Introduction
Cellular senescence is a persistent cell-cycle
arrest of actively proliferating cells that contributes to
physiological and pathological processes including tumor
suppression, inflammatory response and tissue repair (1,2).
Senescence is not a chief strategy of cancer therapy but it may be
a key antitumor response to counteract carcinogenic stress.
Cellular senescence is a primary response against tumorigenesis,
and senescent cells can be eliminated by the innate immune system
(3). Chemotherapy-induced sublethal
stress can accelerate a specific form of senescent phenotype in
cells which is termed as stress-induced premature senescence (SIPS)
(4), so we focused primarily on this
approach.
Growth arrest of proliferating cells is often
triggered by a persistent DNA damage response (DDR) and is
classically executed by activation of the p16-Rb (retinoblastoma)
and/or p53-p21 pathways (5–7). The ataxia telangiectasia mutated (ATM)
protein acting as a damage sensor is recruited to damaged foci,
stabilizing p53 and upregulating p53 transcriptional target p21
which inhibits cyclin-dependent kinase 2 (CDK2) and activates Rb to
prevent entry into cell cycle (8–10). On the
other hand, p16 inhibits CDK4 and CDK6, leading to inactivation of
Rb to block cell cycle progression (6).
CDKs are a family of serine/threonine kinases that
mainly play an important role in regulating cell cycle progression
(11). The ultimate DDR targets are
CDKs, key regulators of cell cycle progression (12). CDKs phosphorylate Rb tumor suppressor
and the related ‘pocket-proteins’ p107 and p130 to maintain them in
an inactive state. Phosphorylated (inactive) pRb and
pocket-proteins facilitate E2F family transcription factors and
promote genes expression related to DNA replication and cell
division, eventually dysregulating cell cycle progression (13). In response of DNA replication stress,
active p53 accelerates expression of the CDK inhibitor p21 which is
an essential mediator of senescence (12). Except regulating cell cycle
progression, the CDK family also has other physiological functions.
For example, CDK5, known as a key regulator in neuronal cells, has
also been found to participate in several physiological and
pathological processes of extra neuronal activities, such as gene
expression, cell migration, apoptosis, and senescence (14,15).
Disorder of cell cycle regulation and aberrant activation of CDKs
occur in almost all human cancers (16,17) and
for the important role of CDKs in many biological processes
commonly abnormally regulated in cancer cells, targeting CDKs
chemically is considered to be a potential therapy for cancer
treatment (18).
Dinaciclib is a potent and specific inhibitor of CDK
that interacts with the acetyl-lysine recognition site of
bromodomains (19). Dinaciclib
specifically targets CDK1/2/5/9 (20). Preclinical studies have shown it to be
a potent growth inhibitor in a murine xenograft model of human
cancers (21,22) and a promoter of apoptosis in most
cancer cells via suppression of Rb phosphorylation (22,23). Here
we investigated antineoplastic effects and mechanisms underlying
dinaciclib plus DOX in preclinical models of multiple myeloma
(MM).
Materials and methods
Cell culture and reagents
Human MM RPMI-8226 cell lines were generously gift
from Dr. Xinliang Mao (The Cyrus Tang Hematology Center of Soochow
University, Suzhou, China) and maintained in RPMI-1640 medium
containing 10% fetal calf serum (FBS), 100 U/ml penicillin and 100
ng/ml streptomycin. Cells were incubated at 37°C with 5%
CO2. Doxorubicin (DOX) was purchased from Sigma-Aldrich;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA; D1515). CDK
inhibitor dinaciclib and ATM kinase inhibitor KU-55933 was
purchased from Selleckchem (Houston, TX, USA; S2768 and S1092).
Anti-ATM (ab32420), anti-p-ATM (S1981, ab81292), anti-Chk2
(ab109413), anti-p-Chk2 (T68, ab32148), anti-p53 (ab32389),
anti-γ-H2AX (S139, ab2893), anti-p16 (ab08349), anti-p21 (ab109199)
anti-CDK1 (ab133327), anti-CDK2 (ab32147), anti-CDK9 (ab76320) and
anti-CDK5 (ab40773) were from Abcam (Cambridge, UK). Anti-p-p53
(S15) was from Immunoway. Anti-GAPDH (TA505454) was obtained from
ZSBG-BIO (Beijing, China).
Cell viability assay
Cells were seeded in a 96-well plate (10,000
cells/well), and incubated with different concentrations of DOX in
three wells for 24 h. Cell viability was measured using an MTT kit
(Sigma-Aldrich; Thermo Fisher Scientific, Inc.). Plates were
detected at 570 nm on a microplate reader (DNM-9602; PERLONG,
Beijing, China).
Cell cycle analysis
Cells were seeded in a 6-well plate
(10×104 cells/well) and incubated with/without 100 nM
DOX for 24 h. Cells were harvested and washed twice with cold PBS,
then fixed with cold 70% ethanol for 1 h at 4°C. Fixed cells were
washed twice with PBS and resuspended with 0.4 ml PBS containing PI
(20 µg/ml; Sigma-Aldrich; Thermo Fisher Scientific, Inc.) and
DNase-free RNaseA (50 µg/ml; Amresco LLC, Solon, OH, USA), and
measured using flow cytometry after 30 min incubation at room
temperature in the dark. Data were analyzed using Flowjo7.6
software (FlowJo LLC, Ashland, OR, USA).
Senescence associated β-galactosidase
(SA-β-Gal) staining
Cells were treated as described and incubated on
poly-L-lysine coated slides (P9155; Sigma-Aldrich; Thermo Fisher
Scientific, Inc.) for 1 h at 37°C. SA-β-Gal staining was conducted
according to the manufacturers' instructions (cat. no. 9860) from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Cells positive
for SA-β-Gal staining were quantified in 100 individual cells for
each sample and analyzed (n=3).
Immunofluorescence staining
Cells were treated as described and incubated on
poly-L-lysine coated slides (P9155; Sigma-Aldrich; Thermo Fisher
Scientific, Inc.) for 1 h at 37°C. Then, cells were fixed in 4%
paraformaldehyde for 20 min at room temperature, permeabilized with
0.5% Triton X-100 for 15 min and blocked with 1% BSA dissolved in
PBS for 1 h. Cells were then incubated with anti-γ-H2AX
(phospho-S139) (1:250; S139; ab2893) overnight at 4°C, and labeled
with TRICT-conjugated goat anti-rabbit secondary antibodies for 1 h
at RT in the dark. Nuclei were stained with 0.1 µg/ml DAPI for 5
min. Cells were viewed under an Olympus (BX51; Olympus Corp.,
Tokyo, Japan) fluorescent microscope. γ-H2AX foci in nuclei were
counted in 50 individual cells for each sample.
Western blot analysis
Cells were lysed in RIPA buffer and lysates were
sonicated and then boiled and centrifuged (447.2 × g). Proteins
were resolved with 8–15% SDS-PAGE and transferred to PVDF
membranes. After blocking, membranes were incubated with the
indicated primary antibodies and then horseradish
peroxidase-conjugated secondary antibodies (1:4,000; ab136817;
Abcam). Blots were visualized using ECL and SuperSignal reagent
(WBKLS0500; EMD Millipore, Billerica, MA, USA). Protein
quantification was conducted using image J software.
Apoptosis assay
Cells were washed and stained with Annexin V-FITC
and PI according to instructions (BD Bioscience, Franklin Lakes,
NJ, USA). At least 1,000 cells were counted to calculate percent
apoptotic in each sample. Cells with Annexin V+/PI- and Annexin
V+/PI+ were defined as apoptotic cells.
Statistical analyses
Data were analyzed using GraphPad Prism 5 (GraphPad
Software, Inc., La Jolla, CA, USA). Differences between multiple
groups was confirmed using one-way analysis of variance followed by
Tukey's post hoc test. The significance of differences between two
groups was determined using the Student's t-test. Data were
obtained from at least three independent experiments and P<0.05
was considered to indicate a statistically significantly
differenence.
Results
DOX inhibited proliferation, induced
cell cycle arrest, and senescence in MM RPMI-8226 cells
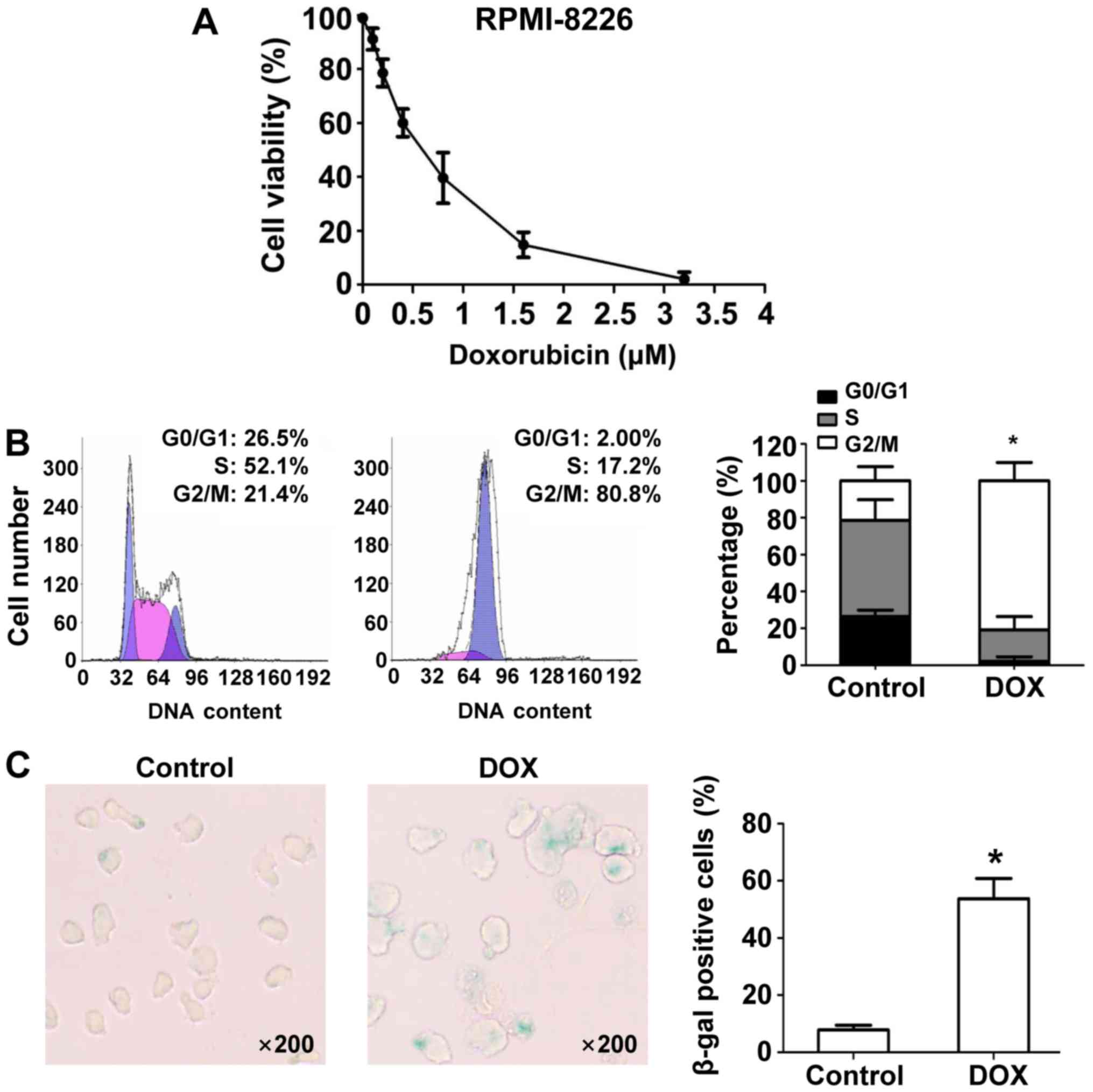
Data from an MTT assay showed that DOX inhibited
RMPI8226 cell growth in a dose-dependent manner (Fig. 1A). Subsequent experiments were
conducted with 100 nM DOX as a representative of DNA damaging
agents to study the effects on MM cells. Next, we studied the
effects of DOX on the cell cycle in RPMI-8226 cells. DNA content
analysis with flow cytometry after DOX treatment for 24 h showed
that DOX induced marked accumulation of G2/M DNA content (21.4% vs.
80.8%; P<0.05) in RPMI-8226 cells compared to DMSO-treated
control cells (Fig. 1B). Increased
nuclear and cell body size, and staining for β-Gal, a molecular
marker of cell senescence, revealed that DOX induces more
senescence in RPMI-8226 cells at 24 h compared with DMSO (Fig. 1C).
Senescence of RPMI-8226 cells caused
by DOX accompanied with DSBs and activation of ATM/Chk2/p53/p21
signaling
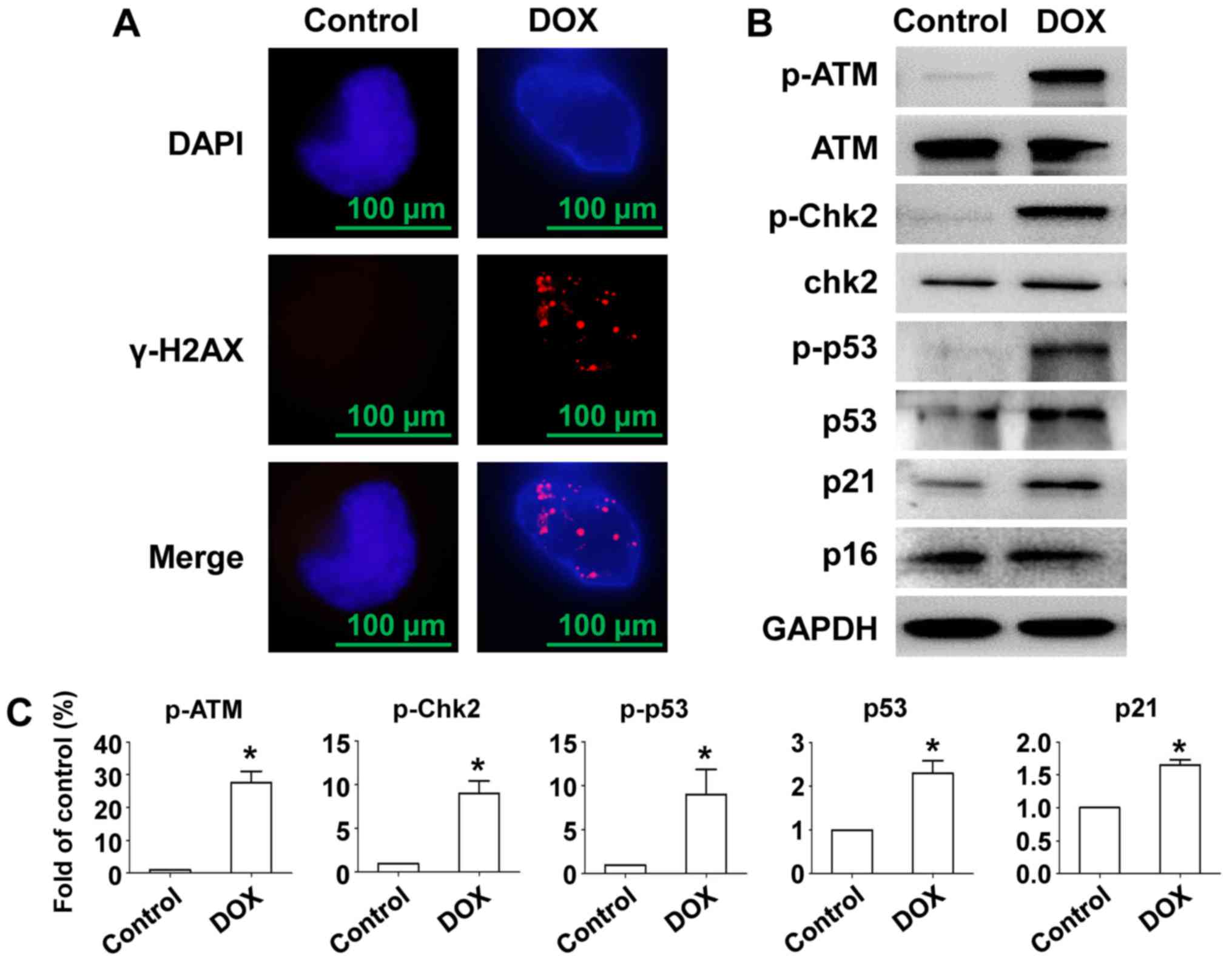
To investigate whether DOX-induced senescence of MM
cells and DNA damage as well as the pathway activated,
immunofluorescent staining of γ-H2AX and Western blot was used.
Data show that DOX induced γ-H2AX foci formation in the nucleus in
RPMI-8226 cells (Fig. 2A). After
treatment with DOX, an intense ATM phosphorylation response was
observed and phosphorylation level of p53 and Chk2 was also
elevated, resulting in stabilization and accumulation of p53
(24). Expression of the CDK
inhibitor p21 was also stimulated by DOX induced oncogenic stress
but not p16 (Fig. 2B and C). Thus,
cell cycle exit and senescence after DOX exposure in RPMI-8226
cells was accompanied by DNA damage and activation of the
ATM/Chk2/p53/p21 pathway, but not the p16 pathway.
Low-dose dinaciclib increased
DOX-induced senescence of MM RPMI-8226 cells but not apoptosis
Dinaciclib has shown promise for treating non-small
cell lung cancer, breast cancer and MM in some I/II phase clinical
trials (18) but combination therapy
deserves attention as a novel cancer treatment strategy. To
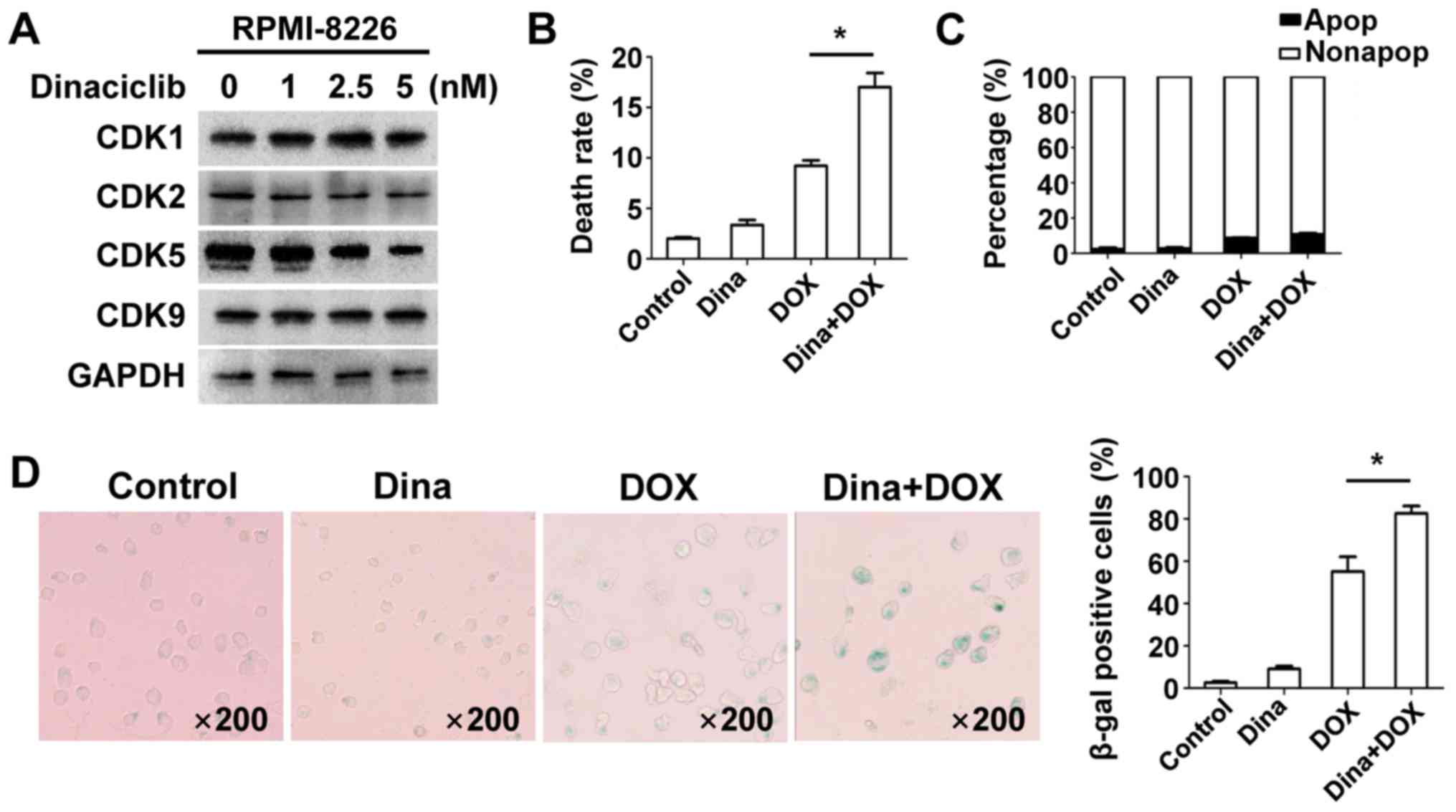
evaluate anti-MM effects of dinaciclib plus DOX, we incubated
RPMI-8226 cells with various concentrations of dinaciclib for 24 h
and expression of CDK1/2/5/9 was measured. The minimum dose (5 nM)
of dinaciclib that decreased CDK5 was chosen for subsequent
experiments to regulate expression of related proteins regardless
of other involved CDKs as well as reduce toxicity clinically
(Fig. 3A). Cell viability, apoptosis,
and senescence were measured in RPMI-8226 cells and data showed
that dinaciclib enhanced DOX-induced inhibition of cell viability
(Fig. 3B), whereas apoptosis did not
change (Fig. 3C). Co-treatment
induced more senescent phenotype (Fig.
3D). We speculated that low-dose dinaciclib elevated anti-MM
activity with DOX by promoting senescence but not apoptosis.
Dinaciclib transforms cells from the
p21 to the p16 pathway in the senescence model induced by DOX in
myeloma RPMI-8226 cells
Now that dinaciclib enhanced DOX-induced senescence
in RPMI-8226 cells, it was easy to assume that whether this
phenomenon was accompanied by elevated p21 expression.
Interestingly western blot for expression of ATM-related proteins
showed that dinaciclib partly reduced phosphorylated ATM and
phosphorylation of ATM downstream Chk2/p53 proteins and level of
p21 (Fig. 4A). We then measured
activation of alternative executor p16 and its expression of
combination was greater compared with DOX alone in RPMI-8226 cells
(Fig. 4A), suggesting that dinaciclib
enhanced DOX triggered senescence of RPMI-8226 cells by switching
from p21 to p16 pathways.
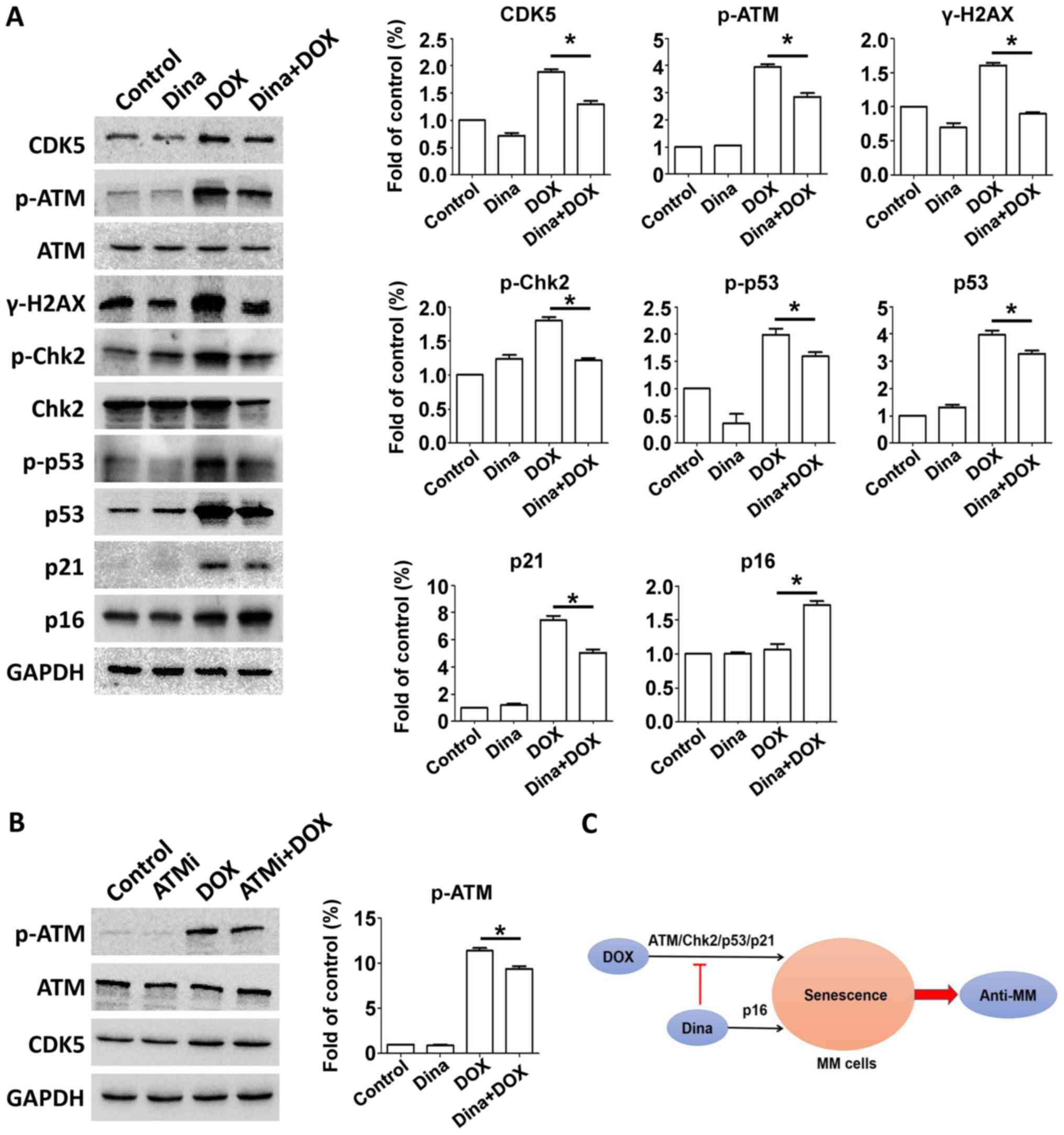 | Figure 4.Dinaciclib transforms the p21 to the
p16 pathway in a senescence model induced by DOX in MM RPMI-8226
cells. (A) RPMI-8226 cells were treated with DMSO, 100 nM DOX, 5 nM
dinaciclib, or DOX and dinaciclib for 24 h. Cells were extracted
and proteins were incubated with primary antibodies against p-ATM,
ATM, γ-H2AX, p-Chk2, Chk2, p-p53, p53, p21 and p16. GAPDH was used
as a loading control. Protein quantification was conducted. (B)
RPMI-8226 cells were treated with DMSO, 10 µM KU-55933, 100 nM DOX,
or DOX and KU-55933 for 24 h. Cells were extracted and proteins
were incubated with primary antibodies against p-ATM, ATM, CDK5.
GAPDH was used as a loading control. Protein quantification was
conducted. (C) Low-dose of dinaciclib enhanced anti-MM effects
mediated by DOX via transformation of the p21-p16 pathways,
accelerating senescence rather than apoptosis induced by DOX. Data
were obtained from at least three independent experiments.
Differences between multiple groups was confirmed using one-way
analysis of variance followed by Tukey's post hoc test. *P<0.05
as indicated. MTT, multiple myeloma; DOX, doxorubicin; ATM, ataxia
telangiectasia mutated; DOX, doxorubicin; CDK, cyclin-dependent
kinase. |
Next we explored the preliminary mechanism of this
transformation. Our data showed that dinaciclib inhibited
expression of CDK5 and phosphorylation of ATM. After incubating
RPMI-8226 cells with 10 µM ATM inhibitor KU-55933 and DOX for 24 h,
we measured expressions of CDK5 and p-ATM and found inhibition of
ATM kinase did not lead to change of CDK5 expression (Fig. 4B). Hence targeting CDK5 restrained
DOX-triggered upregulation of p21 protein by orchestrating ATM
phosphorylation, whereas meanwhile p16 expression was elevated.
Given that inhibiting CDK5 or upregulating p16 brought about
decreased phosphorylation of Rb protein (6,13). We
speculated that inhibition of CDK5 might lead to an increased
expression of p16 protein due to the negative feedback
regulation.
Discussion
MM is neoplastic and characterized by clinical
symptoms, which means its pathogenesis is not clear and a cure does
not exist. At this time, MM treatment chiefly consists of
proteasome inhibitors, immunomodulatory drugs (IMiDs), epigenetic
drugs, monoclonal antibody and immunotherapy. The emergence of so
many new drugs on the one hand brings gospels to more and more MM
patients; on the other hand, it also indicates that MM is still an
incurable disease and the development of new drug targets is always
in progress. Better MM treatment may arise from combinations of
existing chemotherapeutics. In this study we focused on a new CDK
inhibitor dinaciclib and evaluated its effects of combination with
DOX on MM.
DNA damaging agents combined with proteasome
inhibitor (such as bortezomib) are highly effective in the
treatment of newly diagnosed or relapsed MM (25). Regardless, DNA damage agents are still
important drugs in MM treatment, even if new classes of drugs are
widely used. DNA damaging agents can trigger DDR in various types
of cancer cells and activate multiple signaling pathways to inhibit
cell growth, manifested as senescence, apoptosis or necrosis. While
senescent cells are metabolic, antineoplastic roles of senescence
in cancer treatment is to inhibit proliferation of cancer cells and
activate the immune system to eliminate the senescent cells. We
proved that low dose of DOX, a topoisomerase II inhibitor,
generated DNA double strand breaks (DSBs) in MM RPMI-8226 cells,
evidenced by the formation of γ-H2AX foci in the nucleus. And DOX
also enhanced phosphorylation level of ATM protein, mediated
downstream phosphorylation cascade of ATM, including Chk2 and p53,
leading to an increasing expression of p21, an inhibitor of CDK.
Low dose of DOX lead to cell cycle arrest and senescent phenotypes
in RPMI-8226 cells. However, the p16-pRb pathway was not
activated.
Most tumors are highly proliferative due to abnormal
expression of CDKs and cell cycle check point dysregulation. So,
targeting CDKs may be an antitumor strategy. Dinaciclib, a specific
inhibitor of CDK1/2/5/9, has been proven effective in an early
clinical trial as monotherapy for treating refractory and relapsed
MM (20). Thus, we focused on the
efficacy of DOX and dinaciclib. To evaluate the effect of
dinaciclib on its target molecules, we incubated RPMI-8226 cells
with different concentrations of dinaciclib for 24 h and detected
expression of CDK1/2/5/9. We found that low dose (5 nM) of
dinaciclib could only influence activity of CDK5 but not the
others. So subsequent experiments about the effect of
pharmacological inhibition of CDK5 combined with DOX on RPMI-8226
cells were conducted.
Over the last decade Cdk5 has been shown to be
essential for neuronal functions (26), but it may have roles outside the
nervous system. Cdk5 is generally dysregulated in various types of
cancer (15), and targeting it may
become a novel and promising therapeutic strategy for cancer
treatment. CDK5 participates in DDR by phosphorylating ATM and
downstream proteins (27–31), so we treated RPMI-8226 cells with the
lowest concentration of dinaciclib that caused a decline in CDK5
and DOX for 24 h, assuming dinaciclib could influence DDR and
senescence induced by DOX in MM cells. Results showed that compared
to DOX alone, the combination brought about decreased cell
viability and increased senescence in RPMI-8226 cells, but
apoptosis was unchanged. Thus, dinaciclib at least partly
strengthened the effect with DOX against MM by enhancing
DOX-triggered senescence rather than apoptosis. Given that
DOX-induced senescence of RPMI-8226 cells was accompanied by
activation of the p21 pathway, we speculated whether dinaciclib
accelerated DOX-induced senescence of RPMI-8226 cells by enhancing
expression of p21 protein. So we measured expression of related
proteins in ATM/Chk2/p53/p21 pathway after combination treatment in
RPMI-8226 cells and unexpectedly we noted that p21 protein slightly
decreased. It was speculated that there could be alternative
pathway involved in this senescent model. And elevated p16
expression was confirmed in combination dinaciclib with DOX
compared to DOX alone in RPMI-8226 cells. Thus, dinaciclib partly
enhanced senescence induced by DOX in RPMI-8226 cells via a switch
from the p21 to the p16 pathway.
In our study, phosphorylated ATM declined
simultaneously with inhibition of expression of CDK5. According to
other work, CDK5 participates in DDR by phosphorylating ATM. To
study this effect in MM cells, we incubated RPMI-8226 cells with
KU-55933 to block ATM kinase, and DOX for 24 h simultaneously,
andassayed the expression of CDK5. Data showed that the expression
of phosphorylated ATM decreased and expression of CDK5 was
unchanged after inhibiting ATM kinase activity. The result revealed
that CDK5 participated in ATM-related DDR at least partly by
regulating phosphorylated ATM. Targeting CDK5 restrained
DOX-triggered upregulation of p21 protein by orchestrating ATM
phosphorylation, whereas meanwhile p16 expression was elevated. But
how does this transformation work? It had been proved that both
CDK5 and p16 are upstream regulators of Rb protein. Inhibiting CDK5
or upregulating p16 brought about decreased phosphorylation of Rb
protein (6,13). We speculated that after inhibition of
CDK5 activity by dinaciclib, the phosphorylation level of Rb
protein declined, leading to an increased expression of p16 protein
due to the negative feedback regulation. And the deep mechanism is
our subsequent research direction.
Besides both RPMI-8226 and H929 cell lines were
tested in our study, but H929 cells failed to gain good results as
well as RPMI-8226 cells (data not shown), so we speculate that the
conclusions of this study may be associated with type of MM cell
line. In future studies we will order other MM cell lines from
ATCC, such as U226 or MM.1S to further verify our conclusion.
In conclusion, dinaciclib enhances anti-MM effects
of DOX by transformation of the p21-p16 pathways, accelerating
senescence rather than apoptosis induced by DOX in MM RPMI-8226
cells (Fig. 4C). These data may help
to provide tailored treatment for MM patients.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Social
Development Science and Technology Fund of Shaanxi Province (grant
no. 2016SF-071) and the Natural Science Foundation of Shaanxi
Province (grant no. 2017JM8025).
Availability of data and materials
All data generated or analyzed in this study are
included in this manuscript.
Authors' contributions
HT and GG designed the study. HT, LX and XL
conducted the experiments and performed the statistical analysis.
HT and GG reviewed and provided final approval of the version to be
published. All authors read and approved the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rodier F and Campisi J: Four faces of
cellular senescence. J Cell Biol. 192:547–556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
He S and Sharpless NE: Senescence in
health and disease. Cell. 169:1000–1011. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xue W, Zender L, Miething C, Dickins RA,
Hernando E, Krizhanovsky V, Cordon-Cardo C and Lowe SW: Senescence
and tumour clearance is triggered by p53 restoration in murine
liver carcinomas. Nature. 445:656–660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brack C, Lithgow G, Osiewacz H and
Toussaint O: EMBO WORKSHOP REPORT: Molecular and cellular
gerontology Serpiano. Switzerland, September 18–22, 1999. EMBO J.
19:1929–1934. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campisi J: Aging, cellular senescence, and
cancer. Annu Rev Physiol. 75:685–705. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Childs BG, Durik M, Baker DJ and van
Deursen JM: Cellular senescence in aging and age-related disease:
From mechanisms to therapy. Nat Med. 21:1424–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Muñoz-Espín D and Serrano M: Cellular
senescence: From physiology to pathology. Nat Rev Mol Cell Biol.
15:482–496. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alcorta DA, Xiong Y, Phelps D, Hannon G,
Beach D and Barrett JC: Involvement of the cyclin-dependent kinase
inhibitor p16 (INK4a) in replicative senescence of normal human
fibroblasts. Proc Natl Acad Sci USA. 93:13742–13747. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takahashi A, Ohtani N, Yamakoshi K, Iida
S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H and Hara E:
Mitogenic signalling and the p16INK4a-Rb pathway cooperate to
enforce irreversible cellular senescence. Nat Cell Biol.
8:1291–1297. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Beauséjour CM, Krtolica A, Galimi F,
Narita M, Lowe SW, Yaswen P and Campisi J: Reversal of human
cellular senescence: Roles of the p53 and p16 pathways. EMBO J.
22:4212–4222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: Several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gire V and Dulic V: Senescence from G2
arrest, revisited. Cell Cycle. 14:297–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Burkhart DL and Sage J: Cellular
mechanisms of tumour suppression by the retinoblastoma gene. Nat
Rev Cancer. 8:671–682. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arif A: Extraneuronal activities and
regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5.
Biochem Pharmacol. 84:985–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Contreras-Vallejos E, Utreras E and
Gonzalez-Billault C: Going out of the brain: Non-nervous system
physiological and pathological functions of Cdk5. Cell Signal.
24:44–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Konecny GE: Cyclin-dependent kinase
pathways as targets for women's cancer treatment. Curr Opin Obstet
Gynecol. 28:42–48. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hall M and Peters G: Genetic alterations
of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human
cancer. Adv Cancer Res. 68:67–108. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen XX, Xie FF, Zhu XJ, Lin F, Pan SS,
Gong LH, Qiu JG, Zhang WJ, Jiang QW, Mei XL, et al:
Cyclin-dependent kinase inhibitor dinaciclib potently synergizes
with cisplatin in preclinical models of ovarian cancer. Oncotarget.
6:14926–14939. 2015.PubMed/NCBI
|
|
19
|
Martin MP, Olesen SH, Georg GI and
Schönbrunn E: Cyclin-dependent kinase inhibitor dinaciclib
interacts with the acetyl-lysine recognition site of bromodomains.
ACS Chem Biol. 8:2360–2365. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar SK, LaPlant B, Chng WJ, Zonder J,
Callander N, Fonseca R, Fruth B, Roy V, Erlichman C and Stewart AK:
Mayo Phase 2 Consortium: Dinaciclib, a novel CDK inhibitor,
demonstrates encouraging single-agent activity in patients with
relapsed multiple myeloma. Blood. 125:443–448. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feldmann G, Mishra A, Bisht S, Karikari C,
Garrido-Laguna I, Rasheed Z, Ottenhof NA, Dadon T, Alvarez H,
Fendrich V, et al: Cyclin-dependent kinase inhibitor Dinaciclib
(SCH727965) inhibits pancreatic cancer growth and progression in
murine xenograft models. Cancer Biol Ther. 12:598–609. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parry D1, Guzi T, Shanahan F, Davis N,
Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R,
et al: Dinaciclib (SCH 727965), a novel and potent cyclin-dependent
kinase inhibitor. Mol Cancer Ther. 9:2344–2353. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fu W, Ma L, Chu B, Wang X, Bui MM, Gemmer
J, Altiok S and Pledger WJ: The cyclin-dependent kinase inhibitor
SCH 727965 (dinacliclib) induces the apoptosis of osteosarcoma
cells. Mol Cancer Ther. 10:1018–1027. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stracker TH and Petrini JH: The MRE11
complex: Starting from the ends. Nat Rev Mol Cell Biol. 12:90–103.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kumar SK, Rajkumar V, Kyle RA, van Duin M,
Sonneveld P, Mateos MV, Gay F and Anderson KC: Multiple myeloma.
Nat Rev Dis Primers. 3:170462017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dhavan R and Tsai LH: A decade of CDK5.
Nat Rev Mol Cell Biol. 2:749–759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ajay AK, Upadhyay AK, Singh S, Vijayakumar
MV, Kumari R, Pandey V, Boppana R and Bhat MK: Cdk5 phosphorylates
non-genotoxically overexpressed p53 following inhibition of PP2A to
induce cell cycle arrest/apoptosis and inhibits tumor progression.
Mol Cancer. 9:2042010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ogara MF, Belluscio LM, de la Fuente V,
Berardino BG, Sonzogni SV, Byk L, Marazita M and Cánepa ET:
CDK5-mediated phosphorylation of p19INK4d avoids DNA damage-induced
neurodegeneration in mouse hippocampus and prevents loss of
cognitive functions. Biochim Biophys Acta. 1843:1309–1324. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian B, Yang Q and Mao Z: Phosphorylation
of ATM by Cdk5 mediates DNA damage signalling and regulates
neuronal death. Nat Cell Biol. 11:211–218. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Huang E, Qu D, Zhang Y, Venderova K, Haque
ME, Rousseaux MW, Slack RS, Woulfe JM and Park DS: The role of
Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation
in neuronal death. Nat Cell Biol. 12:563–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qu D, Rashidian J, Mount MP, Aleyasin H,
Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, et
al: Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity
and Parkinson's disease. Neuron. 55:37–52. 2007. View Article : Google Scholar : PubMed/NCBI
|