Introduction
Neuroblastoma (NB) is a pediatric cancer with the
highest incidence during pre-scholar age. Patient can onset with
localized or metastatic tumor. High-risk (HR) patients usually are
older than one year of age with a metastatic tumor involving bone
marrow, liver and skeletal bone (1).
The overall survival (OS) of HR patients at 5-years from diagnosis
is near 40%. On the contrary, patients with localized tumors,
usually fall in the Low-Risk or Intermediate-Risk group, and have a
5-years OS ranging between 75 and 98% (2).
Tumor cells show several gross cytogenetic
abnormalities. The most aggressive tumor in children older than 1
years of age, shows numerous copy number variations (CNVs) such as
deletion, gains and gene amplification with homogenously staining
region and/or double min and/or entire chromosome extra-copies
(3). Two type of CNVs can occur in
tumor cells: Structural CNVs that shows structural chromosome
changes such as deletion, amplification and gross chromosome
rearrangement, and numerical CNVs that involves gain or loss of
whole chromosomes (4). Tumor
aggressiveness has been observed strongly associated with such
structural CNVs whereas localized tumors, belonging to patients at
low- and intermediate-risk, are less aggressive and they have
numerical CNVs, prevalently.
Recently, another phenomena observed in NB tumors is
the chromotripsys at chromosome 5, particularly (5). Moreover, NB tumor cells show different
ploidy features: near-diploidy and near-tetraploidy are observed in
HR tumors characterized by structural abnormalities, instead
near-triploidy cells are usually present in tumors of low-risk
patient (6); these tumors show
extra-chromosomes and few structural chromosome abnormalities.
Consistently, patients with the near-triploidy tumor cells have
favorable outcome with a good OS, whereas unfavorable prognosis is
observed in patients who have near-diploidy or near-tetraploidy
tumors (7,8). All together these findings show evidence
of chromosome instability (CIN) in NB cells.
CIN and tumorigenesis
In the past years, enormous interest among the
scientists has stimulated the search for gene mutations as
causative of tumorigenesis (9–11). The
development of next generation technique (NGS) has greatly improved
the understanding of several cancer molecular mechanisms, with the
prospective to improve the treatment by targeting cancer mutated
proteins. However, prior to the interest for gene mutation
research, there was a long period dominated by the search of
numerical and structural chromosomal abnormalities associated to
the genesis of several tumors (12).
In 1914, Boveri first suggested the hypothesis of aneuploidy in
cancer, but only later the relevant role of CIN in the majority of
tumors was demonstrated by Cahill et al (13) and by Heng et al (14). To date, there is a debate if the CIN
is the cause of genomic instability (GIN) developing the cancer
(15,16) or if CIN process is the consequence of
abnormal function of mutated gene encoding for important mitotic
proteins leading to tumor development (17,18).
CIN with chromosome aberrations in copy number is a
form of GIN that make prone the cancer cells to acquire mutations
conferring them rapid tumor progression, aggressiveness and
drug-resistant phenotypes. GIN is characterized by an increased
frequency of genetic alterations deregulating specific biological
signaling associated with cellular cell cycle homeostasis and
induces genomic diversity in cancer cells (19). This genomic chaos gives to cancer cell
populations the properties to adapt themselves at the stimuli of
the tumor microenvironment (20).
For long time, the chromosome missegregation
together with others mechanisms such as gene mutations, chromosomal
rearrangements and epigenetic factors were considered responsible
for tumor growth and tumor heterogeneity. Indeed, cell
heterogeneity is a hallmark of tumors lending the ability of
adapting to external pressures (21,22).
Furthermore, many studies are reporting the link among karyotype
alterations, CIN (23) and cancer
(13,24–26)
strongly indicating that CIN takes part in the origin of cancer
(18,27). Moreover, the role of CIN in
tumorigenesis is also supported by experimental observations of
persistent chromosome missegregation in tumor cell lines (28). Altogether these observations indicate
the link between CIN and aneuploidy (29).
There are evidences that CIN and aneuploidy could
promote tumor initiation, acquisition of drug resistance,
metastasis and relapse (30) through
the variation of the copy number of oncogenes/tumor suppressor
genes allowing cells to adapt to environmental stimuli changes such
as nutrient variation and/or hypoxia (20). Aneuploidy may induce genome
instability and lead to acquisition of genetic cell heterogeneity
triggering selective pressures in clones selection. Therefore, the
survival of cells with CIN to the cancer treatment could be due to
higher adaptive potential of these cells in which specific
chromosomal aberrations confer cellular fitness advantages
(31).
The aneuploidy paradox
Despite aneuploidy commonly occurs in many cancers
where it is an indicator of tumor growth, it often leads to a
reduction in the cell proliferation rate (32,33). This
apparent contradiction is known as the: aneuploidy paradox.
The rate of CIN determines the effect of aneuploidy on tumors;
whereas low rates of CIN (missegregation of a small number of
chromosomes per division) are weakly tumor promoting, higher rates
of CIN (missegregation of more than five chromosomes) cause cell
death and tumor suppression (34).
Thus, cell death induced by CIN sufficiently high arises from an
increase in the number of chromosomes missegregated per cell
division. Coherently with this, Komarova described a mathematical
model showing that a low rate of CIN optimizes the tumor
heterogeneity and survival and that increase of CIN rate is
associated with decreases tumor fitness (35). These observations suggest that
increasing the rate of CIN over a critical threshold could be
efficient to stop the tumor cell proliferation.
This paradoxical situation is attracting
considerable attention for therapeutic purpose, although the
therapeutic targeting of CIN in cancer is still at preclinical
stages. Several oncogenes and tumor suppressor genes are known to
localize within centrosomes, which altered function triggers
centrosome abnormalities (36). Thus,
there are many promising inhibitors against associated centrosome
proteins and many of these drugs/compounds are being tested in
preclinical models and in clinical trials. Two of the most studied
centrosomal kinases with oncogenic properties are: AURKA and AURKB,
and more than 30 AURK inhibitors have been developed and used in
clinical studies [reviewed by (37)].
Tubulin is another important target, as this protein
acts during cellular growth, division, and migration. Taxanes
(paclitaxel and docetaxel) and vinca alkaloids (vinblastine,
vincristine, and vinorelbine), well-known FDA-approved compounds
clinically used for targeting tubulin, have been demonstrated to be
successful to induce mitotic arrest (38).
Additional cancer therapy strategy that has
attracting attention in recent years is synthetic lethality
(39), defined as a condition in
which perturbation in two different genes together results in cell
death but mutation of either alone is compatible with cell life.
The first clinically approved drugs designed to exploit synthetic
lethality are poly(ADP-ribose) polymerase inhibitors (PARPis)
(40). PARP is a nuclear protein
important for recognizing DNA damage and repairing DNA
single-strand breaks (SSBs). It is proposed that inhibition of PARP
results in the accumulation of unrepaired SSBs that are converted
into double-strand breaks during DNA replication resulting in gross
GIN and cell death.
Methods of measuring CIN
Although the evaluation of CIN rate in tumor samples
is not routinely performed in the clinical setting, direct and
indirect methods to measure CIN have been adopted; these methods
are based on both the determination of cell-to-cell variability in
chromosome number and structure within the tumor cell population,
as well as on the assessment of the rate at which these chromosomal
changes occur (41). Therefore, the
methods are able to capture the dynamic nature of CIN. The CIN rate
is directly related to the estimation of the mitotic error
frequency in fixed cells or in formalin-fixed and paraffin-embedded
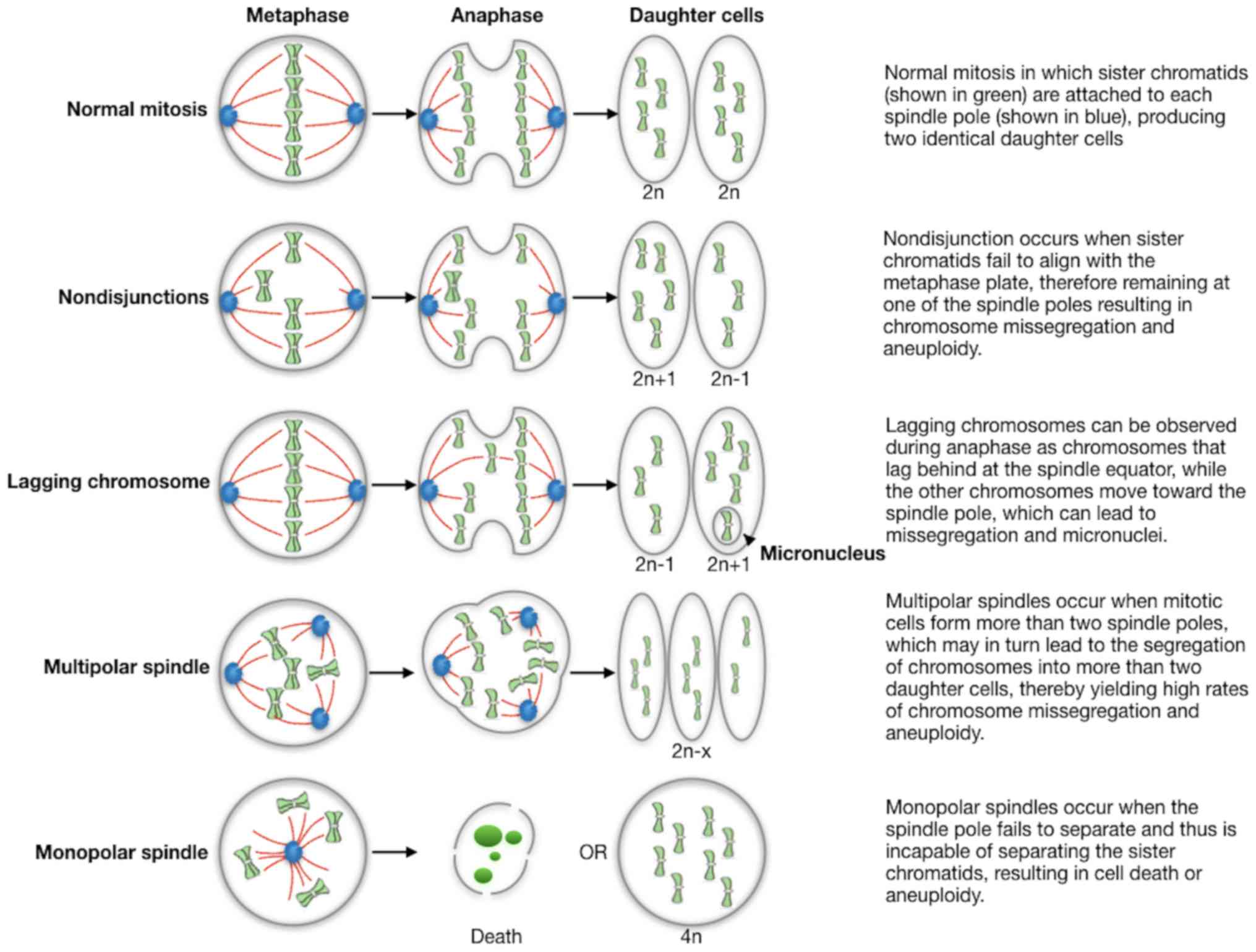
(FFPE) tumor tissues. Some of main defects are summarized in
Fig. 1: abnormalities in chromosome
structure and function resulting in chromosomes that lag in
anaphase or exhibit incomplete separation of sister chromatids;
spindle abnormalities such as multipolar spindles and defects in
cytokinesis are additional sources of abnormal chromosome
segregation; finally, errors in cell cycle regulation, including
delays during division and defects in cell cycle checkpoints, also
leading to missegregation. However, these technical approaches are
difficult to apply in tumors with a low proliferation index and in
tumors in which the anaphases are not clearly observed.
CIN is not only a consequences of compromised
mitotic apparatus but it may also arise after DNA damage or as a
consequence of impaired replication fork progression (42). Defective DNA damage response and
repair results in chromosomal aberration such as deletions,
amplifications, inversions and translocations (43).
The main method for the assessment of both numerical
and structural CIN in tumor cells is the fluorescence in
situ hybridization (FISH). It allows to quantify the variations
in chromosome copy number across the cell population by using
fluorescently labeled DNA probes that bind to centromeres of
specific chromosomes. Thus, FISH evaluates the chromosomal state of
hundreds of cells, inferring the rate of change in chromosome
number from the cell-to-cell variability (44). Another important method is the
single-cell comparative genomic hybridization (CGH) (45). This assay allows the selection of
individual cancer cells based on their deviation from normal cells.
However, single-cell CGH is not amenable to high-throughput
analysis and it is characterized by a considerable economic burden.
Moreover, flow cytometry and DNA image cytometry can be used to
measure cellular DNA content through the use of dyes that bind the
DNA, allowing the assessment of DNA cell cycle distribution and
cellular ploidy. Then, CIN status can be measured by the stemline
scatter index (SSI), which is a measure of the clonal heterogeneity
of the constituent tumor cells (46).
Finally, a more detailed picture of the genomic landscape can be
obtained through Next Generation Sequencing (NGS) systems, which
assume massively parallel sequencing techniques (47).
Recently, different models have been used to measure
CIN in vivo like mouse models that were engineered to mimic
genetic alteration driving CIN, mouse embryonic fibroblast (MEFs)
from mouse models of genetically induced CIN (48) and organoid cultures, that allow to
monitoring chromosome segregations using three-dimensional
live-cell imaging (49). Measuring
CIN in vivo would more accurately show the effect of CIN for
instance during development, the possible role of the immune system
and inter-tissue interactions. However, drawbacks of in vivo
CIN measurement are the limited time available for imaging, the
high cost and the relatively low rate of cell division in
vivo (50).
Despite the studies performed so far, none of the
methods used to study CIN is entirely satisfactory, thus novel
approaches for an accurate detection and assessment of CIN will be
critical both in clinical setting and to therapeutic targeting of
CIN in the future.
CIN takes part in NB development and
aggressiveness
NB is an embryonic tumor that can be present in
fetus. Indeed, some newborn patients exhibit tumors after only a
few days of life; usually, these patients have a very good OS.
Cytogenetic analysis of tumor cells reveals a triploid DNA content
with several numerical CNVs. This, of course, is in contrast with
the presence of structural CNVs in tumor of HR patients older than
one year of age, suggesting an evolution of tumor aggressiveness
associated with CIN. The lapse time between fetus life and infant
at one age of year suggest a time-dependent increase of chromosome
damages (51). As mentioned above,
cell replication errors and abnormal chromosome segregation during
mitosis could originate the abnormal chromosome pattern triggering
the cell to increase their aggressiveness. Masecchia et al
(52) used a learning machine
algorithm to show that numerical chromosome aberrations occurs
early than structural ones.
It is to note that NB is originating from neural
crest cells, a group of cells located on the neural tube and
undergoing to epithelial to mesenchymal transition (EMT) during the
embryonic life. These cells are migrating in the early phases of
embryonic development and some of them take part to the formation
of gastric ganglia and adrenal gland, two sites in which NB growths
and develops. Mouse and zebrafish models have been developed
demonstrating that MYCN oncogene is one of the major actors
in the NB development (53).
Furthermore, it has been shown that ALK and LIN28
genes can participate together MYCN oncogene to the NB
tumorigenesis (54). The role of CIN
in the embryonic phases of NB origin is still to clarify. It is
questionable if some CIN-related genes are involved in the early
phases of NB tumorigenesis. This study needs a more accurate animal
model.
CIN gene signature and NB
Considering the strong relationship between CIN and
aneuploidy, about a decade ago, a computational method was
developed to represent aneuploidy in relation to the expression of
genes localized in aberrant chromosomal region (functional
aneuploidy profile). Thus, the functional aneuploidy as a measure
of the total status of chromosomal imbalance, was inferred using
gene expression data of a given tumor (55). Carter et al (55) showed 70 genes whose expression was
correlated with total functional aneuploidy in several cancer
types: the CIN70 gene signature able to measure the state of
karyotype and to predict clinical outcome in several human cancers.
The CIN70 signature was obtained including most of genes involved
in cellular processes critical for genome integrity maintenance
such as DNA replication, chromosomal condensation, segregation,
de-condensation and structure and genes of cell cycle, spindle
apparatus and mitosis. Carter's study provided a means to assess
the potential role of CIN in tumors initiation. However, in this
study NB tumor was not investigated and CIN signature for NB has
not been identified until now. Since this information is lacking,
we explored gene expression profiles of 504 NB derived from public
dataset E-MTAB-161 (EMBL ArrayExpress database). This dataset
provided expression data of 45 genes out of 70 genes included in
the CIN70 signature. Patients clinical information was used to
define two risk groups: HR group including samples with stages 2,
3, 4 and 4s MYCN-amplified, stage 4 MYCN not
amplified >12 months at diagnosis; low/intermediate-risk (LIR)
group including samples with stages 2, 3 and 4S without MYCN
gene amplified, stage 4 without MYCN amplification <12
months at diagnosis. We observed 31/45 genes differently expressed
between HR and LIR groups (Fig. 2).
These genes show association with cell-cycle regulation (CCNB1,
PRC1 and TPX2) and chromosomal segregation
(TPX2). Furthermore, we found overexpression of key
regulators genes involving in the correct processes of chromosomes
replication, regulation of chromatin status, cytokinesis and
segregation; namely: AURKB, CCNB1, CCNB2, NEK2 and
ZWINT and likely associated with CIN in HR-NB patients.
Interestingly, we observed genes with the highest CIN score
reported in Carter's study, among genes with high expression in
HR-NBs, such as TPX2 and PRC1.
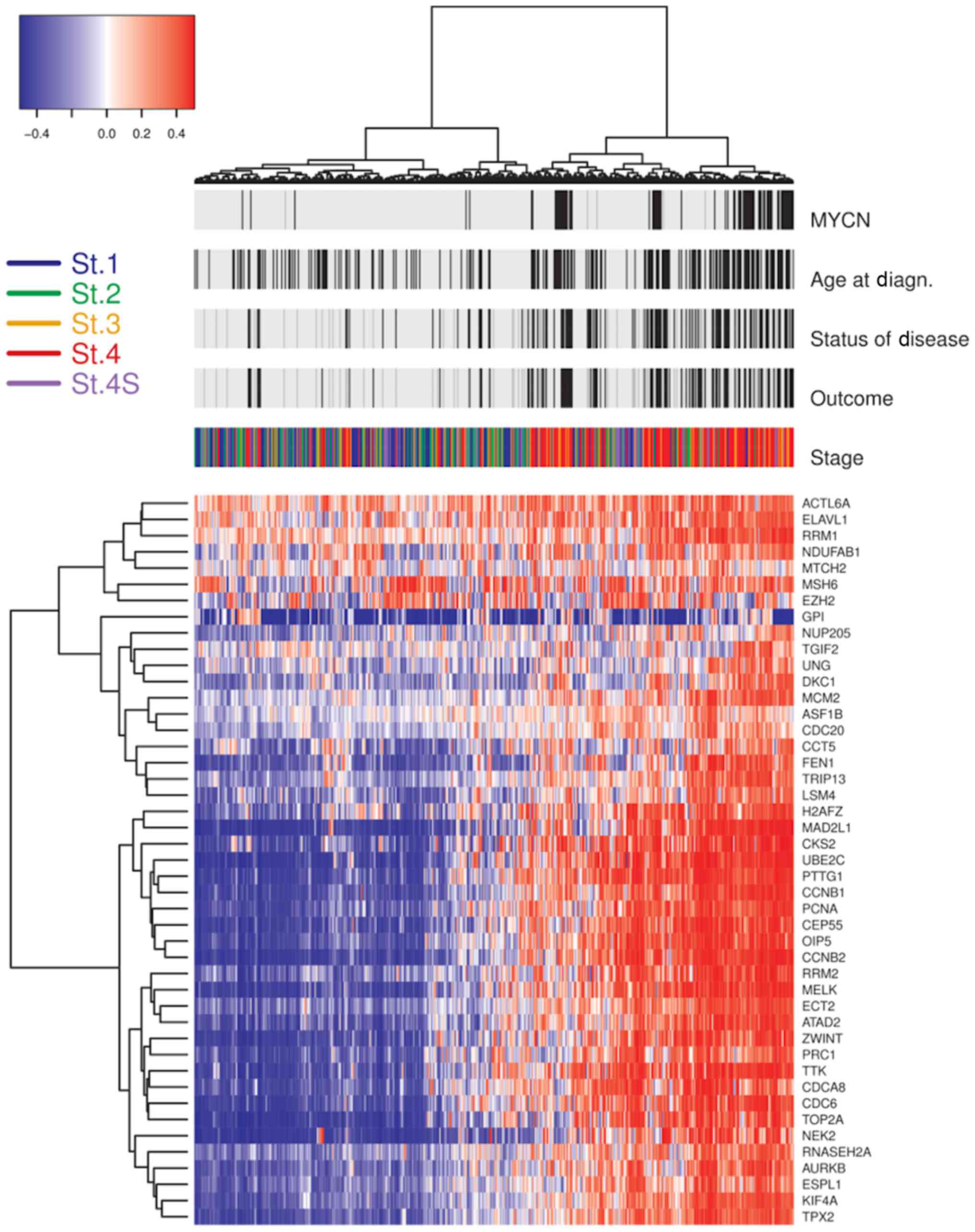 | Figure 2.Unsupervised clustering of 45 genes
with CIN70 in NB. The heatmap of the unsupervised clustering
presents 30 genes with differential expression between NB-high risk
patients (stages 2, 3, 4, and 4S with MYCN gene amplification;
stage 4 patients with an age at diagnosis >12 months) and
low/intermediate-risk patients (stage 1, 2, 3, 4S without MYCN gene
amplification). Above the heatmap are the indicated patient
clinical features and the patients' stage. Black bars represent
unfavorable scores and light gray bars represent favorable scores
for ‘Status of Disease’ and ‘Outcome’. Black bars for the ‘Age at
Diagn.’ identify cases with ages over 60 months at diagnosis. Black
bars for ‘MYCN’ identify cases with amplification of MYCN gene. On
the left of the panel is a key of the staging colors applied. On
the right of the panel are the 45 genes out of CIN70. CIN,
chromosome instability; NB, neuroblastoma; MYCN, neuroblastoma MYC
oncogene; St., stage. |
Moreover, Carter et al (55) produced another CIN selecting the top
25 genes best predicting clinical outcome. Interestingly, we
observed that 17/25 genes of the CIN25 showed high expression in HR
patients. So that, our preliminary data show that CIN25 gene
signature is strongly associated with NB with poor outcome.
Moreover, this result indicates that CIN is active and several
CIN-related genes are operating in HR-NB.
Finally, we were interested to investigate if
upregulated genes could be druggable by Federal Drugs Approved
(FDA) approved molecules and we explored the Drug Gene Interaction
Database (DGID) (dgidb.genome.wustl.edu/). Interestingly, 3/30 genes
upregulated in HR patients stage 4 are targetable by FDA approved
drugs (Table I), already used for
others disease indicating the possible use of these compounds in NB
therapy.
 | Table I.Food and drug administration approved
drugs targeting chromosome instability-associated genes in
neuroblastoma. |
Table I.
Food and drug administration approved
drugs targeting chromosome instability-associated genes in
neuroblastoma.
Conclusion
CIN is an old genomic aspect that is recently
emerged as causative of cancer. Today with the advent of NGS, there
is an additional possibility to study in deep this phenomenon. So
that, it should be useful to initiate a CIN screening on several
cancers which can be exploited for targeted cancer therapy. While
numerous studies demonstrated that CIN may promote tumorigenesis,
primarily through the functional loss of key players governing
chromosome stability, it has also been shown that CIN beyond
tolerable levels actually leads to cell death and tumor suppression
(56). These observations
collectively suggest elevating CIN as a potential chemotherapeutic
strategy in a genetically sensitized background, but additional
studies would be needed to validate the efficacy and effectiveness
of this approach to cancer treatment.
Acknowledgements
The authors would like to thank Dr. Carlo Zanon
(Neuroblastoma Laboratory, Fondazione Istituto di Ricerca
Pediatrica Città della Speranza, Padua, Italy), for obtaining gene
expression profile analysis from the neuroblastoma public
dataset.
Funding
The present review was supported by Fondazione
Italiana per la Lotta al Neuroblastoma/Fondazione Istituto di
Ricerca Pediatrica Città della Speranza (grant no. IRP-13/02).
Availability of data and materials
The datasets used and/or analysed during the current
study are included in this published article.
Authors' contributions
GPT conceived the present review. PF and MRE
selected the articles and identified useful information within
these articles. PF submitted the manuscript. PF, MRE and GPT
contributed equally in writing the manuscript. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Luksch R, Castellani MR, Collini P, De
Bernardi B, Conte M, Gambini C, Gandola L, Garaventa A, Biasoni D,
Podda M, et al: Neuroblastoma (Peripheral neuroblastic tumours).
Crit Rev Oncol Hematol. 107:163–181. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Haupt R, Garaventa A, Gambini C, Parodi S,
Cangemi G, Casale F, Viscardi E, Bianchi M, Prete A, Jenkner A, et
al: Improved survival of children with neuroblastoma between 1979
and 2005: A report of the Italian Neuroblastoma Registry. J Clin
Oncol. 28:2331–2338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Defferrari R, Mazzocco K, Ambros IM,
Ambros PF, Bedwell C, Beiske K, Bénard J, Berbegall AP, Bown N,
Combaret V, et al: Influence of segmental chromosome abnormalities
on survival in children over the age of 12 months with unresectable
localised peripheral neuroblastic tumours without MYCN
amplification. Br J Cancer. 112:290–295. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tonini GP: Growth, progression and
chromosome instability of Neuroblastoma: A new scenario of
tumorigenesis? BMC Cancer. 17(20)2017.
|
|
5
|
Molenaar JJ, Koster J, Zwijnenburg DA, van
Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J,
Westerman BA, van Arkel J, et al: Sequencing of neuroblastoma
identifies chromothripsis and defects in neuritogenesis genes.
Nature. 483:589–593. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaneko Y, Kanda N, Maseki N and Sakurai M,
Tsuchida Y, Takeda T, Okabe I and Sakurai M: Different karyotypic
patterns in early and advanced stage neuroblastomas. Cancer Res.
47:311–318. 1987.PubMed/NCBI
|
|
7
|
Janoueix-Lerosey I, Schleiermacher G,
Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier
J, Peuchmaur M, Valent A, et al: Overall genomic pattern is a
predictor of outcome in neuroblastoma. J Clin Oncol. 27:1026–1033.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stigliani S, Coco S, Moretti S, Oberthuer
A, Fischer M, Theissen J, Gallo F, Garavent A, Berthold F, Bonassi
S, et al: High genomic instability predicts survival in metastatic
high-risk neuroblastoma. Neoplasia. 14:823–832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hahn WC and Weinberg RA: Modelling the
molecular circuitry of cancer. Nat Rev Cancer. 2:331–341. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vogelstein B and Kinzler KW: The multistep
nature of cancer. Trends Genet. 9:138–141. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Albertson DG, Collins C, McCormick F and
Gray JW: Chromosome aberrations in solid tumors. Nature Genet.
34:369–376. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cahill DP, Kinzler KW, Vogelstein B and
Lengauer C: Genetic instability and darwinian selection in tumours.
Trends Cell Biol. 9:M57–M60. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Heng HH, Bremer SW, Stevens JB, Horne SD,
Liu G, Abdallah BY, Ye KJ and Ye CJ: Chromosomal instability (CIN):
What it is and why it is crucial to cancer evolution. Cancer
Metastasis Rev. 32:325–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Duesberg P, Rausch C, Rasnick D and
Hehlmann R: Genetic instability of cancer cells is proportional to
their degree of aneuploidy. Proc Natl Acad Sci USA. 95:13692–13697.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li R, Sonik A, Stindl R, Rasnick D and
Duesberg P: Aneuploidy vs. gene mutation hypothesis of cancer:
Recent study claims mutation but is found to support aneuploidy.
Proc Natl Acad Sci USA. 97:3236–3241. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instabilities in human cancers. Nature. 396:643–649. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rajagopalan H, Nowak MA, Vogelstein B and
Lengauer C: The significance of unstable chromosomes in colorectal
cancer. Nat Rev Cancer. 3:695–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Aguilera A and García-Muse T: Causes of
genome instability. Annu Rev Genet. 47:1–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giam M and Rancati G: Aneuploidy and
chromosomal instability in cancer: A jackpot to chaos. Cell Div.
10:32015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nowell PC: The clonal evolution of tumor
cell populations. Science. 194:23–28. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tonini GP: Molecular mechanisms involved
in DNA repair, in gene rearrangement and in gene amplification may
be considered as an integrated system in maintaining cellular
homeostasis and cell survival. Anticancer Res. 8:881–884.
1988.PubMed/NCBI
|
|
23
|
Matzke MA, Mette MF, Kanno T and Matzke
AJ: Does the intrinsic instability of aneuploid genomes have a
causal role in cancer? Trends Genet. 19:253–256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gisselsson D: Chromosome instability in
cancer: How, when, and why? Adv Cancer Res. 87:1–29. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marx J: Debate surges over the origins of
genomic defects in cancer. Science. 297:544–546. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sieber OM, Heinimann K and Tomlinson IP:
Genomic instability-the engine of tumorigenesis? Nat Rev Cancer.
3:701–708. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bakhoum SF and Compton DA: Chromosomal
instability and cancer: A complex relationship with therapeutic
potential. J Clin Invest. 122:1138–1143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lengauer C, Kinzler KW and Vogelstein B:
Genetic instability in colorectal cancers. Nature. 386:623–627.
1997. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thompson SL and Compton DA: Examining the
link between chromosomal instability and aneuploidy in human cells.
J Cell Biol. 180:665–672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bloomfield M and Duesberg P: Inherent
variability of cancer-specific aneuploidy generates metastases. Mol
Cytogenet. 9:902016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Targa A and Rancati G: Cancer: A CINful
evolution. Curr Opin Cell Biol. 52:136–144. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sheltzer JM and Amon A: The aneuploidy
paradox: Costs and benefits of an incorrect karyotype. Trends
Genet. 27:446–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weaver BA and Cleveland DW: The aneuploidy
paradox in cell growth and tumorigenesis. Cancer Cell. 14:431–433.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Silk AD, Zasadil LM, Holland AJ, Vitre B,
Cleveland DW and Weaver BA: Chromosome missegregation rate predicts
whether aneuploidy will promote or suppress tumors. Proc Natl Acad
Sci USA. 110:E4134–E4141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Komarova NL and Wodarz D: The optimal rate
of chromosome loss for the inactivation of tumor suppressor genes
in cancer. Proc Natl Acad Sci USA. 101:7017–7021. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rivera-Rivera Y and Saavedra HI:
Centrosome-a promising anti-cancer target. Biologics. 10:167–176.
2016.PubMed/NCBI
|
|
37
|
Cicenas J: The Aurora kinase inhibitors in
cancer research and therapy. J Cancer Res Clin Oncol.
142:1995–2012. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kaur R, Kaur G, Gill RK, Soni R and
Bariwal J: Recent developments in tubulin polymerization
inhibitors: An overview. Eur J Med Chem. 87:89–124. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gavande NS, VanderVere-Carozza PS, Hinshaw
HD, Jalal SI, Sears CR, Pawelczak KS and Turchi JJ: DNA repair
targeted therapy: The past or future of cancer treatment? Pharmacol
Ther. 160:65–83. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bhattacharjee S and Nandi S: Synthetic
lethality in DNA repair network: A novel avenue in targeted cancer
therapy and combination therapeutics. IUBMB Life. 69:929–937. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bayani J, Selvarajah S, Maire G, Vukovic
B, Al-Romaih K, Zielenska M and Squire JA: Genomic mechanisms and
measurement of structural and numerical instability in cancer
cells. Semin Cancer Biol. 17:5–18. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaye JA, Melo JA, Cheung SK, Vaze MB,
Haber JE and Toczyski DP: DNA breaks promote genomic instability by
impeding proper chromosome segregation. Curr Biol. 14:2096–2106.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bakhoum SF, Kabeche L, Murnane JP, Zaki BI
and Compton DA: DNA-damage response during mitosis induces
whole-chromosome missegregation. Cancer Discov. 4:1281–1289. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Heng HH, Spyropoulos B and Moens PB: FISH
technology in chromosome and genome research. BioEssays. 19:75–84.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Imle A, Polzer B, Alexander S, Klein CA
and Friedl P: Genomic instability of micronucleated cells revealed
by single-cell comparative genomic hybridization. Cytometry A.
75:562–568. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kronenwett U, Huwendiek S, Ostring C,
Portwood N, Roblick UJ, Pawitan Y, Alaiya A, Sennerstam R,
Zetterberg A and Auer G: Improved grading of breast adenocarcinomas
based on genomic instability. Cancer Res. 64:904–909. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mullauer L: Next generation sequencing:
Clinical applications in solid tumours. Memo. 10:244–247. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Williams BR, Prabhu VR, Hunter KE, Glazier
CM, Whittaker CA, Housman DE and Amon A: Aneuploidy affects
proliferation and spontaneous immortalization in mammalian cells.
Science. 322:703–709. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Drost J, van Jaarsveld RH, Ponsioen B,
Zimberlin C, van Boxtel R, Buijs A, Sachs N, Overmeer RM, Offerhaus
GJ, Begthel H, et al: Sequential cancer mutations in cultured human
intestinal stem cells. Nature. 521:43–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schukken KM and Foijer F: CIN and
aneuploidy: Different concepts, different consequences. Bioessays.
40:2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Coco S, Theissen J, Scaruffi P, Stigliani
S, Moretti S, Oberthuer A, Valdora F, Fischer M, Gallo F, Hero B,
et al: Age-dependent accumulation of genomic aberrations and
deregulation of cell cycle and telomerase genes in metastatic
neuroblastoma. Int J Cancer. 131:1591–1600. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Masecchia S, Coco S, Barla A, Verri A and
Tonini GP: Genome instability model of metastatic neuroblastoma
tumorigenesis by a dictionary learning algorithm. BMC Med Genomics.
8:572015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schleiermacher G, Janoueix-Lerosey I and
Delattre O: Recent insights into the biology of neuroblastoma. Int
J Cancer. 135:2249–2261. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Schnepp RW, Khurana P, Attiyeh EF, Raman
P, Chodosh SE, Oldridge DA, Gagliardi ME, Conkrite KL, Asgharzadeh
S, Seeger RC, et al: A LIN28B-RAN-AURKA signaling network promotes
neuroblastoma tumorigenesis. Cancer Cell. 28:599–609. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carter SL, Eklund AC, Kohane IS, Harris LN
and Szallasi Z: A signature of chromosomal instability inferred
from gene expression profiles predicts clinical outcome in multiple
human cancers. Nat Genet. 38:1043–1048. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chan SH and Ngeow J: Germline mutation
contribution to chromosomal instability. Endocr Relat Cancer.
24:T33–T46. 2017. View Article : Google Scholar : PubMed/NCBI
|