Introduction
Signal transducer and activator of transcription 3
(STAT3) is implicated in angiogenesis, apoptosis, cell-cycle
progression, cell migration and drug resistance by regulating the
expression of phenotype-associated genes (1,2). Canonical
STAT3-mediated signaling involves the phosphorylation of specific
tyrosine residues that leads to homodimerization and translocation
to the nucleus upon the stimulation of cytokines and growth factors
(3,4).
Nuclear STAT3 fine-tunes autophagy by regulating the
transcriptional expression of autophagy-associated genes and
autophagy-targeting microRNAs, whereas its cytoplasmic counterpart
constitutively suppresses autophagy by sequestering eukaryotic
translation initiation factor 2-α kinase 2 or interacting with
forkhead box O (FOXO)1 and FOXO3 (4,5).
Additionally, STAT3 promotes mitochondrial transcription and
oxidative respiration. The mitochondrial translocation of STAT3
serves a major role in IgE-antigen-mediated mast cell exocytosis
and optimal electron transport chain activity, and protects against
oxidative-stress-induced mitochondrial dysfunction and autophagy
(6–8).
Although the levels of activated STAT3 remain
transient in normal cells, it is overactive in ~70% of human solid
tumors and regulates the expression of numerous oncogenic genes
that control the growth and metastasis of tumor cells (3). A meta-analysis revealed that elevated
phosphorylated (p)-STAT3 levels are significantly associated with
tumor cell differentiation, lymph node metastases and poor overall
and disease-free survival rates inpatients with cancer of the
digestive system (9). Janus kinase
(JAK)-STAT3 signaling promotes oncogenesis by upregulating the
proliferation, survival, invasion of tumor cells and
immunosuppression (10). In recent
years, antitumor approaches targeting STAT3 activity include the
inhibition of tyrosine kinases or STAT3 dimer formation (11,12). The
present study directly silenced STAT3 and observed the effects on
the proliferation and growth of colorectal cancer (CRC) cells.
Additionally, the associated molecular mechanisms were also
clarified.
Materials and methods
Cell culture and transfection
HCT-116 and SW480 cells were obtained from the Cell
Resource Center, Institute of Basic Medicine, Peking Union Medical
University (Beijing, China) and grown in a humidified atmosphere
with 5% CO2 at 37°C in Dulbecco's modified Eagle's
medium (DMEM) with 10% fetal bovine serum (FBS; Corning
Incorporated, Corning, NY, USA). Penicillin (100 U/ml) and
streptomycin (100 µg/ml) were added to the medium. All cells were
collected by trypsinization at 37°C followed by centrifugation
(3,000 × g, 5 min, room temperature), washed in phosphate buffered
saline (PBS) and subjected to total RNA and protein extraction.
Either 1.0×106 HCT-116 or SW480 cells
were transfected with pSilencer3.0-H1-STAT3-siRNA-GFP at 500 ng/µl
(pSH1Si-STAT3 group) or pSH1Si-Scramble at 500 ng/µl (mock group)
following seeding on dishes according to the manufacturer's
instructions of Attractene Transfection Reagent (301005; Qiagen,
Inc., Valencia, CA, USA). The untreated cells were used as control.
After 2 h of transfection, cells were selected by G418 with the
collection of monoclones, and immediately used for following
proliferation assay, Acridine orange (AO)/ethidium bromide (EB)
staining, PI staining and animal experiments were performed
following transfection. The shRNA targets STAT3 nucleotides
2144–2162 (GenBank accession no. NM00315): Forward,
5′-GCAGCAGCTGAACAACATGTTCAAGAGACATGTTGTTCAGCTGCTGCTTTTT-3′ and
reverse,
5′-AATTAAAAAGCAGCAGCTGAACAACATGTCTCTTGAACATGTTGTTCAGCTGCTGCTGCGGCC-3′.
Proliferation assay
HCT-116, SW480 and pSH1Si-STAT3-transfected cells
were seeded at 2.0×103 cells per well in 96-well plates
and maintained in media containing 10% FBS. At 0, 24, 48 and 72 h,
the cells then were incubated with 20 µl MTT (5 mg/ml) for 4 h at
37°C. The medium was then discarded and 150 µl dimethyl sulfoxide
was added to each well to dissolve the precipitates. The number of
viable cells was counted by measuring the absorbance at 490 nm
using a microplate reader. The experiment was repeated three
times.
AO/EB staining
AO is a nucleic-acid-selective fluorescent cationic
dye that can pass through live and dead cells. Upon treatment with
the two reagents, live cells appear uniformly green under a
fluorescence microscope, whereas apoptotic cells stain green and
bright green dots appear in the nuclei. Necrotic cells stain orange
under a fluorescence microscope. EB can only stain cells that have
lost membrane integrity. In brief, HCT-116, SW480, pSH1Si-Scramble-
and pSH1Si-STAT3-transfected cells were resuspended in an AO
solution at 1×1010 cells/l. A total of 2 µl AO solution
(100 mg/ml) and 2 µl EB solution (100 mg/ml) were added into 20 µl
cells for 5 min at room temperature. Next, the cells were seeded
onto glass slides and observed in 5 fields under a fluorescence
microscope (×200 magnification).
Flow cytometry
HCT-116 and SW480, pSH1Si-Scramble- and
pSH1Si-STAT3-transfected cells were washed twice with PBS then
detached by trypsinization with free EDTA. The cells were collected
in a centrifuge tube and incubated with 1 ml precooled 70% ethanol
for ≥12 h. The cells were washed twice with PBS again and incubated
for 1 h at 37°C with 1 ml RNase (0.25 mg/ml, Sigma-Aldrich, Merck
KGaA, Darmstadt, Germany). The cells were resuspended in propidium
iodide (PI) at a concentration of 50 µg/ml, and cell cycle analysis
was performed using a flow cytometer with CytExpert software
(version 2.0; Beckman Coulter, Inc., Brea, CA, USA) used for
analysis.
Xenograft models
A total of 15 male BALB/c nude mice of 6–8 weeks
(17.76±1.83 g) were kept in a specific pathogen-free facility in a
temperature-controlled animal room (22–24°C) with a 12/12 h
light/dark illumination cycle. All of the mice had access to
standard rodent food (Beijing Huafukang Biotech Co., Ltd., Beijing,
China) and water ad libitum. All animal treatments were
performed in accordance with international ethics guidelines and
the National Institutes of Health Care and Use of Laboratory
Animals. This study was approved by the Institutional Animal Care
and Use Committee of The First Affiliated Hospital of Jinzhou
Medical University (Jinzhou, China).
HCT-116, pSH1Si-Scramble- and
pSH1Si-STAT3-transfected cells were grown in the medium as
aforementioned. The cells were detached by trypsinization, and
subsequently washed and resuspended in DMEM medium. Subcutaneous
xenografts were established by injection of 1.0×107
HCT-116 cells/mouse (n=5/group), and the mice were assessed every 4
days for tumor size and body weight. The model was established
successfully when there were nodules with the size of a grain of
rice after 1week and a volume of 50–70 mm3 after 2
weeks.
The nude mice were divided into control, mock and
treatment groups, and each group consisted of 5 mice. The treatment
volume was 40 µl, and the plasmid concentration was 0.5 µg/µl,
which was injected intratumorally at several points. At 30 sec
after injection, an electrical impulse was applied twice at
intervals of >1 min. Electrodes were placed at each end of the
tumor's long and short axes (electric field strength, 200 V/cm;
electric pulse duration, 50 msec and pulse frequency, 1Hz). This
process was repeated on days 10 and 17. Tumor volume and mouse body
weight were measured every 4 days. On day 20, the tumors were
removed, and the tumor weight and volume were measured. Tumor
growth was calculated as follows: Length × width2 ×
0.52. At the end of the experiment, the mice from each group were
anesthetized, their images were captured and the mice were
sacrificed. The tumors were weighed and subjected to RNA or protein
extraction for subsequent experiments.
Reverse transcriptase-polymerase chain
reaction (RT-PCR)
Total RNA was isolated from CRC cells or tissues
using TRIzol (Takara Bio, Inc., Otsu, Japan) and quantified using a
NanoDrop spectrophotometer (NanoDrop Technologies; Thermo Fisher
Scientific, Inc., Wilmington, DE USA). RT-PCR was performed with 2
µg total RNA using AMV reverse transcriptase and random primers at
42°C for 1 h (Takara Bio, Inc.). PCR primers were designed and are
listed in Table I. The amplification
of cDNA was performed using the Hot Start Taq Polymerase kit
(Takara Bio, Inc.) with GAPDH as an internal control. The PCR
conditions were as follows: Denaturation at 95°C for 10 min;
followed by 30 cycles of denaturation at 95°C for 15 sec, annealing
at 55°C for 30 sec and extension at 72°C for 30 sec; and as a
termination step, the extension time of the last cycle was
increased to 7 min. The amplicons were electrophoresed in 2%
agarose and visualized under UV light. Densitometric quantification
was performed with β-actin as a control using ImageJ version 1.48
(National Institutes of Health, Bethesda, MD, USA). The value of
the control was set to 1.
 | Table I.Primers used for reverse
transcription-semi-quantitative polymerase chain reaction. |
Table I.
Primers used for reverse
transcription-semi-quantitative polymerase chain reaction.
| Gene | Primer sequence | AT (°C) | Product size
(bp) | Extension time
(sec) |
|---|
| STAT3 |
| 55 | 315 | 30 |
|
Forward |
5′-TTGCCAGTTGTGGTGATC-3′ |
|
|
|
|
Reverse |
5′-AGACCCAGAAGGAGCCGC-3′ |
|
|
|
| Survivin |
|
|
|
|
|
Forward |
5′GAATTCATGGGTGCCCCGACCTTGCC-3′ | 58 | 412 | 30 |
|
Reverse |
5′-AGATCTTTCTTCTTATTGTTGGTTTCC-3′ |
|
|
|
| Caspase-3 |
| 55 | 241 | 30 |
|
Forward |
5′-AGAACTGGACTGTGGCATTG-3′ |
|
|
|
|
Reverse |
5′-TTCTGTTGCCACCTTTCG-3′ |
|
|
|
| β-actin |
|
|
|
|
|
Forward |
5′-AAGTACTCCGTGTGGATCGG-3′ |
|
|
|
|
Reverse |
5′-ATGCTATCACCTCCCCTGTG-3′ | 55 | 600 | 30 |
Western blot analysis
Protein extraction was performed from gastric
carcinoma and epithelial cells or carcinoma tissues by sonication
or homogenization in radioimmunoprecipitation assay lysis buffer
[50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 0.5% Nonidet P-40
(Sigma-Aldrich; Merck KGaA), 5 mM dithiothreitol, 10 mM NaF and
protease inhibitor cocktail (Nacalai Tesque, Inc., Kyoto, Japan)].
A total of 40 µg protein was measured and calculated according to
Kaumas brilliant blue method (13)
from HCT-116, SW480 and pSH1Si-STAT3-transfected cells, and tumors
of HCT-116 and pSH1Si-STAT3-transfected cells per lane. The protein
samples were resolved using 10% SDS-PAGE and transferred to a
polyvinylidene fluoride membrane. The membrane was blocked with 5%
skimmed milk in Tris-buffered saline with Tween 20 (TBST; 10 mM
Tris-HCl, 150 mM NaCl, 0.1% Tween 20) for 1 h at room temperature,
and the primary antibodies were placed on a shaker at 4°C
overnight. The antibodies against β-actin (sc-47778; 1:1,000),
p-STAT3 (sc-56747; 1:500), STAT3 (sc-293151; 1:500), surviving
(sc-101433; 1:500), tumor protein p53 (sc-47698; 1:500) and
caspase-3 (sc-136219; 1:500) were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The cells were rinsed with
TBST and incubated with IgG antibody that was conjugated to
horseradish peroxidase (HRP; Dako, Carpinteria CA, USA) at a
dilution of 1:1,000 at room temperature for 1 h. The bands were
visualized using LAS4010 (GE Healthcare Life Sciences, Little
Chalfont, USA) and ECL-Plus detection reagents (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). The experiment was repeated
for three times. Densitometric quantification was performed with
β-actin as a control using ImageJ version 1.48. The control was
considered to have a value of ‘1’.
Statistical analysis
The results are expressed as mean ± standard
deviation. Statistical evaluation was performed using
Kruskal-Wallis to compare the means of different groups. P<0.05
was considered to indicate statistical significance. SPSS 10.0
software (SPSS, Inc., Chicago, IL, USA) was used to analyze all
data.
Results
Effects and associated mechanisms of
STAT3 silencing on the proliferation and apoptosis of CRC
cells
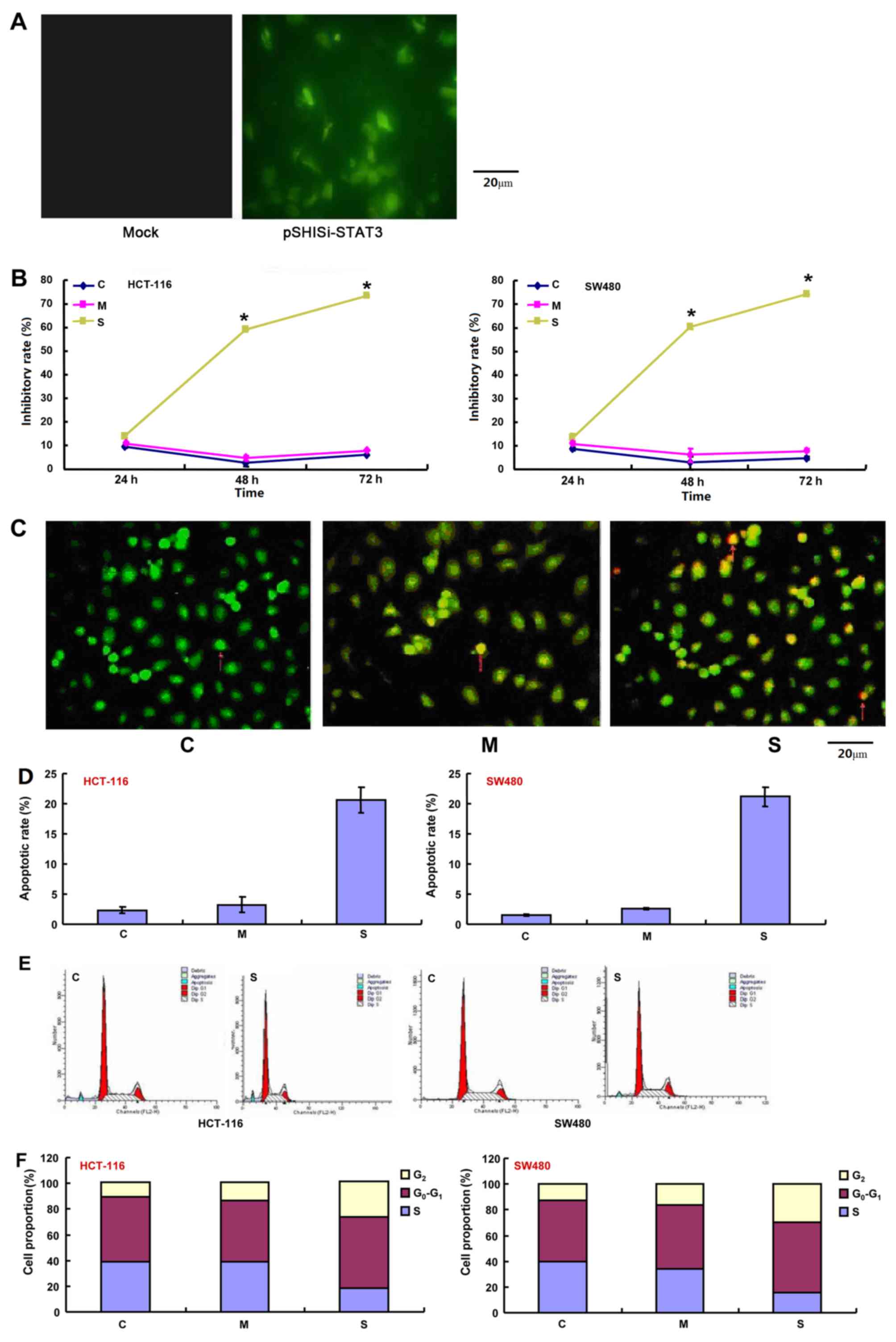
pSH1-Si-STAT3 and pSH1Si-Scramble were successfully
transfected into CRC cells as indicated by the findings from
fluorescence microscopy (Fig. 1A).
The effect of STAT3 silencing on the viability of HCT-116 and SW480
cells was assessed using an MTT assay. As shown in Fig. 1B, the proliferation of the two cell
lines transfected with the pSH1-siRNA-STAT3 plasmid was
significantly suppressed 48 h after transfection compared with the
control and pSH1Si-scramble groups (P<0.05). There was a
significantly higher rate of apoptosis in CRC cells that were
treated with the pSH1-siRNA-STAT3 plasmids than those in the
control and mock groups as indicated by AO/EB and PI staining
(P<0.05; Fig. 1C and D). Cell
cycle analysis revealed a significantly higher proportion of
STAT3-silenced cells in G2/M-phase arrest compared with
the control and mock groups (P<0.05; Fig. 1E and F). The mRNA levels of STAT3 and
survivin were significantly decreased in all cells and tumor
tissues, but the expression of p53 and caspase-3 mRNA was
significantly increased in the pSH1-siRNA-STAT3 group, compared
with the control and mock plasmid groups (P<0.05; Fig. 1G). Western blot analysis revealed that
the expression of p-STAT3, STAT3 and survivin was significantly
lower in cells that were transfected with pSH1-siRNA-STAT3,
compared with the control- and mock plasmid-transfected groups, but
the opposite was true for p53 and caspase-3 (P<0.05; Fig. 1H).
Effects and associated mechanisms of
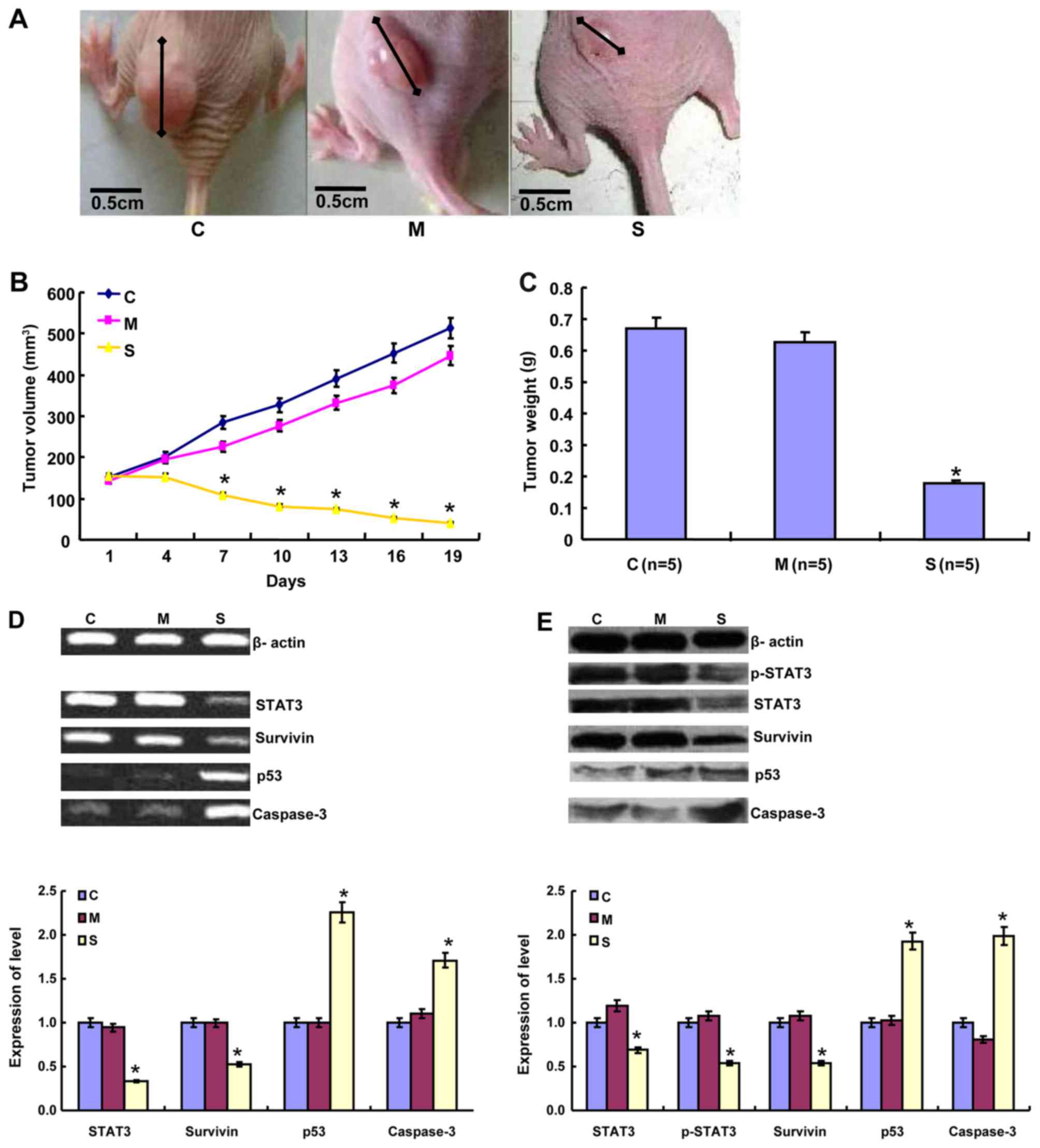
STAT3 silencing on tumor growth of CRC cells
The volume and weight of the mouse tumors in the
pSH1-siRNA-STAT3 group were significantly smaller and lower
compared with the control- and mock plasmid-transfected groups
(P<0.05; Fig. 2A-C). The longest
diameter of a single subcutaneous tumor was 11.2 mm in the present
study. The mRNA levels of STAT3 and survivin were significantly
decreased in all cells and tumor tissues, but the expression of p53
and caspase-3 mRNA was significantly increased in the
pSH1-siRNA-STAT3 group, compared with the control and mock plasmid
groups (P<0.05; Fig. 2D). Western
blot analysis revealed that the expression of p-STAT3, STAT3 and
survivin was significantly lower in cells that were transfected
with pSH1-siRNA-STAT3, compared with the control- and mock
plasmid-transfected groups, but the opposite was true for p53 and
caspase-3 (P<0.05; Fig. 2E).
Discussion
Increased STAT3 activity is common in human
malignancies, including CRC. Reportedly, p-STAT3 immunoreactivity
gradually increases from normal mucosa to adenoma to
adenocarcinoma. p-STAT3 immunoreactivity is positively associated
with the T stage and clinical stage of CRC. In a previous study
involving the analysis of 47 invasive colorectal adenocarcinoma
samples, STAT3 mRNA expression was significantly associated with
tumor size, lymph node and metastasis stage and clinical stage of
the disease (9). In CRC, nuclear
p-STAT3 protein expression was associated with the expression of
its target genes B-cell lymphoma-extra large (Bcl-xL) and survivin
as well as Ki-67. The inactive (UP-STAT3) and active (p-STAT3)
STAT3 proteins are markedly increased in invasive CRC cases. This
increase is associated with Bcl-xL and survivin induction,
increased proliferation and lymph node metastasis (9,14,15). These findings indicate that STAT3
activation contributes to colorectal carcinogenesis and subsequent
disease progression. In the present study, STAT3 knockdown
significantly suppressed cell proliferation and tumor growth, and
induced apoptosis and G2/M arrest in HCT-116 and SW480
cells, suggesting that STAT3 could be employed as a potential
target of gene therapy for patients with CRC.
Hypoxia-inducible factor 3α1 and miR-139-5p deletion
promote the growth of CRC cells by activating the JAK-STAT3
signaling pathway (16,17). The insulin-like growth
factor/STAT3/NANOG/Slug and interleukin-6/STAT3/Fos-related
antigen-1 signaling axes simultaneously control
epithelial-mesenchymal transition and maintenance of stemness in
CRC, which exhibit drug resistance, invasion and metastasis
(18,19). It was documented that STAT3 inhibition
sensitized CRC to chemoradiotherapy in vitro and in
vivo. Furthermore, the overexpression of STAT3 was associated
with the resistance of CRC cells to 5-fluorouracil-based
chemoradiotherapy (20,21). The inhibition of DNA methyltransferase
by miR-124 and miR-874 was demonstrated to suppress growth and
induce apoptosis in CRC by targeting STAT3, where STAT3 is often
hyper-activated (22–24). The overexpression of B7-H3 was able to
induce resistance to apoptosis in CRC cell lines by upregulating
the Jak2-STAT3 signaling pathway, whereas suppressor of cytokine
signaling 3 served as a negative regulator of the JAK-STAT3 pathway
to suppress cell proliferation and cell cycle (25–27). In
addition, the epidermal growth factor/phosphoinositide-3
kinase/STAT3 signaling pathway enhanced the expression of
angiogenic regulators, including vascular endothelial growth factor
and leptin. The inhibition of STAT3 was able to suppress
angiogenesis in CRC (28,29). These data underlie the molecular basis
of gene and drug therapies targeting STAT3.
In the present study, following STAT3 knockdown, the
levels of p-STAT3 and STAT3 were downregulated in HCT-116 and SW480
cells or tumors, indicating that the transfections were effective.
To clarify the molecular mechanisms, the expression of survivin,
p53 and caspase-3 at mRNA and protein levels was detected. The
silencing of STAT3 was able to markedly decrease survivin
expression, but the expression of p53 and caspase-3 was increased.
Survivin is an oncogene that has been implicated in several types
of human cancer. Survivin protein ameliorates caspase activation to
negatively regulate apoptosis or programmed cell death and
localizes to the mitotic spindle by interacting with tubulin during
mitosis to modulate mitosis (30).
Caspase-3 is activated in apoptotic cells by extrinsic (death
ligands) and intrinsic (mitochondrial) pathways, which results in
the cleavage of poly (ADP-ribose) polymerase, actin and the sterol
regulatory element binding protein (31). p53 protects against the development of
cancer, including the induction of apoptosis, cell cycle arrest and
the maintenance of genetic stability (32). Taking the results of the present study
together, it was concluded that STAT3 promoted the proliferation
and growth of CRC cells and suppressed apoptosis by targeting the
expression of survivin, p53 and caspase-3 and could therefore be
considered to be a potential target for gene therapy.
In conclusion, the silencing expression of STAT3
in vitro and in vivo may have a therapeutic effect in
CRC cells, which brings hope of clinical therapy using RNA
interference via oligonucleotide drugs. Future study will continue
to investigate the synergistic effects of STAT3-knockdown and other
therapeutic approaches to inhibit the proliferation and induce the
apoptosis of CRC cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Liaoning Bai
Qian Wan Talents Program, the Scientific Research Fund of Liaoning
Provincial Education Department (grant no. LJQ2014093), Outstanding
Scientific Fund of Shengjing Hospital, Award for Liaoning
Distinguished Professor, Shenyang Science and Technology Grand
(grant no. 18-013-0-59), the Key Scientific and Technological
Project of Liaoning Province (grant nos. 2015408001, 20101064 and
2013225305) and the National Natural Scientific Foundation of China
(grant nos. 81472544 and 81672700).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL, XFY, DFS, YYL, HZS, KQH and HCZ designed the
study, performed the experiment and statistical analysis. JL, KQH
and HCZ prepared reviewed the manuscript. All the authors read the
manuscript, took responsibility for all aspects of the work and
approved the manuscript published without conflict of interest.
Ethics approval and consent to
participate
The ethics of animal performance protocol was
approved by Institutional Animal Care and Use Committee of The
First Affiliated Hospital of Jinzhou Medical University (Jinzhou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wake MS and Watson CJ: STAT3 the
oncogene-still eluding therapy? FEBS J. 282:2600–2611. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao C, Li H, Lin HJ, Yang S, Lin J and
Liang G: Feedback activation of STAT3 as a cancer drug-resistance
mechanism. Trends Pharmacol Sci. 37:47–61. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bromberg JF, Wrzeszczynska MH, Devgan G,
Zhao Y, Pestell RG, Albanese C and Darnell JE Jr: Stat3 as an
oncogene. Cell. 98:295–303. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Srivastava J and DiGiovanni J:
Non-canonical Stat3 signaling in cancer. Mol Carcinog.
55:1889–1898. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
You L, Wang Z, Li H, Shou J, Jing Z, Xie
J, Sui X, Pan H and Han W: The role of STAT3 in autophagy.
Autophagy. 11:729–739. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signaling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Carbognin E, Betto RM, Soriano ME, Smith
AG and Martello G: Stat3 promotes mitochondrial transcription and
oxidative respiration during maintenance and induction of naive
pluripotency. EMBO J. 35:618–634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Erlich TH, Yagil Z, Kay G, Peretz A,
Migalovich-Sheikhet H, Tshori S, Nechushtan H, Levi-Schaffer F,
Saada A and Razin E: Mitochondrial STAT3 plays a major role in
IgE-antigen-mediated mast cell exocytosis. J Allergy Clin Immunol.
134:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Park JK, Hong R, Kim KJ, Lee TB and Lim
SC: Significance of p-STAT3 expression in human colorectal
adenocarcinoma. Oncol Rep. 20:597–604. 2008.PubMed/NCBI
|
|
10
|
Ferguson SD, Srinivasan VM and Heimberger
AB: The role of STAT3 in tumor-mediated immune suppression. J
Neurooncol. 123:385–394. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Furtek SL, Backos DS, Matheson CJ and
Reigan P: Strategies and approaches of targeting STAT3 for cancer
treatment. ACS Chem Biol. 11:308–318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chai EZ, Shanmugam MK, Arfuso F,
Dharmarajan A, Wang C, Kumar AP, Samy RP, Lim LH, Wang L, Goh BC,
et al: Targeting transcription factor STAT3 for cancer prevention
and therapy. Pharmacol Ther. 162:86–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhao Y, Chen S, Gou WF, Niu ZF, Zhao S,
Xiao LJ, Takano Y and Zheng HC: The role of EMMPRIN expression in
ovarian epithelial carcinomas. Cell Cycle. 12:2899–2913. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lassmann S, Schuster I, Walch A, Göbel H,
Jütting U, Makowiec F, Hopt U and Werner M: STAT3 mRNA and protein
expression in colorectal cancer: Effects on STAT3-inducible targets
linked to cell survival and proliferation. J Clin Pathol.
60:173–179. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma XT, Wang S, Ye YJ, Du RY, Cui ZR and
Somsouk M: Constitutive activation of Stat3 signaling pathway in
human colorectal carcinoma. World J Gastroentero. 10:1569–1573.
2004. View Article : Google Scholar
|
|
16
|
Xue X, Jungles K, Onder G, Samhoun J,
Győrffy B and Hardiman KM: HIF-3α1 promotes colorectal tumor cell
growth by activation of JAK-STAT3 signaling. Oncotarget.
7:11567–11579. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zou F, Mao R, Yang L, Lin S, Lei K, Zheng
Y, Ding Y, Zhang P, Cai G, Liang X and Liu J: Targeted deletion of
miR-139-5p activates MAPK, NF-κB and STAT3 signaling and promotes
intestinal inflammation and colorectal cancer. FEBS J.
283:1438–1452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao C, Su L, Shan J, Zhu C, Liu L, Liu C,
Xu Y, Yang Z, Bian X, Shao J, et al: IGF/STAT3/NANOG/Slug signaling
axis simultaneously controls epithelial-mesenchymal transition and
stemness maintenance in colorectal cancer. Stem Cells. 34:820–831.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu H, Ren G, Wang T, Chen Y, Gong C, Bai
Y, Wang B, Qi H, Shen J, Zhu L, et al: Aberrantly expressed Fra-1
by IL-6/STAT3 transactivation promotes colorectal cancer
aggressiveness through epithelial-mesenchymal transition.
Carcinogenesis. 36:459–468. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Spitzner M, Roesler B, Bielfeld C, Emons
G, Gaedcke J, Wolff HA, Rave-Fränk M, Kramer F, Beissbarth T, Kitz
J, et al: STAT3 inhibition sensitizes colorectal cancer to
chemoradiotherapy in vitro and in vivo. Int J Cancer. 134:997–1007.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan LC, Teng HW, Shiau CW, Tai WT, Hung
MH, Yang SH, Jiang JK and Chen KF: Pharmacological targeting
SHP-1-STAT3 signaling is a promising therapeutic approach for the
treatment of colorectal cancer. Neoplasia. 17:687–696. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong H, Chen ZF, Liang QC, Du W, Chen HM,
Su WY, Chen GQ, Han ZG and Fang JY: Inhibition of DNA
methyltransferase induces G2 cell cycle arrest and apoptosis in
human colorectal cancer cells via inhibition of JAK2/STAT3/STAT5
signalling. J Cell Mol Med. 13:3668–3679. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao B and Dong AS: MiR-874 inhibits cell
growth and induces apoptosis by targeting STAT3 in human colorectal
cancer cells. Eur Rev Med Pharmacol Sci. 20:269–277.
2016.PubMed/NCBI
|
|
24
|
Zhang J, Lu Y, Yue X, Li H, Luo X, Wang Y,
Wang K and Wan J: MiR-124 suppresses growth of human colorectal
cancer by inhibiting STAT3. PLoS One. 8:e703002013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu F, Zhang T, Zou S, Jiang B and Hua D:
B7-H3 promotes cell migration andinvasion through the
Jak2/Stat3/MMP9 signaling pathway in colorectal cancer. Mol Med
Rep. 12:5455–5460. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang T, Jiang B, Zou ST, Liu F and Hua D:
Overexpression of B7-H3 augmentsanti-apoptosis of colorectal cancer
cells by Jak2-STAT3. World J Gastroenterol. 21:1804–1813. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei X, Wang G, Li W, Hu X, Huang Q, Xu K,
Lou W, Wu J, Liang C, Lou Q, et al: Activation of the JAK-STAT3
pathway is associated with the growth of colorectal carcinoma
cells. Oncol Rep. 31:335–341. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Qian WF, Guan WX, Gao Y, Tan JF, Qiao ZM,
Huang H and Xia CL: Inhibition of STAT3 by RNA interference
suppresses angiogenesis in colorectal carcinoma. Braz J Med Biol
Res. 44:1222–1230. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cascio S, Ferla R, D'Andrea A, Gerbino A,
Bazan V, Surmacz E and Russo A: Expression of angiogenic
regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3
pathway in colorectal cancer cells. J Cell Physiol. 221:189–194.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soleimanpour E and Babaei E: Survivin as a
potential target for cancer therapy. Asian Pac J Cancer Prev.
16:6187–6191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xiao LJ, Zhao S, Zhao EH, Zheng X, Gou WF,
Takano Y and Zheng HC: Clinicopathological and prognostic
significance of Ki-67, caspase-3 and p53 expression in gastric
carcinomas. Oncol Lett. 6:1277–1284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang X, Simpson ER and Brown KA: p53:
Protection against tumor growth beyond effects on cell cycle and
apoptosis. Cancer Res. 75:5001–5007. 2015. View Article : Google Scholar : PubMed/NCBI
|