Introduction
Lung cancer is the leading cause of
cancer-associated mortalities worldwide (1). There are two main types of lung cancer,
small-cell lung carcinoma (SCLC) and non-SCLC (NSCLC), and the
latter accounts for ~84% of all lung cancer cases in USA in 2018
(2). Lung squamous cell carcinoma
(LSCC) is a subtype of NSCLC, and has a pathogenesis that is
closely correlated with a history of tobacco smoking (3). Despite notable advances in the targeted
treatment of patients with NSCLC, patients with LSCC do not benefit
from these major improvements. For example, patients with the
adenocarcinoma subtype of NSCLC are most likely to respond to
epidermal growth factor receptor (EGFR) kinase inhibitors; however,
patients with LSCC rarely respond to these EGFR kinase inhibitors
(4). Recent studies have identified
genes that may serve important roles in LSCC (5,6); however,
the molecular abnormalities and genetic mechanics of LSCC remain
primarily unknown. Therefore, significant research into the genetic
causes of LSCC has been undertaken, targeting earlier diagnosis and
novel treatment development for the disease.
There have been hundreds of genes associated with
LSCC, reflecting its heterogeneity. Mutations of a number of risk
genes have been frequently reported in LSCC, such as tumor protein
P53 (TP53) (7); however, these genes
may also be biomarkers for multiple other diseases. For example,
TP53 may serve as the diagnostic marker for SCLC (7) and chronic lymphocytic leukemia (8). This decreases the specificity of these
genes as biomarkers for the diagnosis and treatment of LSCC;
however, a number of genes only appear in a limited portion of LSCC
cases, such as ACKR3 and ADGRL2 (9,10).
Furthermore, there are numerous novel genes identified each year
for LSCC. For example, in 2017, fibroblast growth factor receptor
(FGFR) 4 and microRNA 145 were novelly reported as LSCC risk genes
that serve roles in the mechanism of the disease (11,12). These
genes have been identified in limited LSCC studies, which may be
due to the specificity of genome variations in different patients
(13); therefore, early
diagnosis/prediction of LSCC may require multiple genes as
biomarkers. Furthermore, patient-specificity should also be taken
into account when selecting the appropriate individualized
treatment.
To address this issue, the present study first
developed a LSCC genetic database (LSCC_GD), curating all LSCC
target genes available within Pathway Studio (http://pathwaystudio.com), a literature-based pathway
analysis tool used to model associations between proteins, genes,
complexes, cells, tissues and diseases. It has been demonstrated
that Pathway Studio possesses the largest real-time updated
databases in this field of study (14). The disease prediction capability of
these curated genes within LSCC_GD was tested using multiple
independent gene expression datasets, where the selected genes were
used to classify patients with LSCC from healthy controls (LSCC
case/control classification). A sparse representation-based
variable selection (SRVS) algorithm was employed to select the
optimum gene vectors for a given patient group as
biomarkers/features to achieve the highest case/control
classification accuracy. Cao et al (15) demonstrated the effectiveness of the
SRVS algorithm in genetic and imaging variable selection. Instead
of selecting a specific number of variables, the SRVS method
generates a sparse regression weight for each variable, which can
be used for variable ranking.
The results confirmed the specificity of the genomic
variation of different groups of patients with LSCC, and supported
the hypothesis that for a given group of patients with LSCC, there
is a gene vector, from the curated LSCC target genes, that
possesses significant predication power to distinguish LSCC cases
from healthy controls.
Materials and methods
Development of LSCC_GD
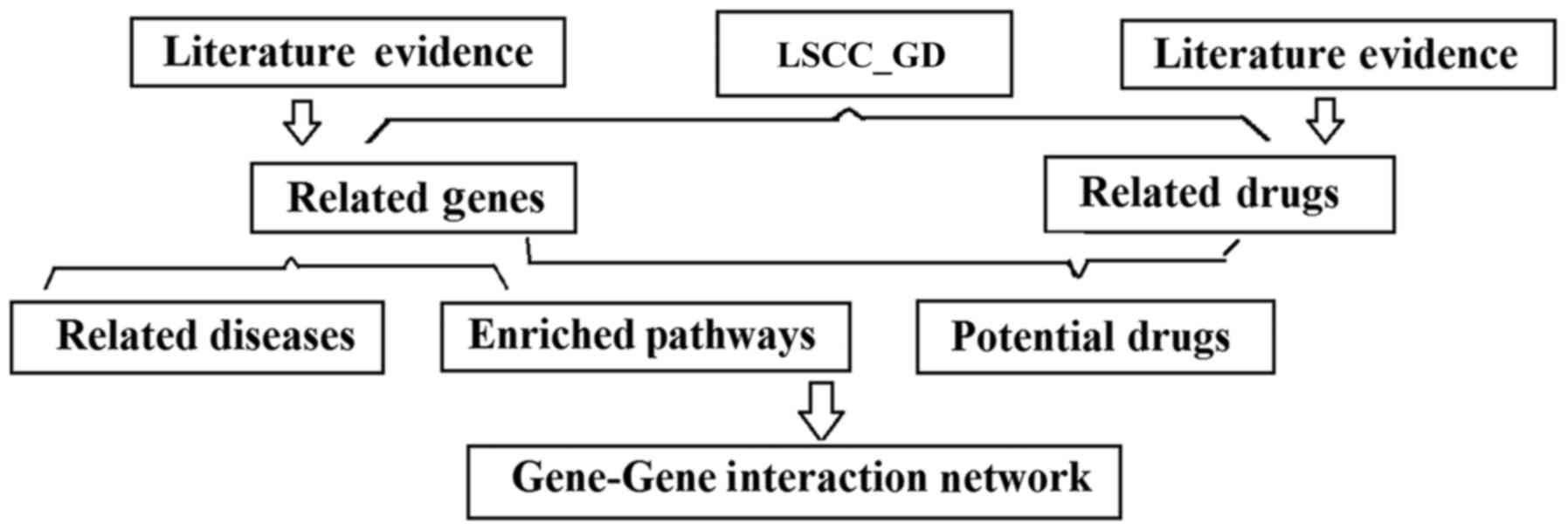
The LSCC_GD database contains 260 genes
(LSCC_GD→Related Genes). These genes were identified from
previously demonstrated associations with LSCC, which are supported
by 685 references (LSCC→Ref for Disease-Gene Relation). The LSCC_GD
database also includes 56 drugs (LSCC_GD→Related Drugs), 101
diseases (LSCC_GD→Related Diseases) and 100 pathways
(LSCC_GD→Related Pathways) associations with LSCC. These
LSCC-associated entities were acquired using Pathway Studio.
LSCC_GD also includes the information of 658 and 126 supporting
references for LSCC-Gene and LSCC-Drug associations, respectively.
For each association, there are ≥1 supporting references. The
reference information includes the title of and the associated
sentences from the source where an association was identified. The
current LSCC_GD database has been deposited into the
‘Bioinformatics Database’ (http://gousinfo.com/database/Data_Genetic/LSCC_GD.xlsx).
It is scalable and will be updated monthly or upon request.
Fig. 1 presents the database schema
of the curated database LSCC_GD.
Gene Set Enrichment Analysis (GSEA) and Sub-Network
Enrichment Analysis (SNEA) were conducted to identify pathways,
diseases, and drugs (small molecules) associated with the 260 LSCC
genes. Fisher's exact test was employed for GSEA and SNEA to
measure the gene-enrichment in annotation terms and the
significance of the overlap between a selected gene group and a
given pathway/sub-network. The top 100 pathways (LSCC→Related
Pathways) were acquired using the Pathway Enrichment Analysis (PEA)
module of Pathway Studio. The 260 LSCC genes were significantly
enriched within these pathways [P<4.4×10−05; q=0.001
for false discovery rate (FDR)]. The 101 diseases (LSCC→Related
Diseases) were identified using the SNEA module (http://pathwaystudio.gousinfo.com/SNEA.pdf). The 260
LSCC target genes were significantly overlapped with the genes
associated with each of these 101 diseases
(P<1.9×10−33; q=0.001 for FDR). A number of the
pathways and diseases have been previously implicated in LSCC,
indicating the pathological association between these target genes
and LSCC.
The Gene-Gene Interaction (GGI) network (LSCC→GGI
Network) was generated based on the enriched pathways. Two genes
were identified as connected if they shared ≥1 pathways. The number
of shared pathways was the edge weight. A 7-by-7 GGI network was
presented as an example, and the full network was presented in the
form of an adjacent matrix in the LSCC→GGI Network. The 105
potential drugs (LSCC→Potential Drugs) were identified using the
SNEA module of Pathway Studio. The drugs/small molecules were
significantly associated with the LSCC target genes, and the
majority of these have not been identified in clinical trial
(92/105). The identified drugs/small molecules may represent
potential drug candidates for LSCC. It is notable that these drugs
demonstrated significant overlap with the drugs/small molecules
associated with LSCC directly (LSCC_GD→Related drugs;
P=1.31×10−18).
SRVS for gene vector selection
The SRVS algorithm (15) was used to rank the 260 LSCC target
genes according to a given experiment dataset. For each gene, a
sparse weight will be assigned by SRVS. The gene vector composed of
the top n genes ranked by SRVS will be the genetic marker for the
LSCC case/control classification, where n is the number of genes
corresponding to the maximum classification ratio (CR) as defined
as:
CR=#correctly classified subjects#total
subjects
The premise of the SRVS algorithm is to select an
optimum number of features, according to sparse representation
theory, when there were notably more features than samples. In the
present study, the figures to be selected were genes. The input of
the SRVS algorithm were the values of gene expression, and the
output were the weight for each gene. These weights were defined as
the SRVSScore, which was used to rank the genes. A detailed
description of the SRVS algorithm was produced by Cao et al
(15).
Gene expression data
In the present study, two Homo sapiens RNA gene
expression datasets [GEO ID: GSE18842 (16) and GSE1987 (17)] were employed to evaluate the
classification performance using the LSCC target genes. These
datasets were selected from the Illumina BaseSpace Correlation
Engine (http://www.illumina.com) and are
publicly available at the National Center of Biotechnology
Information Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/). The data selection
criteria were as follows: i) They were human experiments; ii) the
data were RNA expression datasets; iii) the data were presented as
LSCC case vs. normal control studies; and iv) the tissue was from
the lung. From each dataset, expression data of normal controls and
patients with LSCC were extracted and used for LSCC case/control
classification. Genes of each dataset were limited to LSCC target
genes curated within the database LSCC_GD. The key statistics of
the two datasets are summarized in Table
I.
 | Table I.Statistics of two gene expression
datasets. |
Table I.
Statistics of two gene expression
datasets.
| NCBI GEO ID | GSE18842 | GSE1987 |
|---|
| #LSCC
case/control | 46/45 | 17/9 |
| #Genes from
LSCC_GD | 232 | 702 |
The gene expression profiles of the two gene
expression datasets are also included in LSCC_GD (LSCC_GD→GSE18842
and LSCC_GD→GSE1987). Within each dataset, the SRVS-generated
weights (SRVSScore) and analysis of variance (ANOVA)-generated
P-value score [PValueScore; logic transferred P-values:
−10×log(P-value)] were also presented. For a given gene, the
input of the ANOVA was two vectors of gene expression values, one
vector was for the case group subjects and the other was for the
control group. The gene expression data were provided in
LSCC_GD→GSE18842 and LSCC_GD→GSE1987. The P-value for a gene was
generated from the one-way ANOVA of the case/control comparison
using the corresponding expression data. A SRVSScore or a
PValueScore represented the significance of a gene to the dataset
according to SRVS or ANOVA methods, respectively.
LSCC case/control classification
To identify the best gene vector resulting the
highest CR and the corresponding CR, the LSCC target genes were
ranked by SRVSScore in descending order. Subsequently, a Euclidean
distance-based multivariate classification (14) was performed for each dataset, followed
by a leave-one-out (LOO) cross validation (18). For each run of LOO, the gene
expression data of one subject was used for testing and the rest
for training. The inputs of the classifier were the expression
values of the top n (n=1, 2 …) genes, such that the
CR of the top n genes could be determined. A permutation of
5,000 runs was then conducted to test the hypothesis, that randomly
selected gene sets of the same size can reach equal or higher CR.
The permutation P-values (number of runs with equal or better CRs
over the number of total runs) were calculated. The gene vector
that generated the highest CR was considered the best gene vector,
and was selected for the dataset according to the SRVS method.
Following the same process, the best gene vector
according to the ANOVA approach was identified for each dataset.
For comparison purposes, a CR baseline was also generated using
randomly selected gene sets of n (n=1, 2 …) genes. For each
point of the CR baseline, the value was the mean of 300 CRs, which
were from randomly selected genes within the dataset.
Results
Target genes from LSCC_GD
Due to the lack of space, a small GGI network was
presented as an example in Fig. 2.
The size of the example network was 7-by-7 (49/260 LSCC target
genes). The full GGI network composed of 201/260 LSCC target genes
has been presented in the form of adjacency matrix in LSCC_GD→GGI
Network. An association between two genes indicated that these two
genes shared ≥1 pathways (LSCC_GD→Related Pathways). There were 59
genes not included in the LSCC associated pathways, and therefore
were not presented in the GGI network.
LSCC case/control classification
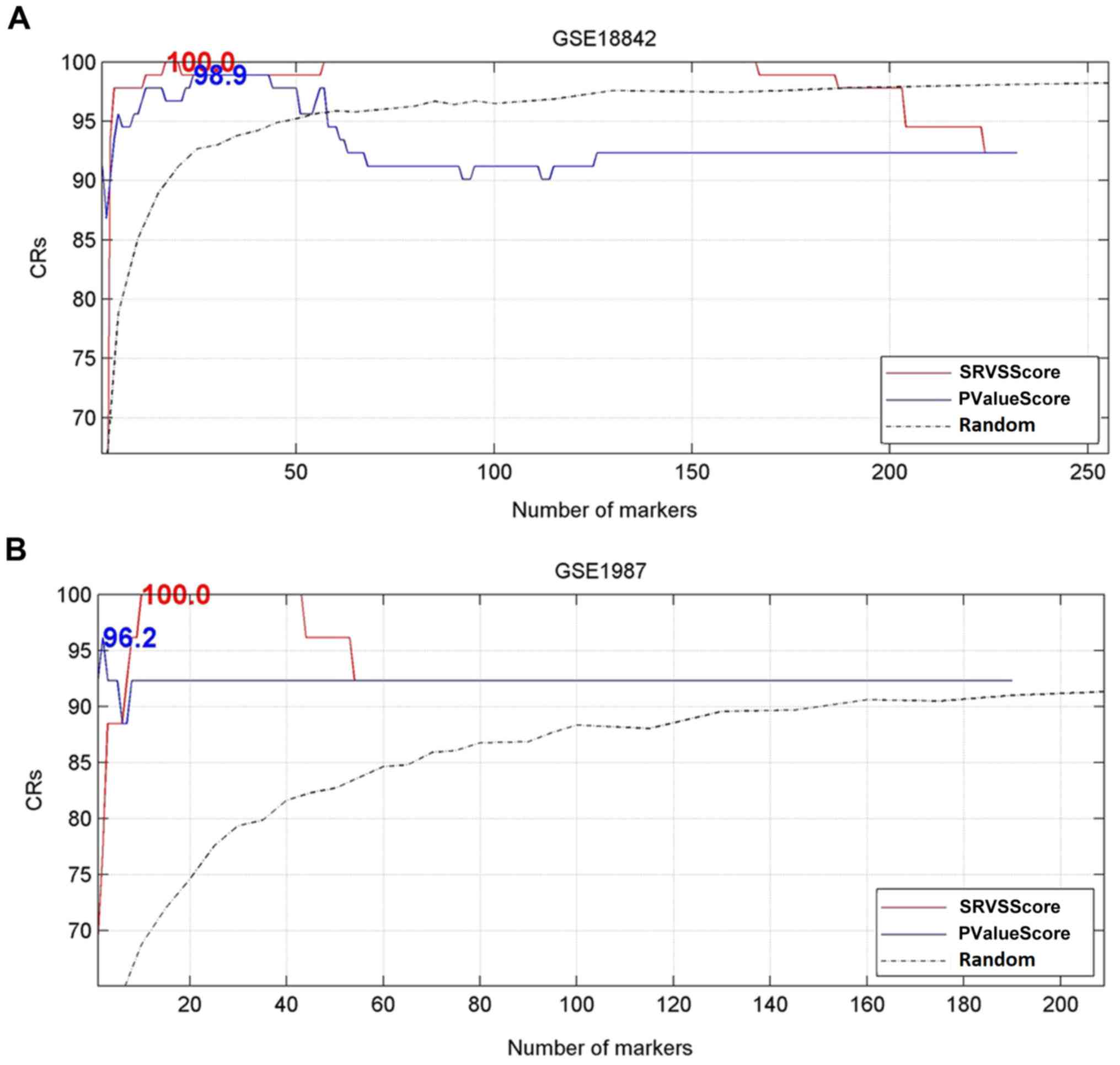
The maximum CRs are marked at the position of the
corresponding number of genes. As depicted in Table II, the results of LOO
cross-validation of the two gene ranking method on two datasets
were summarized, including the maximum CRs, the corresponding
number of top genes and the permutation P-values of the two
methods.
| Table II.Leave-one-out cross validation and
permutation results. |
Table II.
Leave-one-out cross validation and
permutation results.
|
| GSE18842
(case/control: 46/45) | GSE1987
(case/control: 17/9) |
|---|
|
|
|
|
|---|
| Analysis
results | SRVS | ANOVA | SRVS | ANOVA |
|---|
| Maximum CR | 100.00 | 98.90 | 100.00 | 96.15 |
| #Selected
Genes | 17 | 24 | 10 | 2 |
| P-value |
5.0×10−4 |
4.9×10−2 |
1.8×10−3 |
2.0×10−3 |
| Unique genes from
all datasets (%) | 94.11% (16/17) | 91.67% (22/24) | 90.00% (9/10) | 0.00% (0/2) |
| Overlapping genes
of two methods (%) | 23.53% (4/17) | 16.67% (4/24) | 10.00% (1/10) | 50.00% (1/2) |
Fig. 3 displays the
classification results and establishes that compared with the CRs
generated by randomly selected gene sets, the genes selected from
LSCC target genes by SRVSScore and PValueScore may result in
significantly higher classification accuracies. Notably, using only
the top genes with the highest SRVSScore/PValueScore, the highest
CRs were acquired (Fig. 3 and
Table II). These results
demonstrated the effectiveness of the selected top genes by SRVS
and ANOVA in the differentiation of patients with LSCC from
controls, and those selected genes for a specific patient with LSCC
group should be the main genetic targets for the prognosis and
treatment of the patients in the group. Additionally, as displayed
in Table II, the SRVS method
outperformed ANOVA method in terms of CR in all datasets
(5.0×10−4 vs. 4.9×10−2 and
1.8×10−3 vs. 2.0×10−3 for GSE18842 and
GSE1987, respectively).
Table II also depicts
that, for each dataset, the top genes selected by each method may
be significantly different. For the SRVS method, the unique genes
selected for the two datasets ranged from 90–94.11%. For the ANOVA
method, the two dataset (GSE18842 and GSE1987) demonstrated 91.67
and 0% unique genes, respectively [Table
II→Unique genes from all datasets (%)]. These results indicated
the group specificity of the genome variation of the patients
within the two datasets.
Notably, the optimum gene markers for distinct
datasets selected by SRVS and ANOVA may be considerably different
(Table II). The genes selected by
the SRVS method demonstrated a <23.53% overlap with that of the
ANOVA method for both datasets [Table
II→ Overlap genes of two methods (%)]. The results indicated
that SRVS performs differently and more effectively than ANOVA.
Discussion
LSCC is an aggressive cancer type, and the overall
prognosis for patients with LSCC is poor. Previously, numerous
molecular therapy targeted studies have been conducted (19,20), with
hundreds of risk genes identified for the disease. The majority of
these genes serve roles within LSCC-associated genetic pathways,
and a number of them were indicated as drug targets for the
disease, such as FGFR1 and discoidin domain receptor tyrosine
kinase 2 (DDR2) (20–22).
Only a limited number of genes have been frequently
detected in LSCC cases. For instance, focal FGFR1 amplification
occurs in up to 22% of LSCC cases (21). FGFR1 amplification in cells is
dependent on FGFR signaling and is sensitive to FGFR inhibitors
(21). Somatic mutations in the DDR2
kinase gene were also indicated as the potential targets for the
treatment of a portion of patients with LSCC (20,22). These
results reflected the heterogeneity of human tumor types (23), and explain the large size of the LSCC
risk gene pool curated through previous studies (4–12).
Whilst novel LSCC genes are being actively
investigated in continuous genetic and genomic studies performed,
significantly less studies have been conducted to test the validity
of the existing LSCC risk genes as a whole for their diagnostic and
predictive capabilities for LSCC. We hypothesized that, if the
current LSCC gene pool is sufficient to cover the majority of the
genes underlying the genetic pathogeneses of LSCC, then for a given
group of patients with LSCC, ≥1 gene vector from the LSCC gene pool
exists that possesses significance in the classification/prediction
of patients with LSCC from controls. If this hypothesis were true,
then another issue would arise, in how the optimum gene combination
from the target pool for a specific patient group would be
identified.
To test this hypothesis, comprehensive literature
data mining using Pathway Studio was conducted, which identified
260 LSCC target genes. Pathway Studio covers >40,000,000
scientific papers. Each association between these genes and LSCC
was supported by ≥1 literature reports (LSCC_GD→Ref for
Disease-Gene Relation). Within LSCC_GD, there are also 100 pathways
(LSCC_GD→Related Pathways), 101 disease-subnetworks
(LSCC_GD→Diseases) and 105 potential drugs/small molecules
(LSCC_GD→Potential Drugs) present when these genes were
significantly enriched.
PEA demonstrated that the majority of these genes
(201/260) were significantly enriched within multiple genetic
pathways implicated in LSCC (P<4.4×10−5; q=0.001 for
FDR). For instance, there are 66 genes significantly enriched
within four cell apoptosis pathways (P<3.0×10−7;
q=0.001 for FDR) (24,25), including negative regulation of the
apoptotic process [Gene Ontology (GO): 0006916;
P=1.1×10−14]; the apoptotic process (GO: 0008632;
P=9.9×10−08); positive regulation of the apoptotic
process (GO: 0043065; P=2.2×10−7); and negative
regulation of cysteine-type endopeptidase activity involved in the
apoptotic process (GO: 0043154; P=3.0×10−7). There were
also 79 genes significantly enriched within 9 pathways associated
with cell growth and proliferation (P<2.8×10−5)
(26) and 72 genes enriched within
protein kinases (P<3.7×10−6) (27,28). More
pathways can be identified from LSCC_GD→Related Pathways.
Disease SNEA demonstrated that, 250/260 genes were
significantly overlapped with the risk genes of 101 diseases
(P<1.9×10−33; q=0.001 for FDR). The majority of these
101 diseases are different cancers types, and a number of them were
associated with LSCC, including gastric (29) and breast cancer (30). More results from SNEA can be
identified at LSCC_GD→Related Diseases.
Within LSCC_GD, there were 56 known LSCC drugs
(LSCC_GD→Related Drugs) that underwent clinical trials and
demonstrated effectiveness in treating LSCC. These 56 drugs
demonstrated significant overlap (13 overlapped drugs;
P=1.31×10−18) with the top 105 potential drugs/small
molecules (LSCC_GD→Potential Drugs), whose gene sub-networks were
significantly enriched with the 260 LSCC genes. Additionally, a
number of the 260 LSCC genes were target genes of known LSCC drugs.
For instance, AZD4547, a FGFR inhibitor, is a key drug in the
treatment of LSCC. This may be explained due to AZD4547 exhibiting
a highly selective profile across a lung cell line panel, potently
inhibiting cell growth only in those lines harboring amplified
FGFR1 (31). These results
demonstrated that the 260 LSCC genes were associated with LSCC and
therefore may possess classification/prediction power for the
disease; however, due to the heterogeneity of LSCC and the
specificity of human genome variation (13), the significance of these genes being
used as markers for disease diagnosis and personalized treatment
still requires testing.
To address this issue, LSCC case/control
classification was conducted on two independent gene expression
datasets, with two algorithms (SRVS and ANOVA) for gene selection
from the 260 LSCC genes. The basic theory for gene selection is
that mutations of these 260 genes will not be exhibited in a number
of patients with LSCC, and therefore are not effective as
biomarkers for all patients.
Compared with randomly selected genes, those
selected by SRVS and ANOVA generated significantly higher
predication power (permutation P<1.8×10−3 for SRVS,
and P<5.0×10−2 for ANOVA), with improved high
classification accuracy (SRVS vs. ANOVA: 100% vs. 98.9%, and 100%
vs. 96.15%, for datasets GSE18842 and GSE1987, respectively), as
depicted in Table II. These results
indicated that, for a given dataset, a gene vector, from the 260
LSCC gene pools, that could be used as biomarker vector for the
diagnosis and prognosis of the disease exists. Notably, SRVS
outperforms ANOVA in terms of CR on both datasets. This indicated
the effectiveness of the SRVS method for feature selection. It is
notable that both datasets used LSCC lung tissue as sample source,
which may partially explain the high classification results of both
methods.
Cross analysis on the genes selected demonstrated
that optimum biomarkers are dataset specific, as depicted in
Table II. These results indicated
the specificity of the genomic variations of different patients
(12), and highlighted the necessity
of genomic variable selection in the diagnosis and treatment of
patients with LSCC.
To conclude, the present study indicated that the
260 curated LSCC genes from previous studies demonstrated a strong
association with the pathogenesis of LSCC, and possessed
significant diagnostic power as a biomarker network. Furthermore,
SRVS is an effective method to select the optimum gene sub-set for
personalized diagnosis and treatment for a specific group of
patients with LSCC.
Acknowledgements
Not applicable.
Funding
The present study is partially supported by
non-small cell lung cancer research funding of Jiangsu Provincial
Planning Commission, clinical diagnosis and treatment of small lung
lesions and normative research of Wuxi City Health Planning
Commission (grant no. MS201625) and 2017 Fifth Provincial ‘333
Project’ Research Project.
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
BH and NZ contributed to the design and data
collection of the study, HC and GY contributed to the data
collection and analysis. All authors contributed to the writing and
revising of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Heist RS, Mino-Kenudson M, Sequist LV,
Tammireddy S, Morrissey L, Christiani DC, Engelman JA and Iafrate
AJ: FGFR1 amplification in squamous cell carcinoma of the lung. J
Thorac Oncol. 7:1775–1780. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
American Cancer Society: Cancer Facts and
Figures 2018. Atlanta, Ga: American Cancer Society; 2018, Available
online. Last accessed. April 27–2018
|
|
3
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Alberg AJ, Brock MV and Samet JM:
Epidemiology of lung cancer: Looking to the future. J Clin Oncol.
23:3175–3185. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shigematsu H, Lin L, Takahashi T, Nomura
M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, et
al: Clinical and biological features associated with epidermal
growth factor receptor gene mutations in lung cancers. J Natl
Cancer Inst. 97:339–346. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lynch TJ, Bondarenko I, Luft A,
Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H,
Cuillerot JM and Reck M: Ipilimumab in combination with paclitaxel
and carboplatin as first-line treatment in stage IIIB/IV
non-small-cell lung cancer: Results from a randomized,
double-blind, multicenter phase II study. J Clin Oncol.
30:2046–2054. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gibbons DL, Byers LA and Kurie JM:
Smoking, p53 mutation, and lung cancer. Mol Cancer Res. 12:3–13.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stilgenbauer S, Schnaiter A, Paschka P,
Zenz T, Rossi M, Döhner K, Bühler A, Böttcher S, Ritgen M, Kneba M,
et al: Gene mutations and treatment outcome in chronic lymphocytic
leukemia: Results from the CLL8 trial. Blood. 123:3247–3254. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenfeld MR, Malats N, Schramm L, Graus
F, Cardenal F, Viñolas N, Rosell R, Torà M, Real FX, Posner JB and
Dalmau J: Serum anti-p53 antibodies and prognosis of patients with
small-cell lung cancer. J Natl Cancer Inst. 89:381–385. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Azad Behnam B, Lisok A, Chatterjee S,
Poirier JT, Pullambhatla M, Luker GD, Pomper MG and Nimmagadda S:
Targeted imaging of the atypical chemokine receptor 3 (ACKR3/CXCR7)
in human cancer xenografts. J Nucl Med. 57:981–988. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zheng CX, Gu ZH, Han B, Zhang RX, Pan CM,
Xiang Y, Rong XJ, Chen X, Li QY and Wan HY: Whole-exome sequencing
to identify novel somatic mutations in squamous cell lung cancers.
Int J Oncol. 43:755–764. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Quintanal-Villalonga Á, Carranza-Carranza
A, Meléndez R, Ferrer I, Molina-Pinelo S and Paz-Ares L: Prognostic
role of the FGFR4-388Arg variant in lung squamous-cell carcinoma
patients with lymph node involvement. Clin Lung Cancer.
18(667-674): e12017.PubMed/NCBI
|
|
13
|
Lu YF, Goldstein DB, Angrist M and
Cavalleri G: Personalized medicine and human genetic diversity.
Cold Spring Harb Perspect Med. 4:a0085812014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lorenzi PL, Claerhout S, Mills GB and
Weinstein JN: A curated census of autophagy-modulating proteins and
small molecules: Candidate targets for cancer therapy. Autophagy.
10:1316–1326. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cao H, Duan J, Lin D, Shugart YY, Calhoun
V and Wang YP: Sparse representation based biomarker selection for
schizophrenia with integrated analysis of fMRI and SNPs.
Neuroimage. 102:220–228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sanchez-Palencia A, Gomez-Morales M,
Gomez-Capilla JA, Pedraza V, Boyero L, Rosell R and Fárez-Vidal ME:
Gene expression profiling reveals novel biomarkers in nonsmall cell
lung cancer. Int J Cancer. 129:355–364. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dehan E, Ben-Dor A, Liao W, Lipson D,
Frimer H, Rienstein S, Simansky D, Krupsky M, Yaron P, Friedman E,
et al: Chromosomal aberrations and gene expression profiles in
non-small cell lung cancer. Lung Cancer. 56:175–184. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kohavi R: A study of cross-validation and
bootstrap for accuracy estimation and model selectionProceedings of
the Fourteenth International Joint Conference on Artificial
Intelligence. San Mateo, CA: Morgan Kaufmann; 2. pp. 1137–1143.
1995
|
|
19
|
Gan TQ, Xie ZC, Tang RX, Zhang TT, Li DY,
Li ZY and Chen G: Clinical value of miR-145-5p in NSCLC and
potential molecular mechanism exploration: A retrospective study
based on GEO, qRT-PCR, and TCGA data. Tumour Biol.
39:10104283176916832017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hammerman PS, Sos ML, Ramos AH, Xu C, Dutt
A, Zhou W, Brace LE, Woods BA, Lin W, Zhang J, et al: Mutations in
the DDR2 kinase gene identify a novel therapeutic target in
squamous cell lung cancer. Cancer Discov. 1:78–89. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Weiss J, Sos ML, Seidel D, Peifer M,
Zander T, Heuckmann JM, Ullrich RT, Menon R, Maier S, Soltermann A,
et al: Frequent and focal FGFR1 amplification associates with
therapeutically tractable FGFR1 dependency in squamous cell lung
cancer. Sci Transl Med. 2:62ra932010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sos ML and Thomas RK: Genetic insight and
therapeutic targets in squamous-cell lung cancer. Oncogene.
31:4811–4814. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Loeb LA, Loeb KR and Anderson JP: Multiple
mutations and cancer. Proc Natl Acad Sci USA. 100:776–781. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ramsey MR, Wilson C, Ory B, Rothenberg SM,
Faquin W, Mills AA and Ellisen LW: FGFR2 signaling underlies p63
oncogenic function in squamous cell carcinoma. J Clin Invest.
123:3525–3538. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ling B and Wei GZ: p53: Structure,
function and therapeutic applications. J Cancer Mol. 2:141–153.
2006.
|
|
26
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yip PY: Phosphatidylinositol
3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR)
signaling pathway in non-small cell lung cancer. Transl Lung Cancer
Res. 4:165–176. 2015.PubMed/NCBI
|
|
28
|
Davies H, Hunter C, Smith R, Stephens P,
Greenman C, Bignell G, Teague J, Butler A, Edkins S, Stevens C, et
al: Somatic mutations of the protein kinase gene family in human
lung cancer. Cancer Res. 65:7591–7595. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kurishima K, Satoh H, Kagohashi K, Homma
S, Nakayama H, Ohara G, Ishikawa H and Hizawa N: Patients with lung
cancer with metachronous or synchronous gastric cancer. Clin Lung
Cancer. 10:422–425. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Genç B, Solak A, Sahin N and Gülşen A:
Metastasis to the male breast from squamous cell lung carcinoma.
Case Rep Oncol Med. 2013:5939702013.PubMed/NCBI
|
|
31
|
Zhang J, Zhang L, Su X, Li M, Xie L,
Malchers F, Fan S, Yin X, Xu Y, Liu K, et al: Translating the
therapeutic potential of AZD4547 in FGFR1-amplified non-small cell
lung cancer through the use of patient-derived tumor xenograft
models. Clin Cancer Res. 18:6658–6667. 2012. View Article : Google Scholar : PubMed/NCBI
|