Introduction
Lung cancer has been reported as the leading cause
of cancer-associated mortality worldwide, with ~1.8 million new
cases and 1.5 million mortalities attributed to this disease in
2012 worldwide (1). Non-small cell
lung cancer (NSCLC) has been reported to account for ~85% of all
cases of lung cancer (2) with a
5-year overall survival rate of ~15% (3). Lung adenocarcinoma has been reported as
the most commonly diagnosed histological type of NSCLC, with a
mortality rate of >500,000 individuals per year (4).
Therapies targeting a wide variety of activated
oncogenes, translocations or fusions, including epidermal growth
factor receptor (5), ALK receptor
tyrosine kinase (6), KRAS
proto-oncogene GTPase (7) and ROS
proto-oncogene 1 receptor tyrosine kinase (8), have been demonstrated to serve a crucial
role in the progress of treating patients with lung cancer.
Nonetheless, further development of novel and effective therapeutic
targets for treating lung cancer is required. In our previous
study, gene profiles between lung adenocarcinoma tissues and
adjacent healthy lung tissues were compared using whole-genome
sequencing and a number of differentially expressed genes (DEGs)
were identified (9). Among the DEGs,
serine incorporator 2 (SERINC2), a member of the SERINC family of
transmembrane proteins that incorporate serine into membrane lipids
during synthesis, was of particular note (10).
Although the family is highly conserved among
eukaryotes, none of the SERINC family members (SERINC1-5) display
amino acid homology to other proteins. According to Inuzuka et
al (10), SERINC proteins have an
essential function in regulating the biosynthesis of multiple
membrane lipids, including phosphatidylserine and sphingolipid
molecules. Furthermore, phosphatidylserine (11–13) and
sphingolipids (14–16) have been indicated to serve critical
roles in cancer development and progression. Ren et al
(17) demonstrated that small
interfering RNA (siRNA)-mediated knockdown of SERINC1 in
hepatocellular carcinoma inhibits cell cycle progression by
transcriptional activation of p21.
In 2003, Player et al (18) initially identified human SERINC2,
which was referred to as TDE2. The deduced 456-amino acid protein
has 11 putative transmembrane helices, a cysteine-rich region near
the N-terminus, followed by a phenylalanine-rich region and a
central conserved myelocytomatosis oncogene-type helix-loop
dimerization domain. It has been demonstrated by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) that
SERINC2 transcript levels are increased in NSCLC compared with the
levels in non-malignant tissues (18). In addition, in situ
hybridization assays have indicated that lung tumours express
increased expression levels of SERINC2 transcripts compared with
adjacent non-malignant regions (18).
However, the role of SERINC2 in cancer remains unknown. In the
present study, the effects of SERINC2-knockdown on proliferation,
migration and invasion of lung adenocarcinoma cell lines was
investigated.
Materials and methods
Analysis of Oncomine data
The Oncomine database, an online cancer microarray
database that enables multiple comparisons of gene expression
levels in DNA and RNA reported in different studies, was used for
analysing the expression pattern of SERINC2 in lung adenocarcinoma
(https://www.oncomine.org). The SERINC2 gene was
queried and the results were filtered by selecting ‘lung
adenocarcinoma’ and ‘Cancer vs. Normal Analysis’. Normal lung
tissue was used as the control group. Comparative statistical
analysis was conducted using Oncomine algorithms. P<0.01 was
considered to indicate a statistically significant difference.
Details of standardized normalization techniques and statistical
calculations are provided on the Oncomine platform.
Cell line culture and
transfection
Human lung cancer H1650 and A549 cell lines were
purchased from the Shanghai Institutes for Biological Sciences
(Shanghai, China) and cultured in Dulbecco's modified Eagle's
medium, supplemented with 10% foetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 100 U/ml penicillin and
100 mg/ml streptomycin. All cells were maintained at 37°C with 5%
CO2.
Two short hairpin RNAs (shRNAs) targeting SERINC2
were designed and constructed by Sangon Biotech Co., Ltd.
(Shanghai, China). The following sequences were inserted into the
Lentiviral shRNA Vector (Forevergen Biotechnology, Guangzhou,
China): sh1,
5′-TGCGCCTCATCTTCACGTTCTTCTCAAGAGGAAGAACGTGAAGATGAGGCGTTTTTC-3′ and
sh2,
5′-TGTGGTCAGCCCTATCCAGTATCTCAAGAGGATACTGGATAGGGCTGACCATTTTTC-3′.
Lipofectamine 2000® (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for transfection, according to the
manufacturer's protocols. Stable cell lines expressing
SERINC2-shRNA were generated by infection with lentiviruses (1 µl;
Forevergen Biotechnology) produced in 293 cells and selection of
stable clones with 0.5 µg/ml puromycin for 10 days, then subsequent
experiments were performed.
RT-qPCR
Total RNA was extracted from H1650 and A549 cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
The concentration and quality of total RNA samples were determined
by spectrophotometry and agarose gel electrophoresis, respectively.
cDNA templates were synthesized from 1 µg total RNA using a Reverse
Transcription system (Promega Corporation, Madison, WI, USA),
according to the manufacturer's protocols. qPCR was subsequently
performed using GoTaq qPCR Master mix (Promega Corporation),
according to the manufacturer's protocols. The following
thermocycling conditions were used for the qPCR: 95°C for 3 min,
followed by 40 cycles of 95°C for 3 sec and 60°C for 30 sec. The
quality of the PCR products was assessed using a post-PCR melting
curve and β-actin served as the internal control. Relative mRNA
expression levels were quantified using the 2−∆∆Cq
method (19). The following primers
were used: SERINC2 sense, 5′-TGGTGCTGCTCATCGACTTT-3′ and antisense,
5′-TGAAGAAGAAGAGGCCTGCG-3′; and β-actin sense,
5′-AGAAGAGCTACGAGCTGCCTGACG-3′ and antisense,
5′-GGACTCCATGCCACGGAAGGAA-3′.
Immunoblotting
Following harvesting of cells, total cellular
proteins were extracted with radioimmunoprecipitation assay lysis
buffer containing protease inhibitors (Sangon Biotech Co., Ltd.).
Protein concentrations were measured using the Bradford assay
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), according to the
manufacturer's protocols with bovine serum albumin as the standard.
Equal amounts of protein extracts (80–120 µg, dependent on
experiment) from all samples were separated by 10% SDS-PAGE and
subsequently transferred to a polyvinylidene fluoride membrane. The
membrane was blocked with 5% skimmed milk dissolved in
Tris-buffered saline containing 0.1% Tween-20 for 1 h at room
temperature followed by incubation using anti-SERINC2 (1:1,000
dilution; cat. no. ab134312; Abcam, Cambridge, MA, USA),
anti-phospho-AKT (1:1,000 dilution; ser 473, cat. no. 4058; Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-AKT (1:1,000
dilution; cat. no. 9272; Cell Signaling Technology, Inc.) and
β-actin (1:2,000 dilution; cat. no. A001041; Sangon Biotech Co.,
Ltd.) primary antibodies overnight at 4°C. β-actin was used as the
loading control. Subsequent to washing, the membranes were
incubated with corresponding secondary antibodies (1:20,000
dilution; cat. no. 14709; Cell Signaling Technology, Inc.) for 60
min at 37°C and washed again. Signals were visualized using an
enhanced chemiluminescence system (Bio-Rad Laboratories, Inc.),
according to the manufacturer's protocols.
Proliferation and colony formation
assays
Proliferation was analysed using the Cell Counting
kit-8 reagent (CCK-8; Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan), according to the manufacturer's protocols. Cells
were cultured in 96-well plates at a density of 1,000 cells per
well for 1, 2, 3, 4 and 5 days at 37°C. The absorbance was
subsequently measured at a wavelength of 450 nm on a spectrometer
(Thermo Fisher Scientific, Inc.). To examine cell colonies, cells
were cultured at a density of 200 cells/plate and grown for 10 days
at 37°C. The cells were then fixed with a methanol/acetic acid
solution (3:1) for 15 min and stained with 0.5% crystal violet in
methanol for 15 min at room temperature. Subsequent to staining,
the visible colonies were counted under a light microscope.
In vitro wound-healing and invasion
assays
For the wound-healing assay, 2×105 cells
were cultured until confluent (100%). The cell monolayer was then
scratched with a 200-µl pipette tip and the cells were washed three
times with PBS. Fresh growth medium was subsequently added. The
wounded area was monitored every 24 h under a microscope (DFC450;
Leica Microsystems, Inc., Buffalo Grove, IL, USA) and the degree of
wound healing induced by cell migration was quantified using
Image-Pro Plus software 6.0 (Media Cybernetics, Inc., Rockville,
MD, USA).
For cell invasion analysis, 5×104 cells
in serum-free DMEM (GE Healthcare Life Sciences, Logan, UT, USA)
were plated in the upper membrane of Transwell inserts (8 mm)
coated with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA).
After 24 h, the cells on the upper membrane were removed with a
cotton swab and the cells that had invaded into the lower chamber
with DMEM plus 10% FBS were stained with Wright-Giemsa. Images of
cells were captured under a light microscope (DS-Ril Nikon; Nikon
Corporation, Tokyo, Japan) and stained cells were counted by
Image-Pro Plus software 6.0 (Media Cybernetics, Inc.).
Flow cytometry
Cells were seeded into 6-well plates, at
5×105 cells/well, and incubated for 24 h. Cells were
collected by trypsinization and washed once with PBS. The cells
were fixed with 70% ethanol overnight at 20°C, washed thoroughly
with ice-cold PBS and incubated with a propidium iodide (PI)
solution (50 mg/ml RNase A, 0.1% Triton X-100, 0.1 mmol/l EDTA and
50 mg/ml PI) for ≥30 min at 4°C. The samples were evaluated with a
FACSort instrument (BD Biosciences) and the data were analysed with
ModFit LT 4.0 software (Verity Software House, Inc., Topsham, ME,
USA).
Statistical analysis
All experiments were performed in triplicate and the
represented data are derived from individual and separate
experiments. Vector control cells (RNAi-Vector) were used as the
control group in all experiments. All data were evaluated with SPSS
version 20.0 (IBM Corp., Armonk, NY, USA) and the results are
presented as the mean ± standard deviation. Comparisons among
groups were performed using an unpaired two-tailed Student's
t-test. One-way analysis of variance was used for multiple
comparisons to test for significant differences, followed by Tukey'
spost hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
SERINC2 mRNA is expressed at high
levels in lung adenocarcinoma tissues
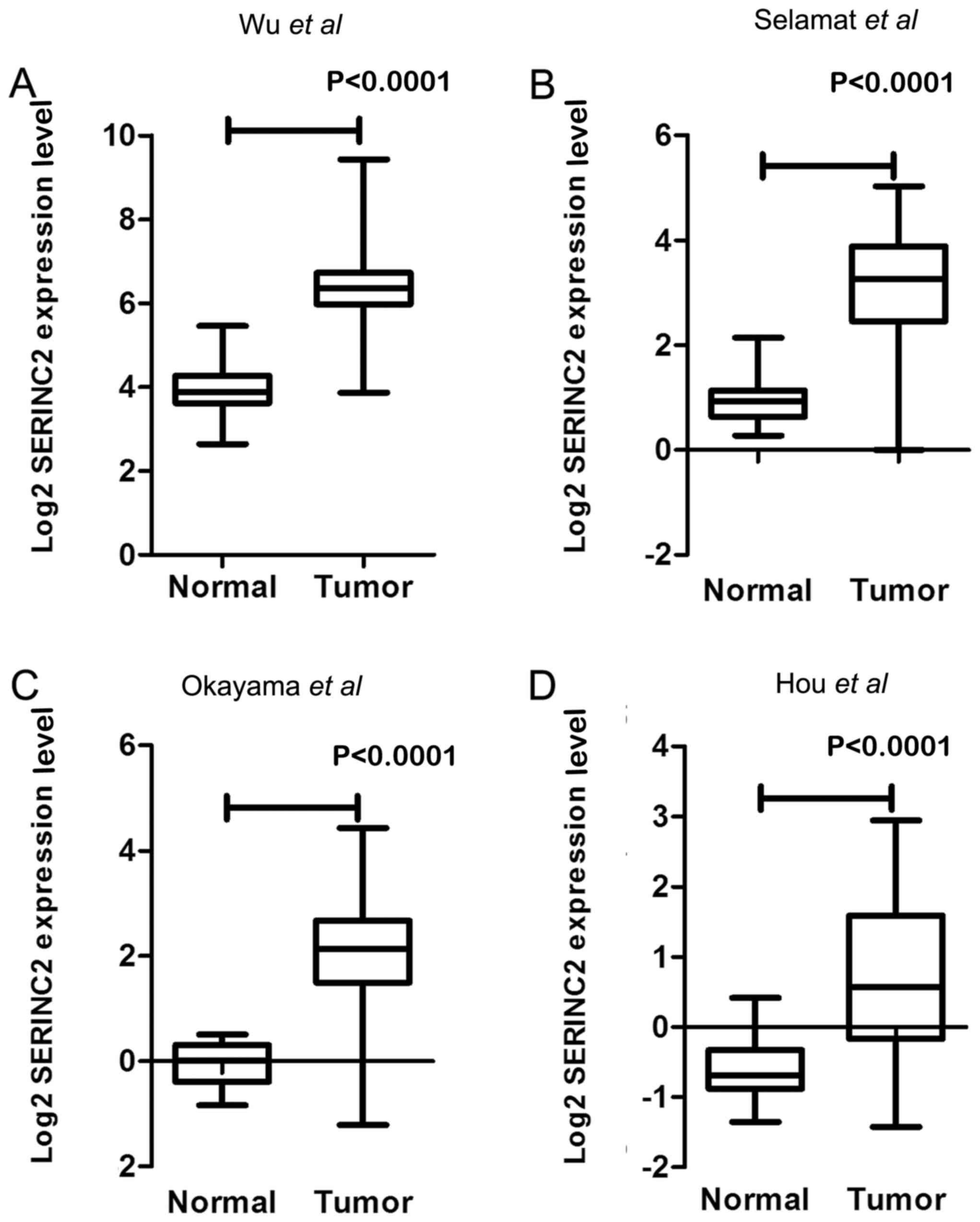
In our previous study (9), the expression levels of SERINC2 mRNA
were evaluated by sequencing 59 pairs of lung adenocarcinoma
tissues and adjacent healthy tissues. SERINC2 was expressed at
significantly higher levels in lung adenocarcinoma tissues compared
with those in corresponding adjacent healthy lung tissues
(fold-change, 5.94; P<0.0001; Fig.
1A). Previous analyses of published microarray datasets
reported in Selamat et al (20) (n=116; fold-change, 4.809; Fig. 1B), Okayama et al (21) (n=246; fold-change, 4.409; Fig. 1C) and Hou et al (22) (n=110; fold-change, 2.497l; Fig. 1D) indicated significantly higher
expression levels of SERINC2 mRNA in lung adenocarcinoma tissues
compared with those in healthy lung tissues.
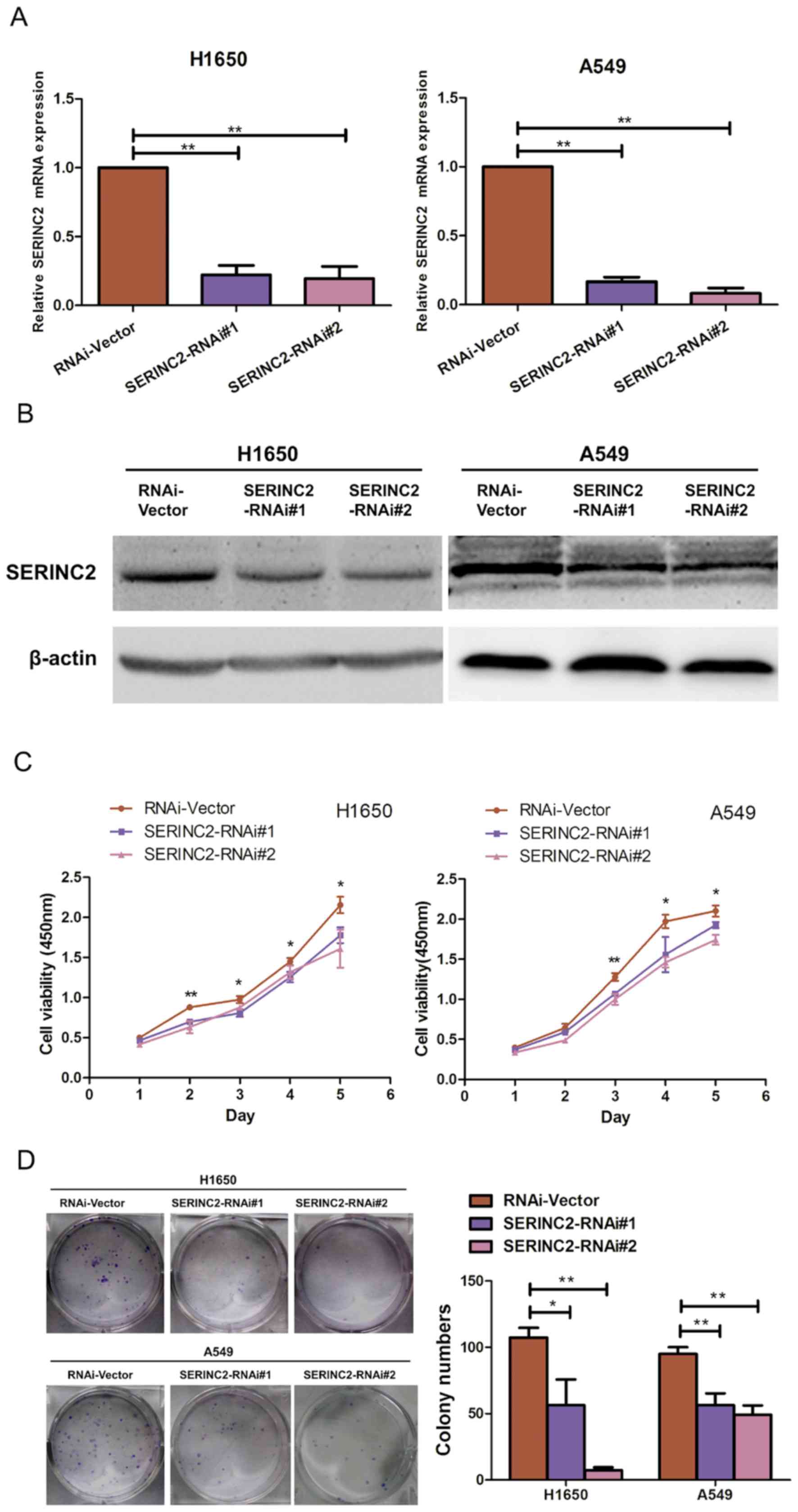
SERINC2 silencing inhibits H1650 and
A549 proliferation
The lung adenocarcinoma H1650 and A549 cell lines
were selected as models to investigate the biological function of
SERINC2. SERINC2 expression in H1650 and A549 cells was
significantly suppressed by two shRNAs (P<0.05; Fig. 2A and B). The effect of SERINC2
silencing on the proliferative activity of H1650 and A549 cells was
assessed using a CCK-8 assay, the results of which revealed that
compared with the control groups, SERINC2-knockdown suppressed the
viability of lung adenocarcinoma cell lines on days 3, 4 and 5 to a
significantly greater extent (P<0.05; Fig. 2C). Furthermore, when SERINC2
expression was reduced in the H1650 and A549 cell lines, colony
numbers were significantly decreased (P<0.05; Fig. 2D). The effect of SERINC2-knockdown on
the cell cycle was also determined. However, based on cell cycle
analysis, there were no significant differences between the
experimental and control groups (data not shown).
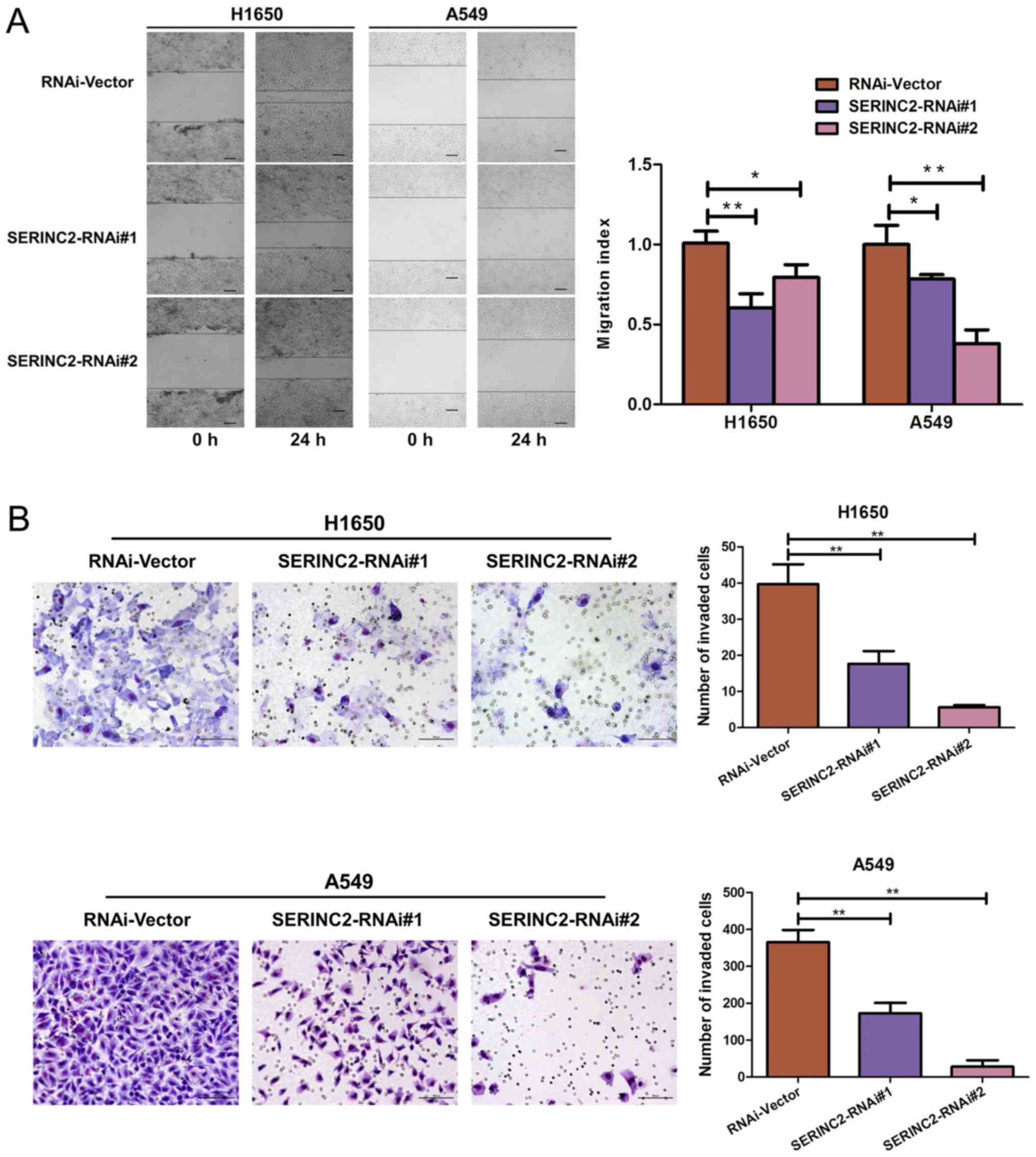
SERINC2 silencing inhibits the
migration and invasion of H1650 and A549 cells
Wound healing and Transwell assays were performed to
investigate the effect of SERINC2-knockdown on the migration and
invasion activities of H1650 and A549 cells, respectively.
SERINC2-knockdown significantly inhibited wound healing (P<0.05;
Fig. 3A), as well as invasive
capacity (P<0.01; Fig. 3B).
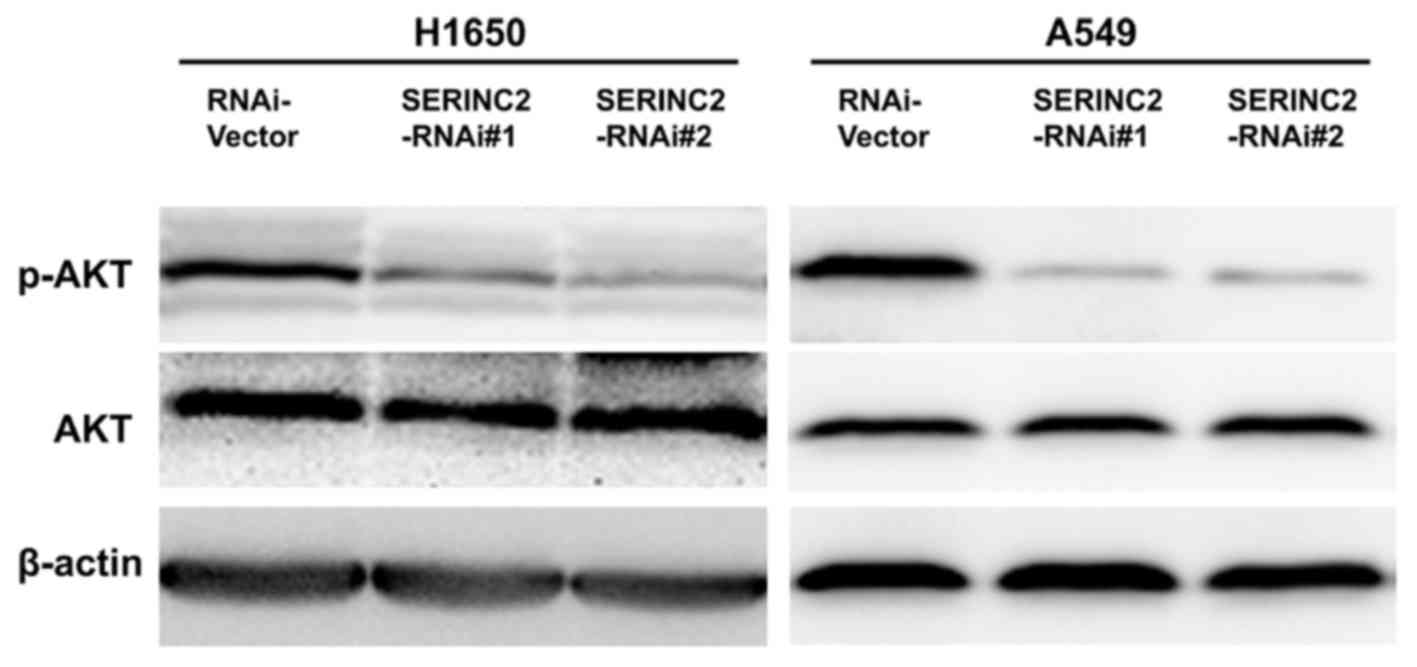
Expression levels of
phosphorylated-protein kinase B (p-AKT) are altered by
SERINC2-knockdown
p-AKT protein expression levels were quantified to
investigate the possible underlying mechanisms of SERINC2 activity.
Levels of p-AKT were significantly decreased when SERINC2 was
knocked down in the H1650 and A549 cells. However, a minimal effect
was observed on total AKT (Fig.
4).
Discussion
In the present study, SERINC2-knockdown using
lentiviral- based shRNAs inhibited the proliferation, migration and
invasion of lung adenocarcinoma cells. In addition, expression
levels of p-AKT were significantly decreased upon
SERINC2-knockdown.
In our previous study, the whole-genome sequencing
approach was employed to distinguish DEGs between lung
adenocarcinoma tissues and adjacent healthy tissues (9). SERINC2 was identified as a DEG that was
significantly upregulated in lung adenocarcinoma. A previous
analysis of published microarray datasets also indicated SERINC2
mRNA to be expressed at significantly higher levels in lung
adenocarcinoma tissues compared with those in healthy lung tissues
(20–22). Player et al (18) observed high SERINC2 expression levels
in the bladder, kidney and muscle; moderate expression in the
stomach, liver, skin, placenta and ovary, and minimal expression in
the brain, spleen and heart. Based on RT-PCR and in situ
hybridization data, an increase in SERINC2 expression was reported
in NSCLC compared with that in healthy lung tissues (18). Regardless, the role of SERINC2 in
cancer requires further investigation.
H1650 cells are derived from metastatic lung
adenocarcinoma (23,24) and A549 cells are a lung adenocarcinoma
cell line (25,26). These lung adenocarcinoma cell lines
were utilized to investigate the role of SERINC2 in the present
study, and it was indicated that SERINC2 silencing inhibited
proliferation based on CCK-8 and colony formation assays.
SERINC1-knockdown has been previously reported to result in cell
cycle arrest and the inhibition of proliferation in hepatocellular
carcinoma cell lines (17). According
to the data of the present study, SERINC2 silencing inhibits the
migration and invasive abilities of H1650 and A549 cells. SERINC1
and SERINC2 belong to the SERINC family of transmembrane proteins
that incorporate serine into phosphatidylserine and sphingolipids
(10). Endoplasmic reticulum
membranes are enriched with SERINC proteins, directly binding to
serine palmitoyltransferase, the key enzyme in sphingolipid
biosynthesis. Therefore, SERINC proteins serve an essential role in
regulating the biosynthesis of lipids, including phosphatidylserine
and sphingolipids (10). Bavituximab
is a phosphatidylserine-targeting monoclonal antibody representing
a novel strategy in cancer therapy (11). As sphingolipid synthesis, including
ceramide and sphingosine-1-phosphate-mediated pathways, has been
reported to serve a vital role in cancer development and
progression (16), SERINC2 may also
be important in cancer development and progression. Overall, the
molecular underlying mechanism of lipid biosynthesis requires
further investigation.
Sterol regulatory element-binding proteins (SREBPs)
are transcription factors associated with cholesterol and fatty
acid synthesis (27), and SREBP-1a
causes G1 cell cycle arrest following the accumulation of
cyclin-dependent kinase inhibitors, including p27, p21 and p16
(28). As indicated in the study by
Ren et al (17),
downregulation of SERINC1 expression enhanced expression of SREBPs
and p21, resulting in impaired cell cycle progression of
hepatocellular carcinoma. However, SERINC2-knockdown was not
reported to cause significant differences in the cell cycle in lung
adenocarcinoma cell lines. In addition, significant changes in
expression of the low-density lipoprotein receptor gene, a target
of SREBPs (27,29), were not observed when SERINC2
expression was knocked down in lung adenocarcinoma cells (data not
shown). Therefore, further research on SREBPs was not conducted.
Differences between SERINC2 and SERINC1 have been observed,
presumably due to their different underlying mechanisms in
regulating cancer.
Phosphatidylinositol 3-kinase (PI3K)/AKT signalling
serves an important role in the proliferation, survival,
differentiation and motility of cancer cells. It has been reported
that constitutive activation of this pathway is a common feature of
human types of cancer and is frequently the outcome upon molecular
alterations of key components of the signalling cascade (30). The focus of such studies has
contributed to the development of novel molecule-targeted
therapies, including PI3K inhibitors (31). In the present study, SERINC2-knockdown
significantly decreased AKT phosphorylation. According to the study
by Huang et al (32),
phosphatidylserine is a critical modulator of AKT activation.
Furthermore, it was indicated in the study by Zhang et al
(33) that phosphatidylserine on the
neuronal membrane may facilitate membrane translocation of AKT in a
phosphoinositide-3,4,5-trisphosphate-dependent manner, thereby
enhancing the subsequent phosphorylation and activation of AKT. As
the SERINC family of transmembrane proteins has been reported to
facilitate the incorporation of serine into phosphatidylserine
(10), SERINC2 may affect the pathway
by modulating lipid synthesis. Therefore, SERINC2-knockdown has
been suggested for inhibiting the proliferative, migratory and
invasive abilities of cells, and it may be associated with the
regulation of the PI3K/AKT pathway. However, further investigations
are required to confirm whether SERINC2-knockdown affects
phosphatidylserine synthesis and directly regulates the PI3K/AKT
pathway. In addition, in vivo studies of the role of SERINC2
are required. Despite these limitations, the present study provides
a unique perspective for evaluating the role of SERINC2 in lung
adenocarcinoma.
In conclusion, the present study provides evidence
that the expression of SERINC2 mRNA is significantly elevated in
tumour tissues derived from patients with lung adenocarcinoma.
Knockdown of SERINC2 expression using a lentiviral-based shRNA
inhibited the proliferation, migration and invasion of lung
adenocarcinoma cells, which may be associated with the PI3K/AKT
pathway. Based on the findings, SERINC2 is required for lung
adenocarcinoma progression. The molecular underlying mechanism
warrants further investigation.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JianH, YZ and DX were responsible for the
conceptualization. YZ, DX and HH performed the methodology. YZ, HP
and YC performed the software analysis. JianH and DX were
responsible for the validation. YZ, DX, JiaxH and WY performed the
formal analysis and interpretation of data. YZ, DX and HH conducted
the investigation. JianH was responsible for the resources and YZ
for the data curation. The manuscript writing (preparation of the
original draft) was performed by YZ and DX. Manuscript preparation
(writing, review and editing) was completed by YZ, DX, JianH,
JiaxH, HP, WY and YC. The present study was supervised by JianH and
administrated by YZ and DX. Final approval of the version was
provided by all authors.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clin Proc. 83:584–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Goldstraw P, Ball D, Jett JR, Le Chevalier
T, Lim E, Nicholson AG and Shepherd FA: Non-small-cell lung cancer.
Lancet. 378:1727–1740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Imielinski M, Berger AH, Hammerman PS,
Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M,
Sivachenko A, et al: Mapping the hallmarks of lung adenocarcinoma
with massively parallel sequencing. Cell. 150:1107–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kwak EL, Bang YJ, Camidge DR, Shaw AT,
Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al:
Anaplastic lymphoma kinase inhibition in non-small-cell lung
cancer. N Engl J Med. 363:1693–1703. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roberts PJ and Stinchcombe TE: KRAS
mutation: Should we test for it, and does it matter. J Clin Oncol.
31:1112–1121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bergethon K, Shaw AT, Ou SH, Katayama R,
Lovly CM, McDonald NT, Massion PP, Siwak-Tapp C, Gonzalez A, Fang
R, et al: ROS1 rearrangements define a unique molecular class of
lung cancers. J Clin Oncol. 30:863–870. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu K, Zhang X, Li F, Xiao D, Hou Y, Zhu S,
Liu D, Ye X, Ye M, Yang J, et al: Frequent alterations in
cytoskeleton remodelling genes in primary and metastatic lung
adenocarcinomas. Nat Commun. 6:101312015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inuzuka M, Hayakawa M and Ingi T: Serinc,
an activity-regulated protein family, incorporates serine into
membrane lipid synthesis. J Biol Chem. 280:35776–35783. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Digumarti R, Bapsy PP, Suresh AV,
Bhattacharyya GS, Dasappa L, Shan JS and Gerber DE: Bavituximab
plus paclitaxel and carboplatin for the treatment of advanced
non-small-cell lung cancer. Lung Cancer. 86:231–236. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin Y, Huang X, Lynn KD and Thorpe PE:
Phosphatidylserine-targeting antibody induces M1 macrophage
polarization and promotes myeloid-derived suppressor cell
differentiation. Cancer Immunol Res. 1:256–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bondanza A, Zimmermann VS, Rouvere-Querini
P, Turnay J, Dumitriu IE, Stach CM, Voll RE, Gaipl US, Bertling W,
Pöschl E, et al: Inhibition of phosphatidylserine recognition
heightens the immunogenicity of irradiated lymphoma cells in vivo.
J Exp Med. 200:1157–1165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kok JW and Sietsma H: Sphingolipid
metabolism enzymes as targets for anticancer therapy. Curr Drug
Targets. 5:375–382. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Proia RL and Hla T: Emerging biology of
sphingosine-1-phosphate: Its role in pathogenesis and therapy. J
Clin Invest. 125:1379–1387. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ogretmen B and Hannun YA: Biologically
active sphingolipids in cancer pathogenesis and treatment. Nat Rev
Cancer. 4:604–616. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ren WH, Yang CY, Yang XM and Yu L:
siRNA-mediated knockdown of hTDE2 retards cell cycle progression
through transcriptional activation of p21. Oncol Rep. 31:1314–1322.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Player A, Gillespie J, Fujii T, Fukuoka J,
Dracheva T, Meerzaman D, Hong KM, Curran J, Attoh G, Travis W and
Jen J: Identification of TDE2 gene and its expression in non-small
cell lung cancer. Int J Cancer. 107:238–243. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Selamat SA, Chung BS, Girard L, Zhang W,
Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, et al:
Genome-scale analysis of DNA methylation in lung adenocarcinoma and
integration with mRNA expression. Genome Res. 22:1197–1211. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Okayama H, Kohno T, Ishii Y, Shimada Y,
Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S,
et al: Identification of genes upregulated in ALK-positive and
EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res.
72:100–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hou J, Aerts J, den Hamer B, van Ijcken W,
den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens
JA, Hoogsteden HC, et al: Gene expression-based classification of
non-small cell lung carcinomas and survival prediction. PLoS One.
5:e103122010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
NCI-H1650 [H-1650, H1650]
(ATCC® CRL-5883™). https://www.atcc.org/~/ps/CRL-5883.ashx
|
|
24
|
NCI-Navy Medical Oncology Branch cell line
supplement. J Cell Biochem Suppl. 24:1–291. 1996.
|
|
25
|
Sexton DW, Blaylock MG and Walsh GM: Human
alveolar epithelial cells engulf apoptotic eosinophils by means of
integrin- and phosphatidylserine receptor-dependent mechanisms: A
process upregulated by dexamethasone. J Allergy Clin Immunol.
108:962–969. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boussat S, Eddahibi S, Coste A, Fataccioli
V, Gouge M, Housset B, Adnot S and Maitre B: Expression and
regulation of vascular endothelial growth factor in human pulmonary
epithelial cells. Am J Physiol Lung Cell Mol Physiol.
279:L371–L378. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shimano H: Sterol regulatory
element-binding proteins (SREBPs): Transcriptional regulators of
lipid synthetic genes. Prog Lipid Res. 40:439–452. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakakuki M, Shimano H, Inoue N, Tamura M,
Matsuzaka T, Nakagawa Y, Yahagi N, Toyoshima H, Sato R and Yamada
N: A transcription factor of lipid synthesis, sterol regulatory
element-binding protein (SREBP)-1a causes G(1) cell-cycle arrest
after accumulation of cyclin-dependent kinase (cdk) inhibitors.
FEBS J. 274:4440–4452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Streicher R, Kotzka J, Muller-Wieland D,
Siemeister G, Munck M, Avci H and Krone W: SREBP-1 mediates
activation of the low density lipoprotein receptor promoter by
insulin and insulin-like growth factor-I. J Biol Chem.
271:7128–7133. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Downward J: Targeting RAS signalling
pathways in cancer therapy. Nat Rev Cancer. 3:11–22. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bedard PL, Tabernero J, Janku F, Wainberg
ZA, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Pérez-García J,
Stathis A, Britten CD, et al: A phase Ib dose-escalation study of
the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with
the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with
selected advanced solid tumors. Clin Cancer Res. 21:730–738. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang BX, Akbar M, Kevala K and Kim HY:
Phosphatidylserine is a critical modulator for Akt activation. J
Cell Biol. 192:979–992. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang W, Liu J, Hu X, Li P, Leak RK, Gao Y
and Chen J: n-3 polyunsaturated fatty acids reduce neonatal
hypoxic/ischemic brain injury by promoting phosphatidylserine
formation and Akt signaling. Stroke. 46:2943–2950. 2015. View Article : Google Scholar : PubMed/NCBI
|