Introduction
Thyroid cancer is the most common type of endocrine
malignancy. The incidence of thyroid cancer has increased markedly
worldwide in the last decade (1).
Papillary thyroid cancer (PTC) is the most common type of thyroid
cancer, accounting for approximately 80% of all cases (2). Understanding of the genetic mechanisms
underlying PTC has improved remarkably in recent years, which
provides more support for molecular diagnosis of the disease
(3,4).
However, predicting PTC as a result of the complicated molecular
interactions participating in the pathogenesis of this type of
cancer remains a clinical challenge (5). In addition, a number of patients with
PTC demonstrate recurrence, invasion and distant metastasis due to
resistance to surgical and radioiodine treatment. Novel
molecular-targeted treatments hold potential for these cases
(6). Therefore, investigation of the
molecular mechanisms involved is expected to aid the identification
of candidate biomarkers and improve molecular diagnosis and
treatment of patients with PTC.
Previous studies of the genetic basis of PTC mainly
focus on a single susceptibility gene, which does not take into
account the interactions of multiple genes. However, complex traits
are modified by the cumulative effect of interactive genes
(7). Elucidation of the interactions
between genes and molecules is essential for understanding the
molecular basis underlying complex diseases. Correlation network
analysis identifies the correlation patterns among genes according
to their co-expression similarities by grouping highly correlated
genes into one module (8). Another
advantage of network-assisted analysis is that transcriptional data
has a more direct impact on protein expression than genomic data,
which provides more biological information regarding the
interactive genes (9). The method
applied in the present study is weighted gene co-expression network
analysis (WGCNA) (8), which has been
successfully used for detection of genetic determinants in many
human diseases, such as osteoporosis (10), obesity (11), familial combined hyperlipidemia
(12) and metaphyseal
chondrodysplasia (13).
A gene harboring changeable interactions with other
genes between diseased and normal conditions indicates that it may
play a crucial role in pathology of the disease. Based on this
hypothesis, Riquelme Medina and Lubovac-Pilav successfully employed
WGCNA to identify genes and pathways involved in the development of
type 1 diabetes (14). In the present
study, WGCNA is employed to generate two networks derived from PTC
and normal tissue control samples, respectively. The two networks
were compared in order to identify the module with the lowest
preservation, which contains genes harboring changeable
interactions. To the best of our knowledge, the present study is
the first to apply WGCNA innovatively in order to identify the
genetic mechanisms of PTC.
Materials and methods
Gene expression data and
processing
The gene expression dataset (accession number:
GSE33630) was downloaded from the NCBI Gene Expression Omnibus
database (http://www.nibi.nih.gov/geo/). Microarray data were
obtained from RNA samples comprised of 49 PTC samples and 45
adjacent normal thyroid samples. The research was approved by Ehics
Committees of the Institute of Endocrinology and Metabolism in
Kiev, Imperial College in London and the Medical Radiological
Research Centre in Obninsk. Details about the data can be found in
the original publication (15). The
platform used for profiling was the Affymetrix U-133 Plus v.2.0
array. The raw CEL data were processed and normalized using affy
package in R software 3.3.0 (16).
The probe IDs were converted into gene symbols using the annotation
file for probes of the platform. The median expression levels of
these probe IDs that correspond to the same gene were selected as
the final expression value of a gene.
Differential gene expression
analysis
In order to identify differentially expressed genes
(DEGs) between the PTC and normal samples, empirical Bayes method
was performed using limma package (17) in R. P-values were adjusted using the
Benjamini and Hochberg method to control the false discovery rate
caused by multiple testing. Genes were selected as DEGs for
subsequent analysis according to the following criteria: logFC
(fold change) >1 and adjusted P-value <0.05. Fold change was
calculated as the ratio of the differential expression.
Construction of weighted gene
co-expression networks
Network construction was performed using the WGCNA R
package (8,18). First, cluster sampling was conducted
for both the PTC and normal samples based on the expression levels
of the DEGs to identify and remove potential outliers. The
parameter beta of power function for network construction was then
set as 8 according to the scale-free topology criterion as
described in a previous study (8),
making the established network satisfy approximate scale-free
topology (linear regression model-fitting index R2 >0.9). WGCNA
identifies modules with highly correlated genes; these genes
usually have similar connectivity patterns with other genes, which
can be defined as the topological overlap measurement (TOM). Genes
were hierarchically clustered and visualized in a dendrogram
according to dissimilarity TOM (1-TOM) (18). The branches of the tree representing
highly correlated genes were grouped into one module marked by a
particular color. Grey donates background genes that belonged to
none of the modules. Different numbers of modules were established
from the dendrogram in the PTC and normal samples,
respectively.
Module preservation between PTC and
normal networks
Module preservation analysis is capable of assessing
whether a module defined in a reference dataset (PTC sample network
in the present study) is also in the test dataset (normal sample
network in the present study). Two composite preservation
statistics, Zsummary (Eq1) and medianRank (Eq2), were
used to compare relative preservation among multiple modules, which
considers both density and connectivity preservation (19). The former is based on the extent of
gene interconnectivity, while the latter is based on the
connectivity pattern of genes.
Zsummary=(Zdensity+Zconnectivity)/2(Eq1)
medianRank=(medianRank.density+medianRank.connectivity)/2(Eq2).
Studies have shown that Zsummary
thresholds distinguish module preservation as follows (19): Zsummary<2, no
preservation; Zsummary>10, strong evidence of module
preservation; and 2<Zsummary<10, weak to moderate
evidence of preservation. Modules with lower Zsummary
values are expected to be less preserved. MedianRank is a
rank-based measurement that relies on a observed preservation
statistic that shows no dependence on modulesize. A higher
medianRank measurement indicates that the module exhibits less
preservation. Modules of poor preservation are significant modules
that perturbate important pathways and biological processes when
comparing diseased and normal networks.
Intramodule gene interaction
analysis
K.in, also known as intramodule connectivity,
describes the connectivity of a gene with other genes in the same
module (10). The k.in rank of a gene
is the ranking of k.in among all of the genes in a particular
module. K.in rank change refers to the difference of the k.in ranks
between the PTC and normal sample networks. A gene with a large
k.in rank change represents a large difference in its interactions
with other genes in PTC and normal samples and indicates that it
has an important role in the development of PTC.
Validation analysis using another gene
expression dataset
In order to partially validate the identified genes
in the significant module, a gene expression validation analysis
was performed using another gene expression profile. The expression
array for the validation was downloaded from the NCBI Gene
Expression Omnibus database (accession number: GSE3678) and was
derived from 7 PTC samples and 7 normal thyroid control samples.
The point-biserial correlation coefficient was employed to estimate
the relationship between PTC and gene expression levels. After 1000
bootstrap replications, the average of the correlation coefficients
was calculated as the final point-biserial correlation coefficient
of each gene.
Functional and pathway enrichment
analyses of the significant module
Functional annotation of the genes in the
significant module was accomplished using the online tool Database
for Annotation Visualization and Integrated Discovery (DAVID)
(http://david.abcc.ncifcrf.gov/)
(20), depending on the GO term
database and pathway database. Pathway enrichment of the module of
interest was performed with another online tool, ConcensusPathDB
(http://cpdb.molgen.mpg.de/) (21), based on the database of KEGG. This
step evaluated the potential function of the genes related to
thyroid carcinomas according to the known databases and known
molecular pathways.
Results
Differential gene expression
analysis
A total of 1062 genes were identified as DEGs for
network construction based on the aforementioned criteria. The 1062
genes consisted of 653 up-regulated genes and 409 down-regulated
genes in the PTC samples compared with the normal samples.
Network construction for the PTC and
normal samples with WGCNA
WGCNA R package was applied in order to generate two
different co-expression networks for the two sample groups with the
1062 DEGs. Hierarchically clustered dendrograms were generated and
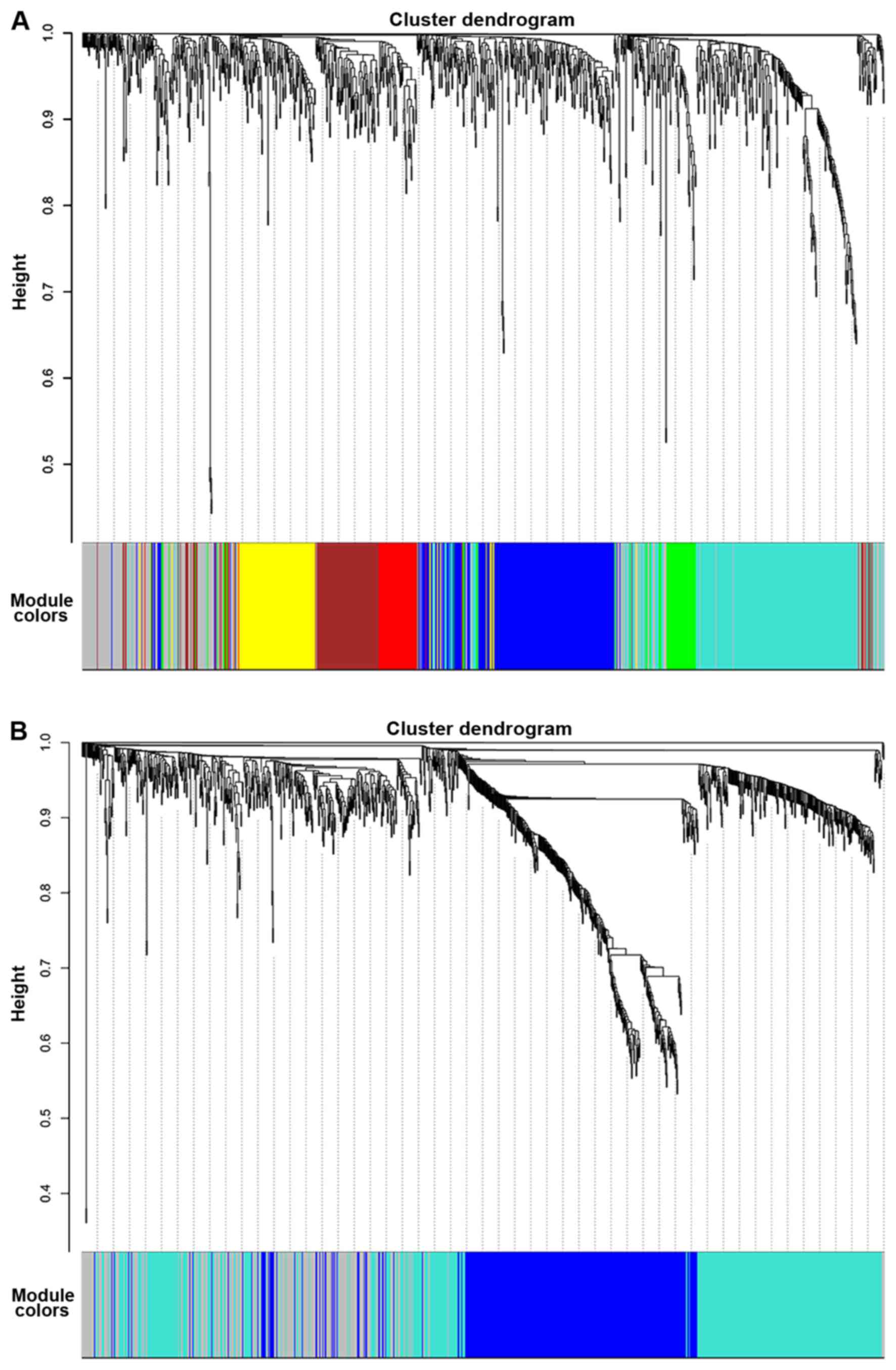
modules containing highly interconnected genes were detected. Seven
modules, represented by blue, brown, green, grey, red, turquoise
and yellow, were detected in the PTC samples, with module sizes of
230, 118, 65, 198, 59, 276 and 116 genes, respectively. Three
modules were identified in the normal samples, represented by blue,
grey and turquoise, which contained 358, 240 and 464 genes,
respectively. The gene cluster dendrograms and detected modules are
presented in Fig. 1.
Comparison of networks between the PTC
and normal samples
In order to visualize the difference between the two
networks, the same color of the module that each gene belonged to
in the PTC sample network was applied to the corresponding gene in
the normal sample network, without changing the hierarchically
clustered dendrogram of the normal sample network. Visual change of
the module structure is presented in Fig.
2. Integrated modules in the PTC sample network were found to
be divided into several parts in the normal sample network.
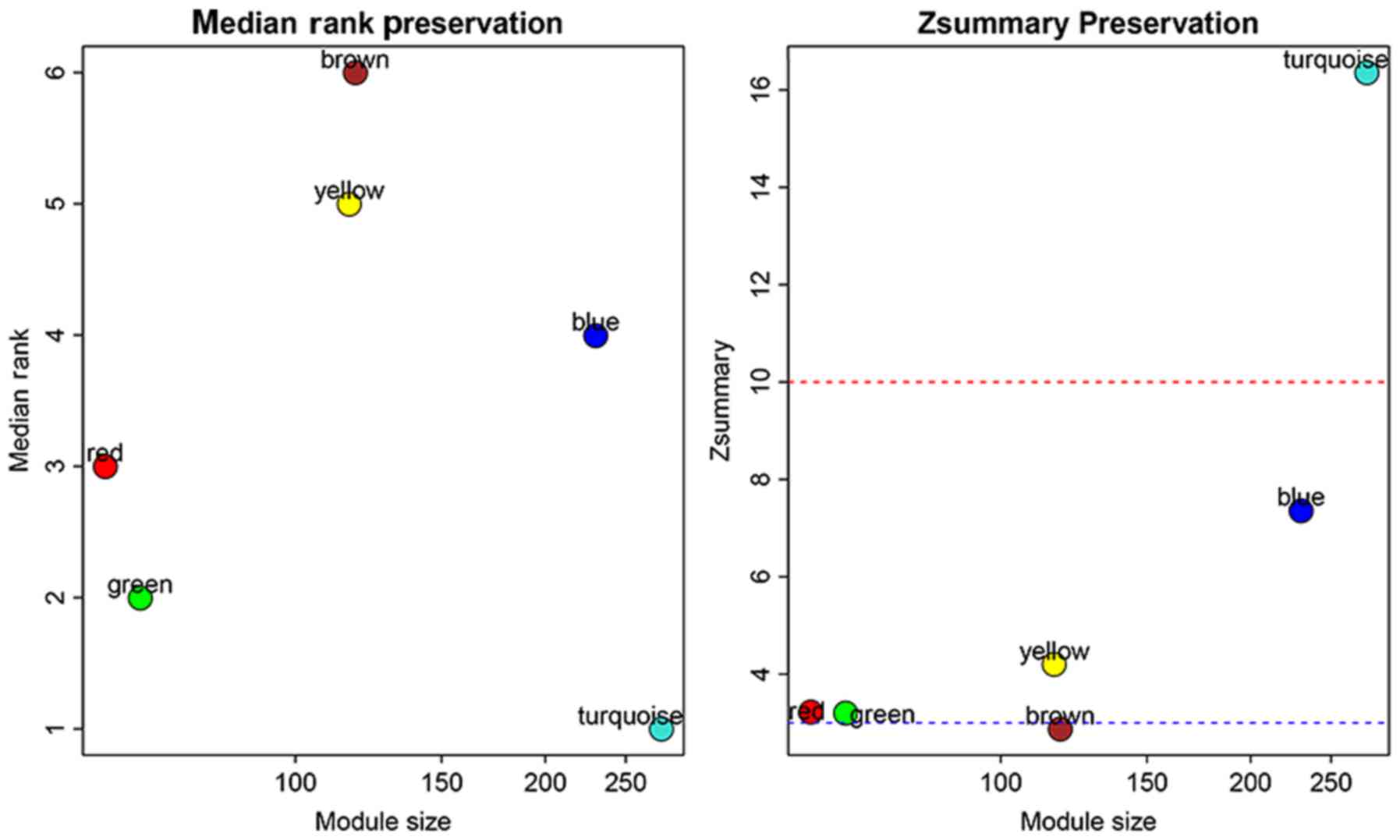
To objectively estimate whether a module is
preserved, Zsummary and medianRank were employed in
order to investigate the module preservation quantitatively. Visual
results of the module preservation analysis including the
statistics medianRank and Zsummary are presented in
Fig. 3. The brown module has the
highest medianRank as well as the lowest Zsummary among
all the modules, indicating that it has the strongest preservation
of all the modules. However, the Zsummaryvalue of the
brown module is <3, which indicates no preservation according to
the criterion of Zsummary. The brown module was selected
for further analysis, when its medianRank and Zsummary
statistics were both taken into account.
Identification of genes associated
with PTC in the brown module
The k.in rank change was calculated for each gene in
the brown module in order to detect gene interaction changes when
PTC occurs. After validation of the 118 genes in the brown module
using point-biserial correlation coefficient, 61 genes were
confirmed to be significantly relevant to PTC with P-value
<0.05. The validation results of the significant genes with the
top20 k.in rank change values are listed in Table I.
 | Table I.Validation results of the significant
genes with top20 k.in rank change values. |
Table I.
Validation results of the significant
genes with top20 k.in rank change values.
| Gene symbol | ra | P-value | k.in rank
changeb |
|---|
| LRP4 | 0.744359743 |
8.71×10−05 | 108 |
| TMEM108 | 0.663751001 | 0.001446 | 94 |
| ETV4 | 0.801399494 |
5.13×10−06 | 89 |
| CAPN3 | 0.751756977 |
6.33×10−05 | 79 |
| KLK7 | 0.808517499 |
3.36×10−06 | 76 |
| TNRC6C-AS1 | 0.864710246 |
5.44×10−08 | 75 |
| FXYD5 | 0.676025828 | 0.001009 | 74 |
| KCNK5 | 0.673939248 | 0.001074 | 73 |
| ST3GAL5 | 0.761898241 |
4.00×10−05 | 72 |
| KLK10 | 0.789168637 |
1.02×10−05 | 71 |
| PRICKLE1 | 0.747557852 |
7.60×10−05 | 69 |
| BCHE | −0.514997779 | 0.031851 | 68 |
| NELL2 | 0.589338467 | 0.008726 | 68 |
| IGFBP6 | 0.565886466 | 0.013735 | 61 |
| LOC102724312 | 0.718083931 | 0.000247 | 61 |
| LPL | 0.833774975 |
6.39×10−07 | 61 |
| SLIT1 | 0.568912151 | 0.012987 | 61 |
| MYH10 | 0.785351749 |
1.25×10−05 | 59 |
| LGALS1 | 0.500150827 | 0.039436 | 57 |
| LRRK2 | 0.843001019 |
3.24×10−07 | 57 |
Functional annotation and enrichment
analyses
The top10 significantly enriched terms in DAVID are
shown in Table II, which includes
extracellular region, integral component of plasma membrane,
negative regulation of ERK1 and ERK2 cascade, extracellular space
and cell-cell signaling.
| Table II.Module functional enrichment in
DAVID. |
Table II.
Module functional enrichment in
DAVID.
| Category | GO ID | GO term | No. of genes | P-value |
|---|
| CC | GO:0005576 | Extracellular
region | 21 |
5.4×10−04 |
| CC | GO:0005887 | Integral component
of plasma membrane | 19 |
8.1×10−04 |
| BP | GO:0070373 | Negative regulation
of ERK1 and ERK2 cascade | 4 |
4.1×10−03 |
| BP | GO:0051965 | Positive regulation
of synapse assembly | 4 |
4.9×10−03 |
| CC | GO:0005615 | Extracellular
space | 16 |
7.9×10−03 |
| BP | GO:0060976 | Coronary
vasculature development | 3 |
8.4×10−03 |
| BP | GO:0016042 | Lipid catabolic
process | 4 |
1.2×10−02 |
| BP | GO:0007411 | Axon guidance | 5 |
1.2×10−02 |
| BP | GO:0007267 | Cell-cell
signaling | 6 |
1.3×10−02 |
| MF | GO:0005198 | Structural molecule
activity | 6 |
1.5×10−02 |
Table III shows the
results of pathway enrichment based on the KEGG database with the
top5 enriched pathways. Several significantly enriched pathways are
expected to be involved in PTC, such as transcriptional
misregulations in cancer and Wnt signaling pathway (22,23).
| Table III.Pathway enrichment in
ConsensusPathDB. |
Table III.
Pathway enrichment in
ConsensusPathDB.
| Pathway name | No. of genes | Genes | P-value |
|---|
| Taste
transduction | 3 | KCNK5, CHRM3,
GABBR2 | 0.0104 |
| Transcriptional
misregulation in cancer | 4 | ETV4, ETV5, PLAU,
DUSP6 | 0.0164 |
| Regulation of actin
cytoskeleton | 4 | MYH10, RRAS, CHRM3,
CYFIP2 | 0.0288 |
| Wnt signal
pathway | 3 | CCND1, PRICKLE1,
WIF1 | 0.0424 |
| Rennin
secretion | 2 | ADORA1, KCNJ2 | 0.0489 |
Discussion
Though studies of genomic events concerning the
genetic mechanisms of PTC have achieved great progress in recent
years (3,4), few of them have noted interactions of
correlated genes thus far. Correlation network-based analysis is a
novel method for investigation of biologically interactive genes
associated with complex disease. Network analysis groups
interactive genes into one module in which genes are enriched for
specific biological pathways. Moreover, expression data used for
gene grouping are from tissues and cells involved in the disease,
so the interconnections between genes are only revealed in the
specific disease conditions.
Previous studies using expression data mainly focus
on the DEGs in order to identify potential genes involved in PTC
(24–27). In the present study, in addition to
performing differential gene expression analysis, co-expression
networks were established for the PTC and normal samples,
respectively, using the 1062 DEGs identified. Comparison of
gene-gene interaction between normal and disease networks is useful
for identifying dysregulated pathways in the process of disease.
This method enhances the ability to detect functional genes with
regards to changing crosstalk with other genes rather than merely
changing expression levels. Module preservation analysis was
applied in the present study and a poorly preserved module
comprised of 118 interactive genes was identified.
Analyzing the genes in the brown module, the k.in
rank change for each gene was calculated in order to detect gene
interaction changes when PTC occurs. Subsequently, 61 genes were
validated to be significantly relevant to PTC in the validation
analysis. LRP4 is a promising candidate gene for PTC as it ranks
first based on the k.in rank changes among the 118 genes in the
brown module and also has a high correlation coefficient (r=0.7444)
with PTC. In addition, it has been demonstrated to be associated
with PTC in previous studies. LRP4 is reported to be overexpressed
in PTC samples (28) and is included
in a molecular classifier that is able to discriminate thyroids
with PTC accurately from normal thyroids (29). KLK7 is also a potential oncogene for
PTC. KLK7 has the fifth largest k.in rank change, as shown in
Table I, as well as a significant
correlation coefficient (r=0.8085) with PTC in the validation
analysis. KLK7 has highly increased levels of expression in PTC
compared with those in normal tissue (30). Furthermore, a previous study suggested
that KLK7 promotes tumorgenesis and progress in PTC in vitro
and in vivo experiments (31).
To further investigate the functional relevance of
the identified genes in PTC, functional annotation and enrichment
analyses were performed and two significant pathways which may play
a role in pathogenesis of PTC were identified. One of the
significant pathways is the Wnt signal pathway, which has been
implicated in the genesis and development of PTC in previous
studies (22,23). CCND1 and PRICKLE1, which are enriched
in this pathway in the enrichment analysis, have both been shown to
be significantly relevant in PTC with high correlation coefficient
values (r=0.8263 and r=0.7476, respectively) in the validation
analysis. CCND1 has been reported to be a vital gene in PTC.
Studies have shown that CCND1 expression is significantly
associated with PTC growth and lymph node metastases through
aberrant activation of the Wnt/beta-catenin pathway (32,33).
PRICKLE1 is potential gene associated with PTC. PRICKLE1 encodes
prickle planar cell polarity (PCP) protein 1and is a core signaling
molecule in the Wnt/PCP signaling pathway (34,35).
Previous studies indicate that the Wnt/PCP signaling pathway plays
critical roles in the proliferation and migration of tumor cells
(36,37). The other pathway identified to be
involved in PTC is transcriptional misregulation in cancer. ETV4,
ETV5 and DUSP6, which are enriched in this pathway, were all
confirmed to be relevant to PTC in the validation analysis. DUSP6
has been reported to play an essential role in the initiation and
progression of PTC by many functional experiments (38–40). ETV4
and ETV5 are both oncogenic E26 transformation-specific family
transcription factors (41) and are
implicated in many other types of cancer, such as colorectal
(42), prostatic (43), endometrial (44) and ovarian (45) cancer. The results of the present study
provide novel direction for better understanding the molecular
mechanisms involved in PTC.
The present study has several limitations. Firstly,
not all of the genes associated with PTC that have been reported in
previous genetic studies were identified as the present study is
only a supplement to the current investigations of the genetic
mechanism of PTC. However, previous studies often focus on the
effect of a single gene on PTC, while the present study highlights
the impact of gene-gene interactions on the tumor pathological
process, which provides a novel insight for detection of potential
functional genes. Secondly, the exact effects of the identified
genes require corroboration with biological experiments. However,
validation and enrichment analyses were used to confirm the
identified genes in the present study. Certain genes, such as CCND1
and DUSP6, were confirmed that have been shown to be involved in
the pathology of PTC by previous functional and experimental
studies.
In conclusion, the present study applied correlated
network analysis in order to concentrate on gene interactions.
Identified genes were validated using another set of expression
data and then functional enrichment analysis was used to identify
more biological associations. A number of novel genes, such as
LRP4, KLK7, PRICKLE1, ETV4 and ETV5, and pathways (Wnt signal
pathway and transcriptional misregulation in cancer) were
identified that may exert important functions in PTC. These results
contribute to the understanding of the genetic basis of PTC and
provide novel insights into the identification of potential
functional genes in PTC.
Acknowledgements
Not applicable.
Funding
The present study partially supported by grants from
The National Institutes of Health (grant nos. R01AR069055, U19
AG055373, R01 MH104680, R01AR059781 and P20 GM109036); The Edward
G. Schlieder Endowment fund from Tulane University; The National
Natural Science Foundation of China (grant no. 81302228); The
Foundation for Pearl River Nova program of Guangzhou (grant no.
2014J2200034); and The Technological Innovation Project of Foshan
City (grant no. 2017AG100102).
Availability of data and materials
The datasets used during the current study are
available in the NCBI Gene Expression Omnibus database, with the
accession numbers are GSE33630 and GSE3678.
Authors' contributions
HWD, JS, ZC and HMX conceived and designed the
study. ZXA, YCC, JML, LPP and XL analyzed the data. ZXA, HWD, YCC,
ZC and HMX wrote the manuscript. CP, CPZ, XFW and RZ collected the
data and preprocessed the raw data.
Ethics approval and consent to
participate
The research was approved by the Ethics Committees
of the Institute of Endocrinology and Metabolism in Kiev, Imperial
College in London and the Medical Radiological Research Centre in
Obninsk.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kitahara CM and Sosa JA: The changing
incidence of thyroid cancer. Nat Rev Endocrinol. 12:646–653. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schneider DF and Chen H: New developments
in the diagnosis and treatment of thyroid cancer. CA Cancer J Clin.
63:374–394. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Research Network, .
Integrated genomic characterization of papillary thyroid carcinoma.
Cell. 159:676–690. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ye L, Zhou X, Huang F, Wang W, Qi Y, Xu H,
Yang S, Shen L, Fei X, Xie J, et al: The genetic landscape of
benign thyroid nodules revealed by whole exome and transcriptome
sequencing. Nat Commun. 8:155332017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hsiao SJ and Nikiforov YE: Molecular
approaches to thyroid cancer diagnosis. Endocr Relat Cancer.
21:T301–T313. 2014.PubMed/NCBI
|
|
6
|
Xing M, Haugen BR and Schlumberger M:
Progress in molecular-based management of differentiated thyroid
cancer. Lancet. 381:1058–1069. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eichler EE, Flint J, Gibson G, Kong A,
Leal SM, Moore JH and Nadeau JH: Missing heritability and
strategies for finding the underlying causes of complex disease.
Nat Rev Genet. 11:446–450. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu W and Ye H: Co-expression network
analysis identifies transcriptional modules in the mouse liver. Mol
Genet Genomics. 289:847–853. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen YC, Guo YF, He H, Lin X, Wang XF,
Zhou R, Li WT, Pan DY, Shen J and Deng HW: Integrative analysis of
genomics and transcriptome data to identify potential functional
genes of BMDs in females. J Bone Miner Res. 31:1041–1049. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kogelman LJ, Cirera S, Zhernakova DV,
Fredholm M, Franke L and Kadarmideen HN: Identification of
co-expression gene networks, regulatory genes and pathways for
obesity based on adipose tissue RNA sequencing in a porcine model.
BMC Med Genomics. 7:572014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Plaisier CL, Horvath S, Huertas-Vazquez A,
Cruz-Bautista I, Herrera MF, Tusie-Luna T, Aguilar-Salinas C and
Pajukanta P: A systems genetics approach implicates USF1, FADS3,
and other causal candidate genes for familial combined
hyperlipidemia. PLoS Genet. 5:e10006422009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang B, He L, Miao W, Wu G, Jiang H, Wu Y,
Qu J and Li M: Identification of key genes associated with
Schmid-type metaphyseal chondrodysplasia based on microarray data.
Int J Mol Med. 39:1428–1436. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Riquelme Medina I and Lubovac-Pilav Z:
Gene Co-expression network analysis for identifying modules and
functionally enriched pathways in type 1 diabetes. PLoS One.
11:e01560062016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tomás G, Tarabichi M, Gacquer D, Hébrant
A, Dom G, Dumont JE, Keutgen X, Fahey TJ III, Maenhaut C and
Detours V: A general method to derive robust organ-specific gene
expression-based differentiation indices: Application to thyroid
cancer diagnostic. Oncogene. 31:4490–4498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Smyth GK: Linear models and empirical
bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article 3. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Langfelder P, Luo R, Oldham MC and Horvath
S: Is my network module preserved and reproducible? PLoS Comput
Biol. 7:e10010572011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Herwig R, Hardt C, Lienhard M and Kamburov
A: Analyzing and interpreting genome data at the network level with
ConsensusPathDB. Nat Protoc. 11:1889–1907. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gilbert-Sirieix M, Makoukji J, Kimura S,
Talbot M, Caillou B, Massaad C and Massaad-Massade L: Wnt/β-catenin
signaling pathway is a direct enhancer of thyroid transcription
factor-1 in human papillary thyroid carcinoma cells. PLoS One.
6:e222802011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ishigaki K, Namba H, Nakashima M, Nakayama
T, Mitsutake N, Hayashi T, Maeda S, Ichinose M, Kanematsu T and
Yamashita S: Aberrant localization of beta-catenin correlates with
overexpression of its target gene in human papillary thyroid
cancer. J Clin Endocrinol Metab. 87:3433–3440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fluge Ø, Bruland O, Akslen LA, Lillehaug
JR and Varhaug JE: Gene expression in poorly differentiated
papillary thyroid carcinomas. Thyroid. 16:161–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qiu J, Zhang W, Xia Q, Liu F, Li L, Zhao
S, Gao X, Zang C, Ge R and Sun Y: RNA sequencing identifies crucial
genes in papillary thyroid carcinoma (PTC) progression. Exp Mol
Pathol. 100:151–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qu T, Li YP, Li XH and Chen Y:
Identification of potential biomarkers and drugs for papillary
thyroid cancer based on gene expression profile analysis. Mol Med
Rep. 14:5041–5048. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu X, Yao J and Tian W: Microarray
technology to investigate genes associated with papillary thyroid
carcinoma. Mol Med Rep. 11:3729–3733. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hucz J, Kowalska M, Jarzab M and Wiench M:
Gene expression of metalloproteinase 11, claudin 1 and selected
adhesion related genes in papillary thyroid cancer. Endokrynol Pol.
57 Suppl A:18–25. 2006.(In Polish). PubMed/NCBI
|
|
29
|
Jarzab B, Wiench M, Fujarewicz K, Simek K,
Jarzab M, Oczko-Wojciechowska M, Wloch J, Czarniecka A, Chmielik E,
Lange D, et al: Gene expression profile of papillary thyroid
cancer: Sources of variability and diagnostic implications. Cancer
Res. 65:1587–1597. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim HS, Kim DH, Kim JY, Jeoung NH, Lee IK,
Bong JG and Jung ED: Microarray analysis of papillary thyroid
cancers in Korean. Korean J Intern Med. 25:399–407. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H, Cai Y, Zheng L, Zhang Z, Lin X
and Jiang N: Long noncoding RNA NEAT1 regulate papillary thyroid
cancer progression by modulating miR-129-5p/KLK7 expression. J Cell
Physiol. 233:6638–6648. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lamba Saini M, Weynand B, Rahier J, Mourad
M, Hamoir M and Marbaix E: Cyclin D1 in well differentiated thyroid
tumour of uncertain malignant potential. Diagn Pathol. 10:322015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lantsov D, Meirmanov S, Nakashima M, Kondo
H, Saenko V, Naruke Y, Namba H, Ito M, Abrosimov A, Lushnikov E, et
al: Cyclin D1 overexpression in thyroid papillary microcarcinoma:
Its association with tumour size and aberrant beta-catenin
expression. Histopathology. 47:248–256. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo D, Yuan Z, Ru J, Gu X, Zhang W, Mao F,
Ouyang H, Wu K, Liu Y and Liu C: A spatiotemporal requirement for
prickle 1-mediated PCP signaling in eyelid morphogenesis and
homeostasis. Invest Ophthalmol Vis Sci. 59:952–966. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dyberg C, Papachristou P, Haug BH,
Lagercrantz H, Kogner P, Ringstedt T, Wickström M and Johnsen JI:
Planar cell polarity gene expression correlates with tumor cell
viability and prognostic outcome in neuroblastoma. BMC Cancer.
16:2592016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vander Vorst K, Hatakeyama J, Berg A, Lee
H and Carraway KL III: Cellular and molecular mechanisms underlying
planar cell polarity pathway contributions to cancer malignancy.
Semin Cell Dev Biol. Nov 4–2017.(Epub ahead of print).
|
|
37
|
Katoh M: WNT/PCP signaling pathway and
human cancer (review). Oncol Rep. 14:1583–1588. 2005.PubMed/NCBI
|
|
38
|
Lee JU, Huang S, Lee MH, Lee SE, Ryu MJ,
Kim SJ, Kim YK, Kim SY, Joung KH, Kim JM, et al: Dual specificity
phosphatase 6 as a predictor of invasiveness in papillary thyroid
cancer. Eur J Endocrinol. 167:93–101. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Degl'Innocenti D, Romeo P, Tarantino E,
Sensi M, Cassinelli G, Catalano V, Lanzi C, Perrone F, Pilotti S,
Seregni E, et al: DUSP6/MKP3 is overexpressed in papillary and
poorly differentiated thyroid carcinoma and contributes to
neoplastic properties of thyroid cancer cells. Endocr Relat Cancer.
20:23–37. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gu Y, Li D, Luo Q, Wei C, Song H, Hua K,
Song J, Luo Y, Li X and Fang L: MicroRNA-145 inhibits human
papillary cancer TPC1 cell proliferation by targeting DUSP6. Int J
Clin Exp Med. 8:8590–8598. 2015.PubMed/NCBI
|
|
41
|
Kedage V, Selvaraj N, Nicholas TR, Budka
JA, Plotnik JP, Jerde TJ and Hollenhorst PC: An interaction with
ewing's sarcoma breakpoint protein EWS defines a specific oncogenic
mechanism of ETS factors rearranged in prostate cancer. Cell Rep.
17:1289–1301. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xiao J, Yang S, Shen P, Wang Y, Sun H, Ji
F and Zhou D: Phosphorylation of ETV4 at Ser73 by ERK kinase could
block ETV4 ubiquitination degradation in colorectal cancer. Biochem
Biophys Res Commun. 486:1062–1068. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qi M, Liu Z, Shen C, Wang L, Zeng J, Wang
C, Li C, Fu W, Sun Y and Han B: Overexpression of ETV4 is
associated with poor prognosis in prostate cancer: Involvement of
uPA/uPAR and MMPs. Tumour Biol. 36:3565–3572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pedrola N, Devis L, Llauradó M, Campoy I,
Martinez-Garcia E, Garcia M, Muinelo-Romay L, Alonso-Alconada L,
Abal M, Alameda F, et al: Nidogen 1 and Nuclear Protein 1: Novel
targets of ETV5 transcription factor involved in endometrial cancer
invasion. Clin Exp Metastasis. 32:467–478. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Llauradó M, Majem B, Castellví J, Cabrera
S, Gil-Moreno A, Reventós J and Ruiz A: Analysis of gene expression
regulated by the ETV5 transcription factor in OV90 ovarian cancer
cells identifies FOXM1 overexpression in ovarian cancer. Mol Cancer
Res. 10:914–924. 2012. View Article : Google Scholar : PubMed/NCBI
|