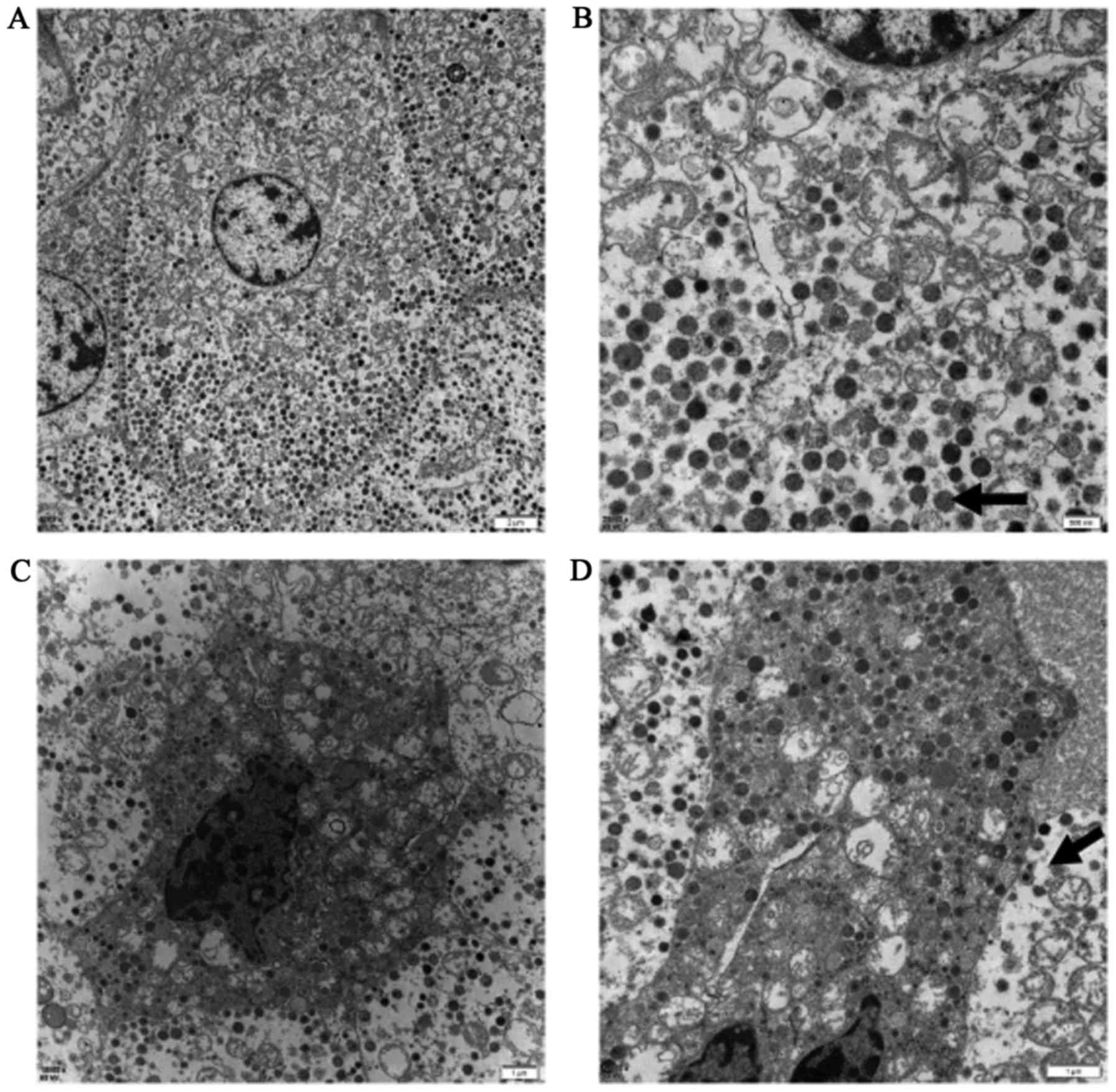
Electron microscopy
Electron microscopy demonstrated that the tumors
consisted of two types of cells containing secretory granules. The
majority of the cells exhibited a lighter cytoplasmic density,
larger secretory granules, finer chromatin and fewer mitochondria,
compared with the other types of cells (Fig. 3A-D). The diameter of these secretory
granules ranged between 170 and 350 nm.
Hormone levels in medium and tissue
homogenates
Although the tumor tissue was negatively stained for
ACTH, ACTH was detected in the medium (440 pg/ml), with a
concentration higher than that in the circulation of the patient
(23.8 pg/ml). The concentrations of norepinephrine, epinephrine and
dopamine in DMEM were 160.0, 1.8 and 26.1 µg/l, respectively.
The ACTH concentration was 410 pg/ml in the tissue
homogenate of the index pheochromocytoma, and was 39.9 (case 1),
43.6 (case 2) and 25.7 (case 3) pg/ml in the tissues of the other
three pheochromocytomas, respectively.
Effect of mechanistic target of
rapamycin (mTOR) inhibitor on hormone secretion
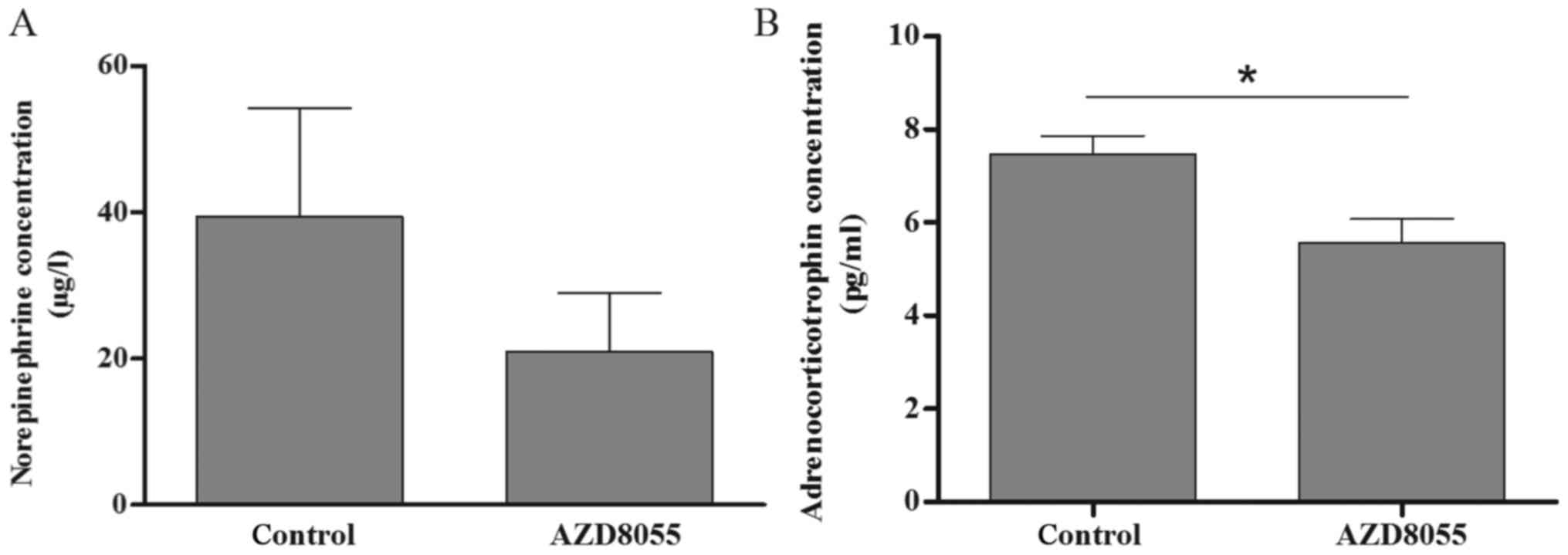
As demonstrated in Fig.
4, in the primary cell culture of the index pheochromocytoma,
AZD8055 significantly inhibited ACTH secretion (P<0.05).
Norepinephrine secretion was also inhibited but the difference was
not significant (P=0.13). No significant changes were detected in
the secretion of epinephrine or dopamine.
Discussion
The current study presented the third case of
ACTH-secreting bilateral pheochromocytoma worldwide. Although the
bilaterality of the pheochromocytoma positively suggested a
germline genetic mutation (5,13), no known genetic change was observed in
this case.
In this case, the high ACTH concentration in the
tissue homogenate supported the diagnosis of ACTH-dependent Cushing
syndrome caused by bilateral pheochromocytomas, although the plasma
ACTH level was not very high and ACTH immunostaining yielded
negative results. Cassarino et al (9) also reported a case of ACTH-secreting
pheochromocytoma with negative ACTH immunostaining. High molecular
weight ACTH precursors or other ACTH-derived peptides may be
responsible for the negative immunostaining for ACTH.
Reports concerning the ultrastructure of ectopic
ACTH-secreting pheochromocytoma were insufficient and the results
were inconsistent. Lamovec et al (18) revealed only one type of cells with
neuroendocrine granules sized between 125 and 216 nm in an
ACTH-secreting pheochromocytoma. By contrast, Sakuma et al
(10) observed two independent cell
populations in the tumor tissue, which were consistent with the
results of the present study. Furthermore, immunohistochemical
results demonstrated that, in the cells, staining for ACTH and
tyrosine hydroxylase (TH) was mutually exclusive (10). It is difficult to differentiate the
cells in terms of the diameter of neuroendocrine granules since
typical catecholamine granules range between 200 and 300 nm and the
size of ectopic ACTH granules is also within this range (18,19). A
co-immunostaining of ACTH and TH was not feasible with the case
presented in the current study since the ACTH immunostaining was
negative.
Although the majority ACTH-secreting
pheochromocytomas are benign in nature and are curable by surgery,
a few cases are malignant or unresectable (20). Novel medical therapies have previously
been trialed for malignant pheochromocytomas. It was reported that
metyrapone could lower catecholamine and ACTH concentrations at a
dose of 1–6 g/day (10,20). It is well-known that metyrapone, a
11β-hydroxylase inhibitor, may reduce cortisol production by the
adrenal gland, but its effect in reducing catecholamine secretion
is impressive (10). In our previous
study, AZD8055 was revealed to be able to inhibit the proliferation
of pheochromocytoma cells (21). The
present study revealed that AZD8055 also exerted a significant
inhibitory effect on the secretion of ACTH-secreting
pheochromocytoma cells. Since AZD8055 is a potent mTOR inhibitor,
mTOR signals may serve a role in the production and secretion of
ACTH and catecholamines.
Notably, a study has reported that plasma ACTH and
catecholamine levels in these patients were increased during a
dexamethasone suppression test. The phenomenon is dubbed
glucocorticoid-driven positive-feedback loop (10), but this did not occur in the case
presented in the study. As of yet, little is known regarding
ACTH-secreting pheochromocytoma. Certain features of ACTH-secreting
pheochromocytoma, including its positive response to metyrapone and
the glucocorticoid-driven positive-feedback loop, is not yet fully
understood.
Presented in this study is the third case of
bilateral ACTH-secreting pheochromocytomas reported globally.
Electron microscopy revealed two types of neuroendocrine cells in
the tumor tissue. The mTOR inhibitor was able to inhibit hormone
secretion in primary cell culture. The pathogenesis of
ACTH-secreting pheochromocytoma remains unclear and warrants
further investigation.
Repeated results are required to prove the effect of
AZD8055 in ACTH-secreting pheochromocytomas. However, the low
incidence of this disease limits opportunities for repeated
experiments or clinical trials (3,5,6–14).
In-depth investigation into the effects of AZD8055 in
ACTH-secreting pheochromocytoma is required in future studies.
Acknowledgements
The authors would like to thank Dr Celso E.
Gomez-Sanchez (Department of Medicine, University of Mississippi
Medical Center, Jackson, Mississippi, USA), for providing
antibodies against CYP11B1. The abstract of the paper was presented
at the 5th International Symposium on Phaeochromocytoma and
Paraganglioma 2017, August 30, 2017 - September 2, 2017, Sydney,
Australia.
Funding
The present study was supported by the Chinese
Academy of Medical Sciences Initiative for Innovative Medicine
(CAMS-I2M; grant no. 2017-I2M-1-001).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FW and CYL performed the primary culture
experiments; ALT and FW analyzed the data; ALT and FW interpreted
the results of the experiments; YXL and ALT designed the research;
YYC and FW prepared the figures; YYC and FW acquired and analyzed
the clinical data; FW drafted the manuscript; JS interpreted the
pathology results; ALS detected the catecholamine concentration in
the medium; ALT and YXL edited and revised the manuscript; and all
authors approved the final version of manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee for Human Research of Peking Union Medical College
Hospital (IRB approval number: S-K222).
Patient consent for publication
Written informed consent for publication was
acquired from the patient.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beard CM, Sheps SG, Kurland LT, Carney JA
and Lie JT: Occurrence of pheochromocytoma in Rochester, Minnesota,
1950 through 1979. Mayo Clin Proc. 58:802–804. 1983.PubMed/NCBI
|
|
2
|
Gimm O, Koch CA, Januszewicz A, Opocher G
and Neumann HP: The genetic basis of pheochromocytoma. Front Horm
Res. 31:45–60. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kirkby-Bott J, Brunaud L, Mathonet M,
Hamoir E, Kraimps JL, Trésallet C, Amar L, Rault A, Henry JF and
Carnaille B: Ectopic hormone-secreting pheochromocytoma: A
francophone observational study. World J Surg. 36:1382–1388. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shanberg AM, Baghdassarian R, Tansey LA,
Bacon D, Greenberg P and Perley M: Pheochromocytoma with
hypercalcemia: Case report and review of literature. J Urol.
133:258–259. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mendonça BB, Arnhold IJ, Nicolau W,
Avancini VA and Boise W: Cushing's syndrome due to ectopic ACTH
secretion by bilateral pheochromocytomas in multiple endocrine
neoplasia type 2A. N Engl J Med. 319:1610–1611. 1988. View Article : Google Scholar
|
|
6
|
Nijhoff MF, Dekkers OM, Vleming LJ, Smit
JW, Romijn JA and Pereira AM: ACTH-producing pheochromocytoma:
Clinical considerations and concise review of the literature. Eur J
Intern Med. 20:682–685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ballav C, Naziat A, Mihai R, Karavitaki N,
Ansorge O and Grossman AB: Mini-review: Pheochromocytomas causing
the ectopic ACTH syndrome. Endocrine. 42:69–73. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li XG, Zhang DX, Li X, Cui XG, Xu DF, Li
Y, Gao Y, Yin L and Ren JZ: Adrenocorticotropic hormone-producing
pheochromocytoma: A case report and review of the literature. Chin
Med J (Engl). 125:1193–1196. 2012.PubMed/NCBI
|
|
9
|
Cassarino MF, Ambrogio AG, Pagliardini L,
De Martin M, Barresi V, Cavagnini F and Giraldi Pecori F:
ACTH-secreting pheochromocytoma with false-negative ACTH
immunohistochemistry. Endocr Pathol. 23:191–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sakuma I, Higuchi S, Fujimoto M, Takiguchi
T, Nakayama A, Tamura A, Kohno T, Komai E, Shiga A, Nagano H, et
al: Cushing syndrome due to ACTH-secreting pheochromocytoma,
aggravated by glucocorticoid-driven positive-feedback loop. J Clin
Endocrinol Metab. 101:841–846. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Folkestad L, Andersen MS, Nielsen AL and
Glintborg D: A rare cause of Cushing's syndrome: An ACTH-secreting
phaeochromocytoma. BMJ Case Rep. 2014:bcr2014205487. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Falhammar H, Calissendorff J and Höybye C:
Frequency of Cushing's syndrome due to ACTH-secreting adrenal
medullary lesions: A retrospective study over 10 years from a
single center. Endocrine. 55:296–302. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Borzouei S, Bahar Mousavi SH, Fereydouni
MA, Salimbahrami SA and Taghipour M: Multiple endocrine neoplasia
type IIa associated with Cushing's syndrome. Arch Iran Med.
17:451–454. 2014.PubMed/NCBI
|
|
14
|
Cohade C, Broussaud S, Louiset E, Bennet
A, Huyghe E and Caron P: Ectopic Cushing's syndrome due to a
pheochromocytoma: A new case in the post-partum and review of
literature. Gynecol Endocrinol. 25:624–627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang LH, Fang XC and Pan GZ: Relationship
between mast cells and nerve fibers in the intestinal mucosa in
patients with irritable bowel syndrome. Chin J Dig. 23:332–335.
2003.
|
|
16
|
Tong A, Liu G, Wang F, Jiang J, Yan Z,
Zhang D, Zhang Y and Cai J: A novel phenotype of familial
hyperaldosteronism type III: Concurrence of aldosteronism and
cushing's syndrome. J Clin Endocrinol Metab. 101:4290–4297. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Du J, Tong A, Wang F, Cui Y, Li C, Zhang Y
and Yan Z: The roles of PI3K/AKT/mTOR and MAPK/ERK signaling
pathways in human pheochromocytomas. Int J Endocrinol.
2016:52869722016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lamovec J, Memoli VA, Terzakis JA, Sommers
SC and Gould VE: Pheochromocytoma producing immunoreactive ACTH
with Cushing's syndrome. Ultrastruct Pathol. 7:41–48. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Corrin B and McMillan M: Fine structure of
an oat cell carcinoma of the lung associated with ectopic ACTH
syndrome. Br J Cancer. 24:755–758. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kakudo K, Uematsu K, Matsuno Y, Mitsunobu
M, Toyosaka A, Okamoto E and Fukuchi M: Malignant pheochromocytoma
with ACTH production. Acta Pathol Jpn. 34:1403–1410.
1984.PubMed/NCBI
|
|
21
|
White A, Ray DW, Talbot A, Abraham P,
Thody AJ and Bevan JS: Cushing's syndrome due to phaeochromocytoma
secreting the precursors of adrenocorticotropin. J Clin Endocrinol
Metab. 85:4771–4775. 2000. View Article : Google Scholar : PubMed/NCBI
|