Introduction
Chronic liver diseases such as cirrhosis and liver
cancers are one of the leading causes of death worldwide. The
development of chronic liver diseases is associated with a wide
range of liver injuries, including virus infection, alcohol
consumption, and metabolic disorders (1,2). Liver
cancer is known to mostly develop under fibrotic background
(3) and hepatic non-parenchymal cells
play central roles in the progression of liver fibrosis (2,4).
Therefore, the progression from liver fibrosis to liver cancer is
thought to be coordinately regulated through the oncogenic
activation of hepatocytes and microenvironmental modulation of
non-parenchymal cells.
A-kinase anchoring proteins (AKAPs)
spatio-temporally regulate cellular signalings by scaffolding
effector proteins (5,6). AKAP12 (also called as
gravin/SSeCKS/AKAP250) interacts with signaling mediators such as
protein kinase A, protein kinase C, and Src, and modulates a
variety of cellular and physiological events, including
cytoskeletal remodeling, cell migration, blood-brain barrier
development, and oncogenic processes (7,8).
Furthermore, AKAP12 is recognized as a tumor suppressor, as evident
from the downregulation of its expression in several human cancers
such as prostate, ovarian, gastric, and breast cancers. AKAP12
downregulation is often caused by chromosomal hypermethylation or
deletion (9–12). Furthermore, its overexpression in
cells is known to inhibit oncogenic and metastatic properties,
whereas its deficiency enhances the susceptibility to oncogenic
transformation (13,14).
Previous studies have reported the downregulation of
AKAP12 in human hepatocellular carcinoma (HCC) and its association
with promoter hypermethylation, chromosomal deletion, and some
specific microRNAs (15–17). Forced overexpression or silencing of
AKAP12 gene in HCC cell lines revealed that AKAP12 inhibits HCC
cell growth and survival, suggestive of its tumor suppressor
activity (17). However, the role of
AKAP12 at the level of liver organ as well as its functions in
hepatic non-parenchymal cells remain to be investigated.
In the present study, we analyzed the expression
pattern of AKAP12 in normal and injured liver tissue and compared
thioacetamide (TAA)-induced liver injuries between wild-type (WT)
and AKAP12-knockout (KO) mice to evaluate the role of AKAP12 in the
progression of liver fibrosis and cancer.
Materials and methods
Animals and TAA-induced liver
injuries
All mouse experiments were reviewed and approved by
the Committee for Care and Use of Laboratory Animals at Seoul
National University and performed according to the Guide for Animal
Experiments edited by the Korean Academy for Medical Sciences. WT
and AKAP12-deficient (AKAP12 KO) C57BL/6 mice were bred and
maintained as previously described (10). Hepatic fibrosis and tumorigenesis were
induced by TAA in weight-matched 8- to 10-week-old WT or AKAP12-KO
male mice. For fibrosis model, TAA was intraperitoneally injected
thrice a week at a dose of 150 mg/kg for 8 weeks (18,19). The
vehicle group received saline thrice a week for 8 weeks. To
generate liver tumor, mice were fed with drinking water containing
TAA at 300 mg/l for 26 weeks (20).
At the end of the experimental period, mice were euthanized via
deep anesthesia and cardiac perfusion.
Human tissue specimens
HCC tissues and adjacent non-tumorous liver samples
were obtained from patients who received surgical resection for HCC
at Asan Medical Center, Seoul, Korea. Informed consent was obtained
from all patients. HCC patients who meet the following criteria are
included for the analysis: at leat 20 years of age and surgical
specimen is histologically confirmed as HCC. Patients who accompany
other malignancy or Child-Pugh class C disease are excluded. To
determine HBV infection, HBsAg were detected using microparticle
enzyme immunoassay (Abbott Laboratories, Chicago, IL, USA) or
immunoradiometric assay kit (DiaSorin S.p.A., Vercelli, Italy).
Patients' clinical characteristics are summarized in Table I. The study was approved by the
Institutional Review Board of the Asan Medical Center, Seoul,
Korea.
 | Table I.Clinical characteristics of human
subjects included in the present study. |
Table I.
Clinical characteristics of human
subjects included in the present study.
| Patient no. | Age | Sex | Etiology | Max. tumor size
(cm) | Edmondson-Steiner
grade | Cirrhosis |
|---|
| 1 | 69 | M | HBV | 7.1 | 3 | No |
| 2 | 42 | M | HBV | 1.8 | 3 | No |
| 3 | 48 | M | HBV | 3.5 | 2 | Yes |
| 4 | 78 | F | HBV | 5.0 | 4 | Yes |
| 5 | 43 | M | HBV | 3.8 | 4 | No |
| 6 | 70 | F | HBV | 5.3 | 3 | Yes |
| 7 | 56 | M | HBV | 2.2 | 3 | No |
| 8 | 55 | F | HBV | 11.8 | 3 | Yes |
| 9 | 51 | M | HBV | 1.9 | 3 | Yes |
| 10 | 59 | M | HBV | 3.2 | 2 | Yes |
| 11 | 57 | F | HBV | 3.0 | 4 | No |
| 12 | 58 | M | HBV | 2.3 | 3 | No |
| 13 | 74 | M | NBNC | 10.5 | 4 | No |
| 14 | 45 | M | HCV | 2.5 | 3 | No |
| 15 | 71 | M | Alcohol | 1.4 | 4 | No |
Histological analysis and
immunohistochemistry
To visualize the deposition of extracellular matrix
(ECM) proteins, paraffin sections were de-paraffinized and stained
with picro-sirius red (Abcam, Cambridge, MA, USA) according to
manufacturer's instructions. Immunohistochemical staining was
performed on paraffin or frozen liver sections. To stain the
paraffin sections, antigen retrieval was performed for 30 min at
95°C in Tris buffer (ph 9.0). After blocking with 5% normal donkey
serum (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in
phosphate-buffered saline (PBS), the sections were overnight
incubated at 4°C with primary antibodies for AKAP12 (I. Gelman,
Roswell Park Cancer Institute), αSMA (Agilent Technologies, Inc.,
Santa Clara, CA, USA), laminin (Sigma-Aldrich; Merck KGaA), SE-1
(R&D Systems, Inc., Minneapolis, MN, USA), and epithelial cell
adhesion molecule (EpCAM; BD Biosciences, Franklin Lakes, NJ, USA).
After extensive washing with PBS containing 0.1% Tween-20 solution,
the sections were treated with Alexa 488 or 546-conjugated
secondary antibodies (1:750; Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) for 1 h at room temperature, followed by
counter staining with Hoechst stain (Sigma-Aldrich; Merck KGaA).
Fluorescent images were taken under a confocal microscope (Carl
Zeiss AG, Oberkochen, Germany) and immuno-positive areas were
quantified by ImageJ software (21).
A total of 5–10 low magnification (×50) images from a single animal
were quantified and the mean value was considered as a
representative value for that animal.
Isolation of RNA and quantitative
polymerase chain reaction (qPCR) analysis
Liver tissues were homogenized using TissueLyser II
(Qiagen, Hilden, Germany) in TRIzol reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) and total RNA was extracted according to
the manufacturer's instructions. Two micrograms of RNA from each
sample was reverse transcribed with Moloney murine leukemia virus
(MMLV) reverse transcriptase (Promega Corporation, Madison, WI,
USA). Quantitative real-time PCR was performed using StepOnePlus
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) with RealHelix qPCR kit (NanoHelix Co., Ltd., Seoul, Korea).
The relative mRNA levels were normalized by a housekeeping gene
encoding glyceraldehyde-3-phosphate (GAPDH). Primer
sequences used in this study are provided in Table II.
| Table II.Primer sequences for qPCR
detection. |
Table II.
Primer sequences for qPCR
detection.
| A, Human
primers |
|---|
|
|---|
| Gene | Sequences
(5′-3′) |
|---|
|
AKAP12αβ |
|
Forward |
CAGAAGTCAGAGCAAGTGCC |
|
Reverse |
ACCTGAGGGGGAACATTTGA |
| AKAP12α |
|
Forward |
AACGGTCAAGGAGCCCTAAA |
|
Reverse |
CATCTTCAGAGTCTCTCTGTCCAA |
| AKAP12β |
|
Forward |
CCGCTAAGCTGATCTCCTGT |
|
Reverse |
CATCTTCAGAGTCTCTCTGTCCAA |
| GAPDH |
|
Forward |
TGAACGGGAAGCTCACTGG |
|
Reverse |
TCCACCACCCTGTTGCTGTA |
|
| B, Mouse
primers |
|
| Gene | Sequences
(5′->3′) |
|
| col1a1 |
|
Forward |
CATGTTCAGCTTTGTGGACCT |
|
Reverse |
GCAGCTGACTTCAGGGATGT |
| αSMA |
|
Forward |
GACACCACCCACCCAGAGT |
|
Reverse |
ACATAGCTGGAGCAGCGTCT |
| Elastin |
|
Forward |
GGGCCCTGGTATTGGAGGTC |
|
Reverse |
ACTCCACCTCTGGCTCCGTA |
| EpCAM |
|
Forward |
AGGGGCGATCCAGAACAACG |
|
Reverse |
ATGGTCGTAGGGGCTTTCTC |
| TGF-β1 |
|
Forward |
TTGCTTCAGCTCCACAGAGA |
|
Reverse |
TGGTTGTAGAGGGCAAGGAC |
| PDGFa |
|
Forward |
GAGATACCCCGGGAGTTGAT |
|
Reverse |
AAATGACCGTCCTGGTCTTG |
| PDGFb |
|
Forward |
CCTCGGCCTGTGACTAGAAG |
|
Reverse |
GGACGAGGGGAACAACATTA |
| GAPDH |
|
Forward |
TGAACGGGAAGCTCACTGG |
|
Reverse |
TCCACCACCCTGTTGCTGTA |
Western blot analysis
Liver tissues were lysed in radioimmunoprecipitation
assay (RIPA) buffer containing 25 mM Tris pH 7.4, 150 mM NaCl, 5 mM
MgCl2, 0.5% NP-40, phosphatase inhibitor cocktail (Sigma) and
proteinase inhibitor cocktail (EMD Millipore, Billerica, MA, USA).
Protein concentrations were determined using a bicinchoninic acid
(BCA) Assay kit (Thermo Fisher Scientific, Inc.). 20 µg of lysates
were resolved on polyacrylamide gel and then immunoblotted for
AKAP12 (I. Gelman, Roswell Park Cancer Institute) and vinculin
(Santa Cruz Biotechnology, Inc., Dallas, TX, USA) as described
previously (22).
Statistical analysis
All data are expressed as mean ± standard error of
the mean (SEM). One-way analysis of variance (ANOVA) followed by
Tukey's tests or two-tailed Student's t-test was used for
statistical analyses. P<0.05 was considered to indicate a
statistically significant difference. All statistical analyses were
performed using GraphPad Prism v.5.00 (GraphPad Software, Inc., La
Jolla, CA, USA).
Results
AKAP12 is downregulated in human HCC
tissues positive for hepatitis B virus (HBV) infection
Previous reports from three independent groups
revealed the significant downregulation in AKAP12 expression in
human HCC samples (15–17). To confirm the reduced AKAP12 gene
expression in Korean HCC patients, we analyzed protein lysates from
13 pairs of HCC and adjacent non-tumor tissues by western blotting.
Of these, 11 HCC samples exhibited reduced level of AKAP12 protein
as compared with the adjacent non-tumor tissues (Fig. 1A). The remaining two HCC samples
showed an increase in the expression of AKAP12 levels and were
negative for HBV infection. On the other hand, the 11 samples with
reduced AKAP12 expression in the tumor were positive for HBV
infection. We analyzed transcript levels of AKAP12 isoforms alpha
and beta separately in 12 HBV-associated HCC tissues and one each
of non-HBV/non-HCV (NBNC)-, alcohol- and HCV-associated HCC tissues
(Fig. 1B and C). Transcript levels of
total AKAP12αβ amplified with common AKAP12αβ primers showed a
significant reduction in AKAP12 expression in HCC tumors as
compared to adjacent non-tumor tissues of HBV groups. We performed
qPCR analysis to detect the expression of AKAP12α and AKAP12β
isoforms using isoform-specific primer sets and found less
significant reduction in AKAP12β gene expression in tumor tissues
(P=0.0594), whereas the reduction in the expression of AKAP12α gene
in tumors was highly significant (P=0.0002) (Fig. 1B). In contrast, HCC samples with other
etiologies showed variable results (Fig.
1C), although we failed to analyze statistical differences by
etiologies owing to the limited number of samples.
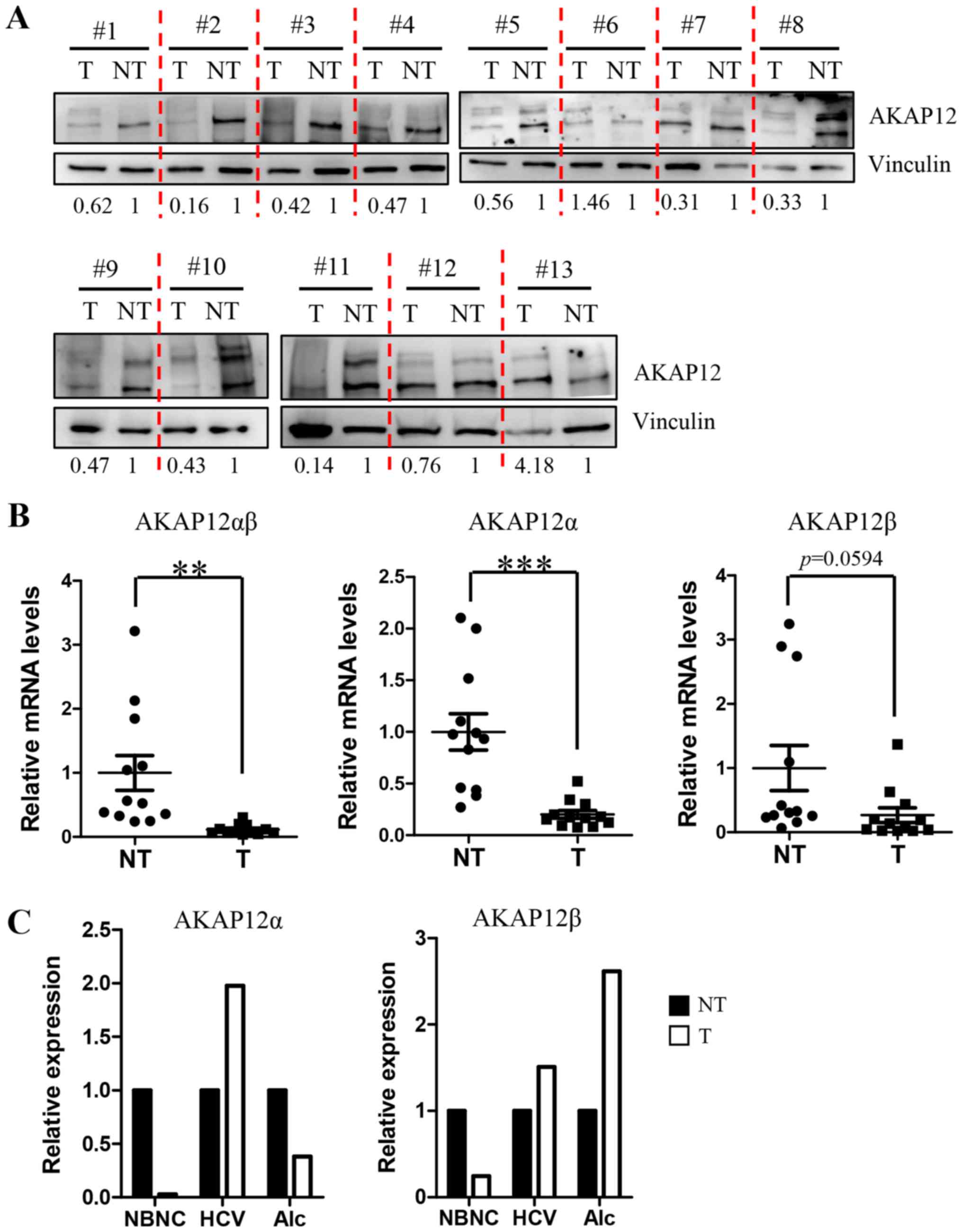 | Figure 1.AKAP12 is downregulated in human HCC.
(A) Western blots were obtained from tissue lysates isolated from
HCC tumors (T) and adjacent non-tumor tissues (NT). Vinculin was
used as a loading control. Relative AKAP12 band intensities of T/NT
pairs are shown under western blot images after normalization with
vinculin intensities. (B) Transcript levels of total AKAP12 (left),
AKAP12α isoform (middle), and AKAP12β isoform (right) were compared
between 13 tumor/non-tumor tissues carrying HBV infection using
qRT-PCR analysis. Data are expressed as mean ± SEM. **P<0.01,
***P<0.001, n=13. (C) Transcript levels of AKAP12α (left) and
AKAP12β (right) isoforms were compared between one each of
non-HBV/non-HCV (NBNC)-, HCV-, and alcohol (Alc)-associated HCC
tissue. Graphs are relative level of mRNAs to non-tumor tissues.
HCC, hepatocellular carcinoma; HBV, hepatitis B virus; HCV,
hepatitis C virus; NBNC, non-HBV and non-HCV; qRT-PCR, quantitative
real-time polymerase chain reaction; mean ± standard error of the
mean (SEM). |
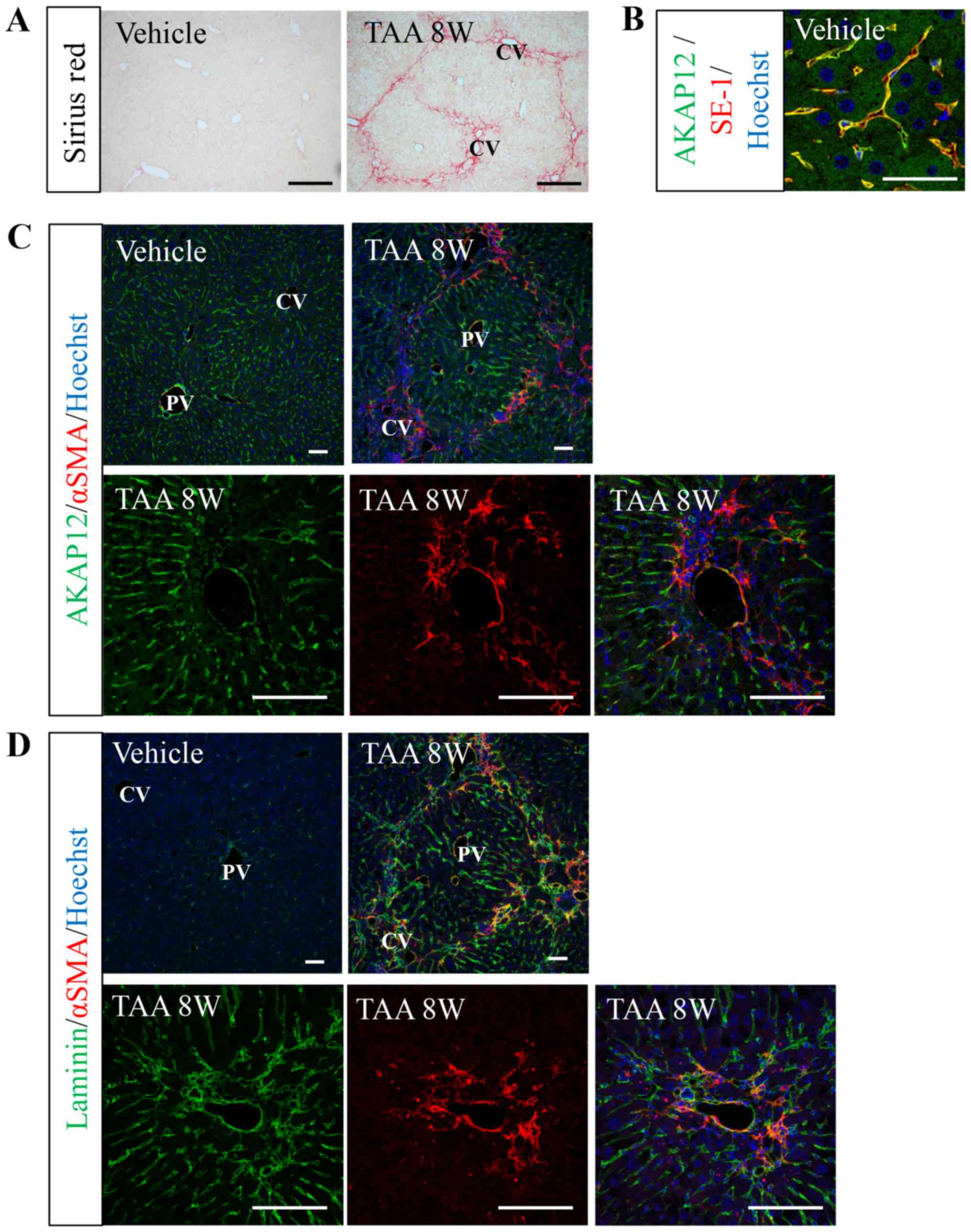
Sinusoidal AKAP12 expression is
reduced in fibrotic regions following TAA administration for 8
weeks
To evaluate the role of AKAP12 in liver fibrosis, we
used TAA-induced liver fibrosis model. Picro-sirius red staining
revealed that 8 weeks of intraperitoneal TAA injection resulted in
the accumulation of ECM from central veins (Fig. 2A). In the vehicle-treated mouse
livers, AKAP12 immunoreactivity was observed in the sinusoidal
endothelial cells (LSECs) as revealed by co-localization with a
LSEC marker, SE-1, whereas hepatocytes were almost negative for
AKAP12 staining (Fig. 2B and C).
After TAA injection, the sinusoidal AKAP12 expression was reduced
in fibrosis regions where an obvious activation of myofibroblasts
was deetected (Fig. 2C). Contrary to
AKAP12 expression pattern in normal and fibrotic livers, laminin
expression was nearly absent in normal liver but increased in
fibrotic livers, especially in the area of myofibroblast activation
(Fig. 2D). These results indicate
that TAA administration results in centrilobular fibrosis that
involves capillarization of sinusoidal endothelium and that
sinusoidal AKAP12 expression is diminished in capillarized
microvessels in the fibrotic area.
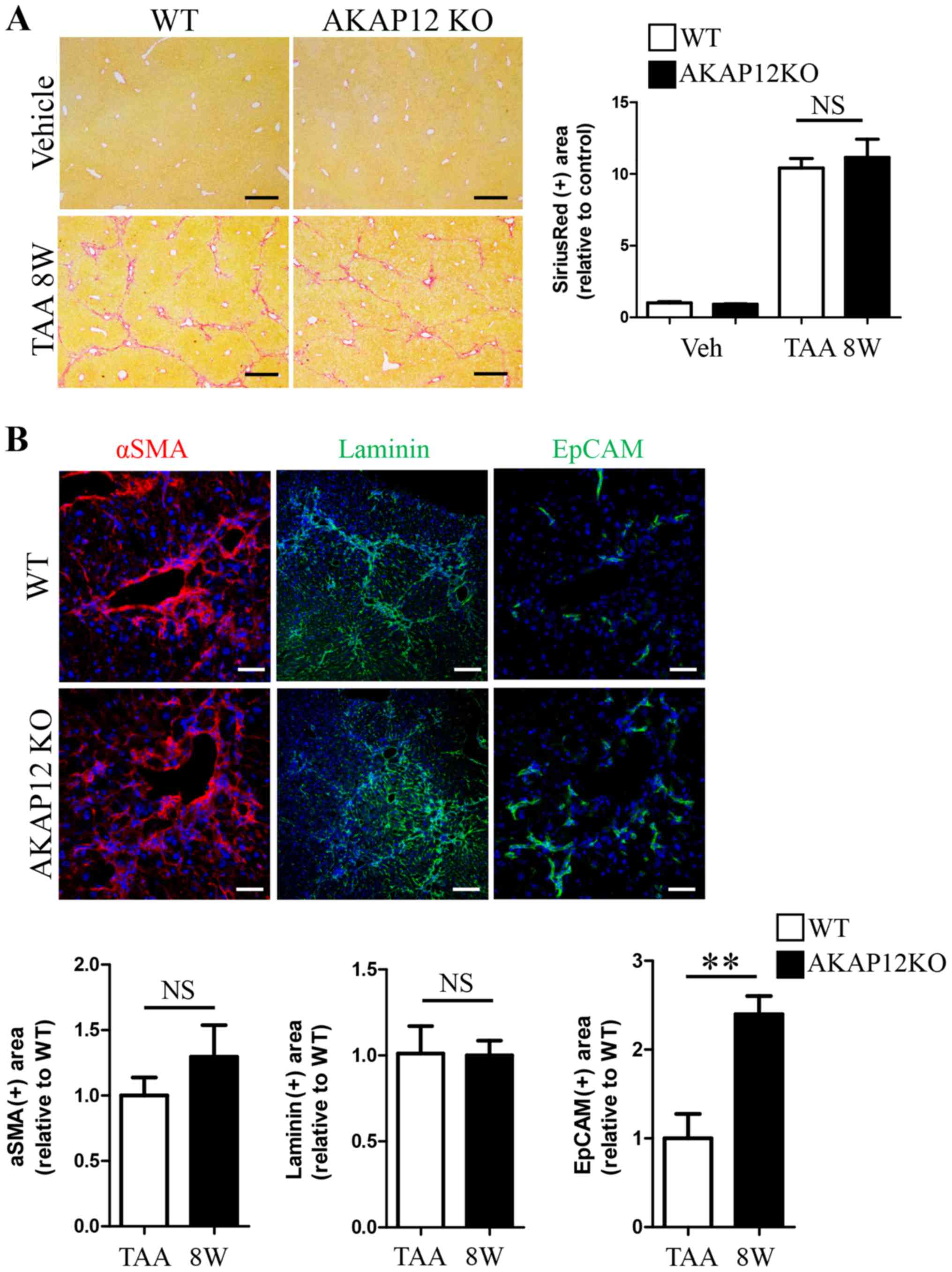
Comparison of liver injuries between
WT and AKAP12-KO mice after short-term TAA injection
We evaluated if AKAP12 deficiency leads to the
alteration in liver injury caused by TAA. After 8 weeks of TAA
administration, the level of ECM deposition was comparable between
livers from AKAP12-KO mice and WT littermates, as revealed by
picro-sirius red staining (Fig. 3A).
Myofibroblastic activation as well as sinusoidal basement membrane
formation were similar between genotypes (Fig. 3B, αSMA and laminin staining). In
contrast, EpCAM-positive cholangiocyte expansion was approximately
2.5-fold higher in AKAP12-KO livers than WT controls (Fig. 3B, EpCAM staining). We compared
transcript levels of specific fibrosis-related genes in WT and
AKAP12-KO livers with qRT-PCR. As evidenced by the results of
immunohistochemical staining, EpCAM mRNA level was higher in
AKAP12-KO mice than in WT control after 8 weeks of TAA injection
(Fig. 4A, left). Moreover, the
transcript levels of certain ECM genes, namely Col1a1 and
elastin, were significantly increased in AKAP12-KO livers
than in WT controls after TAA injection (Fig. 4A, middle and right). However, no
significant difference was observed in the level of myofibroblast
marker αSMA (Fig. 4B, left)
and fibrosis-related soluble factors such as TGF-β1, PDGFa,
and PDGFb (Fig. 4B) between
genotypes.
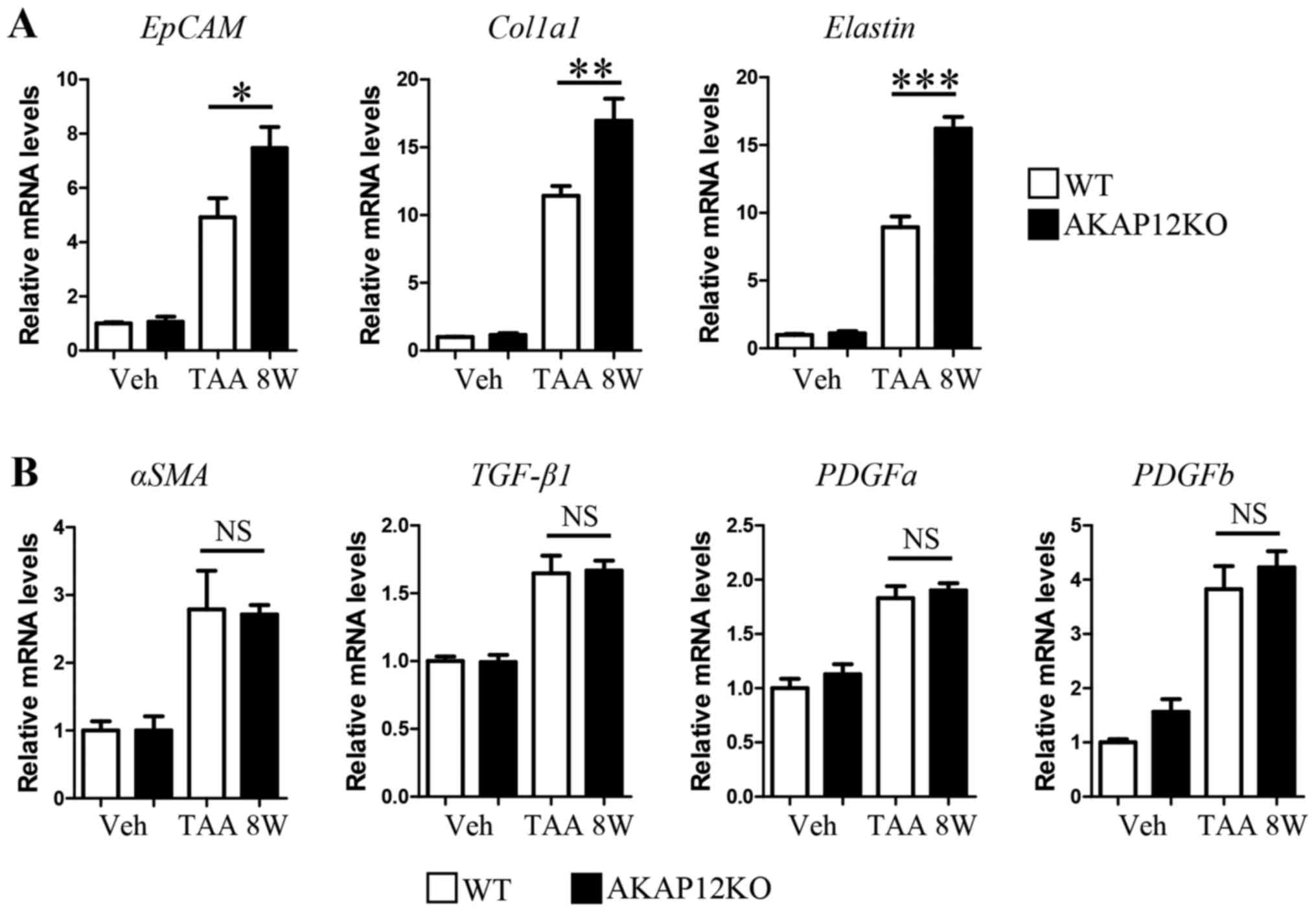 | Figure 4.Comparison of fibrosis-related gene
expression between WT and AKAP12-KO livers after 8 weeks of TAA
administration. (A) Relative mRNA levels of EpCAM, Col1a1,
and elastin in liver tissues were quantified by qRT-PCR with
specific primers. (B) Relative mRNA levels of αSMA, TGF-β1,
PDGFa, and PDGFb in liver tissues were quantified by
qRT-PCR with specific primers. Data are expressed as mean ± SEM.
n=4 mice per group. *P<0.05; **P<0.01; ***P<0.001; NS, not
significant. AKAP, A-kinase anchoring protein; gapdh,
glyceraldehyde 3-phosphate dehydrogenase; Col1a1, collagen type I
alpha 1; αSMA, alpha smooth muscle actin; EpCAM, epithelial cell
adhesion molecule; TGF-β1, transforming growth factor beta 1;
PDGFa, platelet derived growth factor subunit A; PDGFb, platelet
derived growth factor subunit B; gapdh, glyceraldehyde 3-phosphate
dehydrogenase; qRT-PCR, quantitative real-time polymerase chain
reaction. |
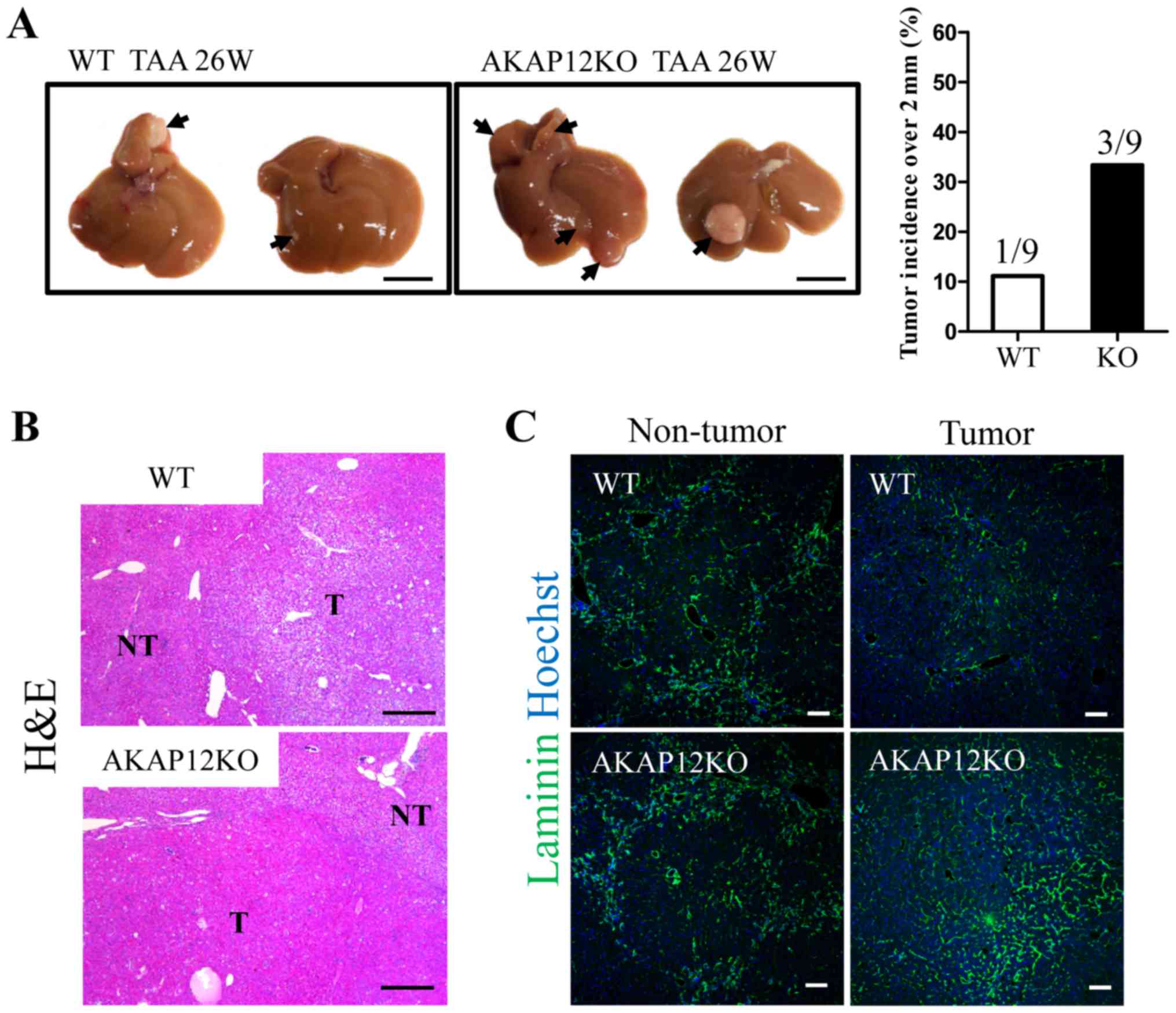
Comparison of hepatic tumorigenesis
between WT and AKAP12-KO mice after long-term TAA
administration
Long-term TAA administration to rodents is known to
generate liver tumors, including hepatocellular adenoma and HCC
(20,23,24). As
AKAP12 is a potential tumor suppressor in human HCC, we compared
the tumorigenesis of WT and AKAP12-KO mice after 26 weeks of TAA
administration. Five of nine WT mice developed tumor nodules that
were visible to the physical eye and one mouse developed a tumor
over 2 mm size with maximum diameter of 7.8 mm (Fig. 5A). In contrast, six of nine AKAP12-KO
mice developed tumor nodules visible to the physical eye and three
mice developed tumors over 2 mm size with maximum tumor diameter of
7.4/8.3/8.6 mm, respectively (Fig.
5A). Histological analysis with hematoxylin and eosin staining
revealed more malignant tumors in AKAP12-KO livers (Fig. 5B). Furthermore, tumors in AKAP12-KO
livers were more vascularized than WT tumors, as revealed by
laminin staining. Laminin immunoreactivity of adjacent fibrotic
area was comparable between genotypes (Fig. 5C). Therefore, AKAP12 seems to exhibit
an inhibitory role in hepatic tumorigenesis and may be associated
with the regulation of hepatic non-parenchymal cells such as liver
sinusoidal endothelial cells.
Discussion
It is now well-recognized that stromal cells in
tumor microenvironment play important roles in tumorigenesis and
metastasis (25,26). Tumor-associated microenvironmental
cells such as cancer-associated fibroblasts and tumor endothelial
cells are profoundly different from their counterparts in normal
tissues (25,26) in terms of morphology, behavior, gene
expressions, and even genetic alterations (27,28).
Evidences suggest that some tumor suppressor genes undergo genetic
alterations not only in the tumor cells but also in
tumor-associated stromal cells. For instance, p53 is often found to
be mutated in stromal cells and is associated with tumor grade and
metastasis (28–30).
Our immunohistochemistry results reveal the
prominent expression of AKAP12 in sinusoidal endothelial cells of
normal liver tissue (Fig. 2B),
suggestive of the possibility of microenvironmental regulation by
AKAP12 in liver injuries. The outcome of short-term and long-term
injuries in AKAP12-KO mice included the increase in ductular
proliferation and liver cancer formation, respectively. These
results indicate that AKAP12 may mediate intercellular
communication between sinusoidal endothelial cells and cells with
epithelial characteristics. Moreover, sinusoidal AKAP12 expression
was reduced following liver injury, indicative of the existence of
a mechanism that alters the expression level of AKAP12 in
sinusoidal endothelium.
Previous studies have highlighted the downregulation
of AKAP12 expression in human HCC, suggesting its possible role as
a tumor suppressor in HCC (13,15,16).
Several mechanisms have been proposed to be involved in the
reduction of AKAP12 expression in HCC. Goeppert et al
demonstrated that AKAP12 reduction in advanced cancer was
associated with chromosomal loss and promoter hypermethylation and
revealed MiR-183/186-dependent regulation of AKAP12 expression in
cirrhosis and dysplastic nodules (15). Hayashi et al showed AKAP12
reduction in HCC by hypermethylation and without chromosomal
deletion (16), whereas Xia et
al proposed MiR-103-mediated regulation of AKAP12 expression as
a mechanism of HCC development (13).
Therefore, multiple mechanisms may be involved in the
downregulation of AKAP12 expression in HCC. Considering our data
demonstrating prominent AKAP12 expression in liver sinusoids, we
suggest that some of the downregulation mechanisms may be
responsible for the reduction in sinusoidal AKAP12 expression in
injured livers. Future studies with isolated tumor cells and
tumor-associated endothelial cells are necessary to address the
cell-type specific mechanism underlying AKAP12 reduction in
HCC.
Liver fibrosis and the development of HCC are linked
pathogenesis as most HCCs develop under fibrotic background
(3). Therefore, the effect of AKAP12
deficiency on HCC development could be caused by its earlier
effects on liver fibrosis. When we addressed the effect of AKAP12
deficiency on liver fibrosis by short term TAA treatment, we did
not observe significant changes in overall ECM accumulation as
revealed by picro-sirius red staining (Fig. 3A). Interestingly, however, transcript
levels of specific ECM genes were elevated in AKAP12 KO livers
(Fig. 4A), suggesting that
AKAP12-deficiency led to the alteration in some fibrosis-related
gene expression by hepatic myofibroblasts. The discrepancy between
transcript levels and histological ECM accumulation is probable
owing to the complexity of ECM remodeling during fibrogenesis.
Under fibrotic circumstances, liver cells produce not only ECM
proteins but also matrix metalloproteases (MMPs) and tissue
inhibitor of metalloproteases (TIMPs), and therefore the tissue
level of ECM accumulation is determined by the balance between ECM
proteins and ECM-modifying enzymes (31). Thus, it is possible that
AKAP12-mediated alteration in certain ECM gene expression is
diluted by ECM modifying enzymes. Further studies including whole
transcriptome analysis of AKAP12-deficient livers will be able to
address detailed mechanisms of AKAP12-mediated ECM remodeling.
Despite minor effect of AKAP12-deficiency on liver
fibrosis, AKAP12 KO mice aggravated tumorigenesis after long term
TAA administration (Fig. 5A). These
results suggest AKAP12's additional tumor suppressing roles in
tumorigenesis under fibrotic background. Considering specific
AKAP12 expression by hepatic non-parenchymal cells, it may play
tumor suppressor functions via the regulation of hepatic tumor
microenvironment.
Taken together, our study suggests a novel mechanism
underlying AKAP12-dependent inhibition of liver injuries that
involves AKAP12 functions in hepatic non-parenchymal cells.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Global Core
Research Center (GCRC) Program (grant no. 2011-00,30001), Bio &
Medical Technology Development Program (grant no.
2015M3A9E6028949), and NRF grant (grant no. 2015R1C1A2A01054446,
received by H.S. Lee) through the National Research Foundation of
Korea (NRF) funded by the Ministry of Education, Ministry of
Science, ICT and Future Planning (MSIP).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HSL designed and performed experiments, and wrote
the manuscript. JC designed and performed experiments. TS performed
statistical analysis. EJL and JGK designed the experiments. SHR,
DL, MKJ, EY and YHC performed human specimen preparation and
analysis. IHG contributed to manuscript drafting and animal
experiment data analysis. KWK designed the experiments and wrote
the manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients. The human study was approved by the Institutional Review
Board of the Asan Medical Center, Seoul, Korea. All mouse
experiments were reviewed and approved by the Committee for Care
and Use of Laboratory Animals at Seoul National University and
performed according to the Guide for Animal Experiments edited by
the Korean Academy for Medical Sciences.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AKAP
|
A-kinase anchoring protein
|
|
HCC
|
hepatocellular carcinoma
|
|
TAA
|
thioacetamide
|
|
ECM
|
extracellular matrix
|
|
WT
|
wild-type
|
|
KO
|
knockout
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
HBV
|
hepatitis B virus
|
|
EpCAM
|
epithelial cell adhesion molecule
|
References
|
1
|
Pellicoro A, Ramachandran P, Iredale JP
and Fallowfield JA: Liver fibrosis and repair: Immune regulation of
wound healing in a solid organ. Nat Rev Immunol. 14:181–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friedman SL: Mechanisms of hepatic
fibrogenesis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sakurai T and Kudo M: Molecular link
between liver fibrosis and hepatocellular carcinoma. Liver Cancer.
2:365–366. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fausther M, Pritchard MT, Popov YV and
Bridle K: Contribution of liver nonparenchymal cells to hepatic
fibrosis: Interactions with the local microenvironment. Biomed Res
Int. 2017:68247622017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carnegie GK, Means CK and Scott JD:
A-kinase anchoring proteins: From protein complexes to physiology
and disease. IUBMB Life. 61:394–406. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Poppinga WJ, Muñoz-Llancao P,
Gonzalez-Billault C and Schmidt M: A-kinase anchoring proteins:
cAMP compartmentalization in neurodegenerative and obstructive
pulmonary diseases. Br J Pharmacol. 171:5603–5623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gelman IH: Suppression of tumor and
metastasis progression through the scaffolding functions of
SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 31:493–500. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee SW, Kim WJ, Choi YK, Song HS, Son MJ,
Gelman IH, Kim YJ and Kim KW: SSeCKS regulates angiogenesis and
tight junction formation in blood-brain barrier. Nat Med.
9:900–906. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu W, Gong J, Hu J, Hu T, Sun Y, Du J,
Sun C, Guan M, Jiang H and Lu Y: Quantitative assessment of AKAP12
promoter methylation in human prostate cancer using
methylation-sensitive high-resolution melting: Correlation with
gleason score. Urology. 77(1006): e1–7. 2011.PubMed/NCBI
|
|
10
|
Akakura S, Huang C, Nelson PJ, Foster B
and Gelman IH: Loss of the SSeCKS/Gravin/AKAP12 gene results in
prostatic hyperplasia. Cancer Res. 68:5096–5103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gelman IH: The role of
SSeCKS/gravin/AKAP12 scaffolding proteins in the spaciotemporal
control of signaling pathways in oncogenesis and development. Front
Biosci. 7:d1782–d1797. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi MC, Jong HS, Kim TY, Song SH, Lee DS,
Lee JW, Kim TY, Kim NK and Bang YJ: AKAP12/Gravin is inactivated by
epigenetic mechanism in human gastric carcinoma and shows growth
suppressor activity. Oncogene. 23:7095–7103. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xia W, Unger P, Miller L, Nelson J and
Gelman IH: The Src-suppressed C kinase substrate, SSeCKS, is a
potential metastasis inhibitor in prostate cancer. Cancer Res.
61:5644–5651. 2001.PubMed/NCBI
|
|
14
|
Lin X and Gelman IH: Reexpression of the
major protein kinase C substrate, SSeCKS, suppresses v-src-induced
morphological transformation and tumorigenesis. Cancer Res.
57:2304–2312. 1997.PubMed/NCBI
|
|
15
|
Goeppert B, Schmezer P, Dutruel C, Oakes
C, Renner M, Breinig M, Warth A, Vogel MN, Mittelbronn M, Mehrabi
A, et al: Down-regulation of tumor suppressor A kinase anchor
protein 12 in human hepatocarcinogenesis by epigenetic mechanisms.
Hepatology. 52:2023–2033. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hayashi M, Nomoto S, Kanda M, Okamura Y,
Nishikawa Y, Yamada S, Fujii T, Sugimoto H, Takeda S and Kodera Y:
Identification of the A kinase anchor protein 12 (AKAP12) gene as a
candidate tumor suppressor of hepatocellular carcinoma. J Surg
Oncol. 105:381–386. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xia W, Ni J, Zhuang J, Qian L, Wang P and
Wang J: MiR-103 regulates hepatocellular carcinoma growth by
targeting AKAP12. Int J Biochem Cell Biol. 71:1–11. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Meyer C, Xu C, Weng H, Hellerbrand
C, ten Dijke P and Dooley S: Animal models of chronic liver
diseases. Am J Physiol Gastrointest Liver Physiol. 304:G449–G468.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wallace MC, Hamesch K, Lunova M, Kim Y,
Weiskirchen R, Strnad P and Friedman SL: Standard operating
procedures in experimental liver research: Thioacetamide model in
mice and rats. Lab Anim. 49 1 Suppl:S21–S29. 2015. View Article : Google Scholar
|
|
20
|
Abe M, Yoshida T, Akiba J, Ikezono Y, Wada
F, Masuda A, Sakaue T, Tanaka T, Iwamoto H, Nakamura T, et al:
STAT3 deficiency prevents hepatocarcinogenesis and promotes biliary
proliferation in thioacetamide-induced liver injury. World J
Gastroenterol. 23:6833–6844. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hartig SM: Basic image analysis and
manipulation in ImageJ. Curr Protoc Mol Biol Chapter. 14:Unit14 15.
2013.doi: 10.1002/0471142727.mb1415s102. View Article : Google Scholar
|
|
22
|
Shin MW, Bae SJ, Wee HJ, Lee HJ, Ahn BJ,
Le H, Lee EJ, Kim RH, Lee HS, Seo JH, et al: Ninjurin1 regulates
lipopolysaccharide-induced inflammation through direct binding. Int
J Oncol. 48:821–828. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sakurai T, Yada N, Watanabe T, Arizumi T,
Hagiwara S, Ueshima K, Nishida N, Fujita J and Kudo M:
Cold-inducible RNA-binding protein promotes the development of
liver cancer. Cancer Sci. 106:352–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Helmy SA, El-Mesery M, El-Karef A, Eissa
LA and El Gayar AM: Chloroquine upregulates TRAIL/TRAILR2
expression and potentiates doxorubicin anti-tumor activity in
thioacetamide-induced hepatocellular carcinoma model. Chem Biol
Interact. 279:84–94. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Balkwill FR, Capasso M and Hagemann T: The
tumor microenvironment at a glance. J Cell Sci. 125:5591–5596.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanahan D and Coussens LM: Accessories to
the crime: Functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bussard KM, Mutkus L, Stumpf K,
Gomez-Manzano C and Marini FC: Tumor-associated stromal cells as
key contributors to the tumor microenvironment. Breast Cancer Res.
18:842016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Campbell I, Qiu W and Haviv I: Genetic
changes in tumour microenvironments. J Pathol. 223:450–458. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fukino K, Shen L, Patocs A, Mutter GL and
Eng C: Genomic instability within tumor stroma and
clinicopathological characteristics of sporadic primary invasive
breast carcinoma. JAMA. 297:2103–2111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wernert N, Löcherbach C, Wellmann A,
Behrens P and Hügel A: Presence of genetic alterations in
microdissected stroma of human colon and breast cancers. Anticancer
Res. 21:2259–2264. 2001.PubMed/NCBI
|
|
31
|
Arriazu E, de Galarreta Ruiz M, Cubero FJ,
Varela-Rey M, de Obanos Pérez MP, Leung TM, Lopategi A, Benedicto
A, Abraham-Enachescu I and Nieto N: Extracellular matrix and liver
disease. Antioxid Redox Signal. 21:1078–1097. 2014. View Article : Google Scholar : PubMed/NCBI
|