Introduction
Histone deacetylase inhibitors (HDACIs) are
compounds that regulate the alteration of chromatin structure and
subsequent gene expression in cancer cells, and are recognized as
potential novel antitumor agents (1).
HDACIs have emerged as a promising class of anticancer agents,
leading to the approval by the USA Food and Drug Administration
(FDA) of vorinostat [suberoylanilide hydroxamic acid (SAHA)] for
the treatment of cutaneous T-cell lymphoma. In addition, other
HDACIs, including panobinostat, belinostat and entinostat, are
currently being investigated as potential monotherapeutic agents
for use in combination with other antitumor therapies in clinical
trials (2). Entinostat (MS-275), a
potent class of HDACIs currently in use in preclinical studies
(3,4),
induces apoptosis through the activation of caspase-3, upregulation
of B-cell lymphoma 2 (Bcl-2)-associated X protein and
downregulation of Bcl-2. A Phase I study of entinostat combined
with sorafenib was conducted in 31 patients with advanced solid
tumors in various types of cancer, including colorectal cancer,
non-small cell lung cancer (NSCLC), sarcoma,
esophageal/gastroesophageal junction cancer, thyroid cancer and
melanoma (5). Although the clinical
benefits of entinostat for hepatocellular carcinoma (HCC) were not
specifically stated in this Phase I study, the combination of
entinostat and sorafenib treatment was effectively tolerated.
However, the benefit of this combinatorial therapy, which functions
via cytotoxic synergy, has been demonstrated in a number of cancer
cell lines including U2-OS sarcoma, H226, MV 522 and H157 NSCLC and
HepG2 (5). Furthermore, a previous
study demonstrated that the combination of SAHA and sorafenib
enhanced anti-proliferative activity in HCC cells in vitro
(6). Notably, the suppression of
autophagy increased the inhibitory effects of SAHA and sorafenib,
alone or in combination on HCC cell growth.
3-(1-benzenesulfonyl-2,3-dihydro-1H-indol-5-yl)-N-hydroxyacrylamide
(MPT0E028) is an orally available
N-hydroxyacrylamide-derived HDACI that has been demonstrated
to elicit an increased degree of anticancer activity compared with
vorinostat (7). The effects of
sorafenib in combination with MPT0E028 synergistically decreased
liver cancer cell viability and significantly improved the tumor
growth delay in a human hepatoma cell line (Hep3B) xenograft model,
by increased apoptotic cell death (8). MPT0E028 altered the modifications of
histone and non-histone proteins, whereas sorafenib blocked
MPT0E028-induced extracellular-signal-regulated kinase (ERK)
activation and its downstream signaling cascades including signal
transducer and activation of transcription 3 phosphorylation and
induced myeloid cell differentiation protein upregulation,
suggesting that the synergistic interaction between MPT0E028 and
sorafenib occurs at least in part through the inhibition of ERK
signaling.
Trichostatin A (TSA) and SAHA are hydroxamate
HDACIs, and have been identified to trigger cell cycle arrest and
apoptosis through the induction of tumor suppressor gene expression
in a number of types of cancer cell (9,10). SAHA
has been approved by the FDA for the treatment of cutaneous T-cell
lymphoma (11). TSA is proposed as a
potential anticancer drug that activates p53, p21 and p27,
subsequently resulting in tumor growth inhibition in liver, breast
and gastric cancers (12–14).
Constitutive activation of nuclear factor-κB (NF-κB)
is observed in a number of types of tumor tissue and is associated
with aggressive tumor progression (15,16). The
expression of matrix metalloproteinase 9 (MMP-9), vascular
endothelial growth factor (VEGF), X-linked inhibitor of apoptosis
protein (XIAP) and B-cell lymphoma 2 (Bcl-2) induced by NF-κB are
suppressed by chemotherapeutic agents. Furthermore, these agents
augment the therapeutic efficacy via the blockade of NF-κB-mediated
chemoresistance in vitro and in vivo (17–19).
Notably, HDACIs increase the activation and expression of
pro-apoptotic proteins including caspases 3/8 and cytochrome
c, and activate NF-κB-mediated acquired resistance to
HDACIs. However, it is possible to suppress acquired resistance to
HDACIs in cancer, using NF-κB inhibitors (9,14,20,21).
Sorafenib, a targeted drug for multi-kinase
inhibition approved by the FDA for renal carcinoma, inhibits NF-κB
activity induced by 12-O-tetradecanoylphorbol-13-acetate and
radiation, in human HCC and colorectal cancer-bearing animal models
(22,23). In a previous study, our group
demonstrated that sorafenib increases the therapeutic efficacy of
SAHA against HCC; through the inhibition of SAHA-induced ERK/NF-κB
signaling pathways in a human HCC-bearing animal model (21). However, whether sorafenib is able to
enhance the antitumor effects of TSA in HCC has not been fully
elucidated. To the best of our knowledge, the present study is the
first to evaluate the potential synergistic effects of sorafenib in
combination with TSA in human HCC in vitro. The purpose of
the present study was to identify the optimal time schedule and
underlying mechanisms of sorafenib treatment in combination with
TSA against human HCC in Huh7/NF-κB-luc2 cells.
Materials and methods
Chemicals
TSA was obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). For in vitro experiments, a 10 mM
solution of TSA in absolute ethanol was prepared and stored at 20°C
until use. Sorafenib was extracted from Nexavar tablet (Bayer
Healthcare Co., Leverkusen, Germany) as described previously
(23).
Cell culture
The human Huh7 hepatocellular carcinoma cell line
was provided by Dr Jason Chia-Hsien Cheng (Department of Radiation
Oncology, National Taiwan University Hospital, Taipei, Taiwan).
Cells were cultured in Dulbecco's modified Eagle's medium (DMEM;
cat. no. SH30022.02; HyClone Laboratories, GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; Hyclone), 100 U/ml penicillin and 100 µg/ml streptomycin at
37°C in a humidified incubator containing 5% CO2. The
Huh7/NF-κB-luc2 stable clone was cultured using the same
method as described above for Huh7 cells, with the addition of 500
µg/ml geneticin (Calbiochem; Merck KGaA).
Construction of NF-κB/luc2 vector
pGL4-luc2 vector (Promega Corporation,
Madison, WI, USA) was digested with AseI and BsAI,
then using the Klenow enzyme to produce the blunted end. The
NF-κB-responsive elements were isolated from a pNF-κB-luc
vector (Clontech Laboratories, Inc., Mountain View, CA, USA) by
MluI and HindIII, and then using the Klenow enzyme to
produce the blunted end. The NF-κB-responsive element was inserted
and ligated into digested pGL4-luc2, resulting in a
pNF-κB-luc2 vector.
Plasmid transfection and stable clone
selection
The transfection of Huh7 was performed using jetPEI™
DNA transfection reagent (Polyplus Transfection, New York, NY,
USA). A total of 2×106 cells were seeded in a 100 mm
diameter dish 24 h prior to transfection. Subsequently, 8 µg
pNF-κB-luc2 vector DNA and 16 µl jetPEI solution were
diluted in 500 µl 145 µM NaCl, and then immediately mixed together
and incubated for 30 min at room temperature. The jetPEI/DNA
mixture was added to the cells in the 100 mm dish, which was then
incubated at 37°C for 24 h. Cells were then trypsinized and
cultured with DMEM containing 1 mg/ml G418 supplemented with 10%
FBS for 2 weeks. The surviving clones were isolated and expanded to
sufficient numbers to satisfy the following culture conditions:
1×106, 5×105, 3×105, 1×105, 5×104, 3×104 and 1×104 cells/well in
96-well plate, in order to calculate the photons emitted from one
cell and select the stable clone to use. Cells were incubated with
500 µM D-luciferin at 37°C for 15 min, and signals were acquired
for 1 min with a IVIS50 Imaging System (Xenogen; Caliper Life
Sciences, Hopkinton, MA, USA). Regions of interest were drawn
around each well and quantified as photon (s)/cell using Living
Image software (Version 2.20, Xenogen, Caliper Life Sciences). The
Luc2 protein expression in each clone was assayed using
bioluminescent imaging (BLI) as later described. Cells in those
clones with ≥3 photons (s)/cell were used in the present study. The
recombinant bioluminescent cell clone was renamed as
Huh7/NF-κB-luc2 cell line (22). A mutant inhibitor of NF-κBα (IκBαM)
vector (p-IκBαM; Clontech Laboratories, Inc.) was used as the
positive control for the suppression of NF-κB activity as described
previously (21).
MTT assay
Huh7/NF-κB-luc2 cells were cultured in DMEM
supplemented with 10% FBS in 96-well plates at a density of
3×104 cells/well for 24 h, followed by TSA treatment at
different concentrations (between 0 and 4 µM) for a further 24 and
48 h at 37°C (24). Following
culture, cells were washed with fresh medium followed by the
addition of MTT solution (100 µl at 5 mg/ml) into each well and
incubated for 4 h at 37°C. Following removal of the MTT solution,
cells were exposed to 100 µl dimethylsulfoxide (DMSO) for 5 min,
and plates were scanned with an ELISA plate reader (PowerWave X340;
BioTek, Winooski, VT, USA) using a test wavelength of 570 nm and a
reference wavelength of 630 nm.
NF-κB luciferase reporter gene assay
using BLI
Huh7/NF-κB-luc2 cells demonstrated ~80%
confluency in a 75-cm2 culture dish at a density of
5×106 cells/dish. Therefore, Huh7/NF-κB-luc2
cells were plated out at a density of 3×104 cells/well
in 96-well plates in the present study. Cells were cultured in DMEM
supplemented with 10% FBS in 96-well plates at 37°C for 24 h, and
then treated with different concentrations of TSA (between 0 and 4
µM) for a further 24 h and 48 h at 37°C. For the initial
experiment, cells were treated with vehicle (DMEM with 10% FBS and
0.01% absolute alcohol), TSA (1 µM), sorafenib (10 µM), and a
combination of sorafenib (10 µM) and TSA (1 µM) for 48 h.
Additionally, to verify which MAPK inhibitor affected the NF-κB
activity, Huh7/NF-κB-luc2 cells were cultured in DMEM
supplemented with 10% FBS in 96-well plates at 37°C for 24 h, then
treated with inhibitors of mitogen-activated protein kinase
(MAPK)/ERK kinase (MEK; PD98059; cat. no. tlrl-pd98; InvivoGen,
Hong Kong, China), protein kinase B (Akt;
1L-6-hydroxymethyl-chiro-inositol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonate,
cat. no. 1701-1, BioVision, Inc., Milpitas, CA, USA), c-Jun
N-terminal kinase (JNK; SP600125, cat. no. 8177s, Cell Signaling
Technology, Inc., Danvers, MA, USA) and p38 (SB203580, cat. no.
5633s; Cell Signaling Technology, Inc.) for another 48 h. The
activity of NF-κB was determined by BLI. D-luciferin (100 µl; 500
µM; Xenogen; Caliper Life Sciences) was added to each well, and
images were captured for 1 min using an IVIS50 Imaging System
(Xenogen; Caliper Life Sciences). Signals were quantified as
photons/sec, and compared with that of DMSO-treated controls to
obtain the relative NF-κB activity using Living Image software
(Version 2.20; Xenogen; Caliper Life Sciences).
The treatment schedules for
Huh7/NF-κB-luc2 cells treated with TSA, sorafenib and combined TSA
and sorafenib
Huh7/NF-κB-luc2 cells were cultured in DMEM
supplemented with 10% FBS in 96-well plates at a density of
3×104 cells/well for 24 h at 37°C, followed by various
treatments: Cells were treated with vehicle (DMEM with 10% FBS and
0.01% absolute alcohol) for 48 h; TSA (1 µM) for 48 h; sorafenib
(10 µM) for 48 h; TSA (1 µM) for 24 h followed by sorafenib (10 µM)
for a further 24 h; or concurrent TSA (1 µM) plus sorafenib (10 µM)
for 48 h; and sorafenib (10 µM) 24 h prior to TSA (1 µM) for 24 h.
All the aforementioned treatments were performed at 37°C. Following
the treatments, cells were assayed for the viability by MTT assay
as follows: Cells were washed with DMEM followed by the addition of
MTT solution (100 µl at 5 mg/ml) into each well and incubated for 4
h at 37°C. Following removal of the MTT solution, cells were
exposed to 100 µl DMSO for 5 min at room temperature, and plates
were scanned with an ELISA plate reader (PowerWave X340) using a
test wavelength of 570 nm and a reference wavelength of 630 nm. The
relative NF-κB activity was determined by BLI. D-luciferin (100 µl;
500 µM; Xenogen; Caliper Life Sciences) was added to each well, and
images were captured for 1 min using an IVIS50 Imaging System
(Xenogen; Caliper Life Sciences). Signals were quantified as
photons/sec, and compared with that of DMSO-treated controls to
obtain the relative NF-κB activity using Living Image software
(version 2.20; Xenogen; Caliper Life Sciences).
Western blotting
Huh7/NF-κB-luc2 cells were seeded (2×106
cells/dish) into 10-cm diameter dishes and incubated for 24 h,
prior to transfection with empty and IκBαM vectors, and treatment
with TSA (1 µM) for 48 h. Additionally, cells were treated with TSA
(1 µM), sorafenib (10 µM), and a combination of TSA (1 µM) and
sorafenib (10 µM) for 48 h at 37°C. Cells from each treatment group
were then harvested for total protein extraction and analyzed by
western blotting. Lysis buffer (50 mM Tris/HCl, pH 8.0, 120 mM
NaCl, 0.5% NP-40 and 1 mM phenylmethanesulfonylfluoride; 4°C for ~1
h) was used for the extraction of total proteins from cells. The
proteins (40 µg/lane) were separated via SDS-PAGE (10% gel) and
transferred onto a polyvinylamide difluroide membrane (Millipore,
Billerica, MA, USA). The membrane was blocked with 5% non-fat milk
in TBST buffer solution (10 mM Tris-base, 150 mM NaCl, 0.1%
Tween-20) for 1 h at room temperature, followed by incubation with
the appropriate primary antibodies against MMP-9 (1:1,000; cat. no.
AB19016), VEGF (1:2,000; cat. no. ABS82), XIAP (1:2,000; cat. no.
ab21278; Abcam, Cambridge, UK), Bcl-2 (1:2,000; cat. no. 05/729),
total (T)-ERK (1:2,000; cat. no. 05-1152), phosphorylated (P)-ERK
(1:5,000; cat. no. 9106; Cell Signaling Technology, Inc.), cleaved
caspase-3 (1:5,000; cat. no. MAB10753) and β-actin (1:1,000; cat.
no. MABT523) overnight at 4°C. Antibodies against the
aforementioned proteins were purchased from EMD Millipore
(Billerica, MA, USA) unless specified otherwise. Membranes were
further incubated for 1 h at room temperature with horseradish
peroxidase-conjugated goat anti-rabbit secondary antibodies.
Protein expression was detected using an enhanced chemiluminescence
system and Image J software (version 1.50i, National Institute of
Health, Bethesda, MA, USA) was used for quantification.
Mitochondrial membrane potential (ΔΨm)
assay
A total of 2×105 cells from different
treatment groups were collected by centrifugation (140 × g, 5 min
at room temperature), washed twice with PBS and resuspended in 500
µl 3,3′-dihexylacarbocyanine iodide (4 µM) and
2′,7′-dichlorofluorescin diacetate (10 µM) prior to incubation at
37°C for 30 min. ΔΨm was measured with a flow cytometer
(BD Biociences, Franklin lakes, NJ, USA) using the Indo-1/AM cell
permeant dye (cat. no. I-1223; Thermo Fisher Scientific, Inc.).
Statistical analysis
Paired Student's t-test was performed for
comparisons between two groups. One-way analysis of variance
followed by Tukey's post hoc test was performed when comparing more
than two groups. All statistical analyses were performed using SPSS
software (version 21.0; IBM Corp., Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
TSA induces cytotoxicity, NF-κB
activity and NF-κB downstream effector protein expression in
Huh7/NF-κB-luc2 cells in a time-dependent manner
Huh7/NF-κB-luc2 cells were treated with TSA
at different concentration (between 0 and 4 µM TSA) for 24 or 48 h.
The viability of Huh7/NF-κB-luc2 cells was significantly
inhibited at 24 and 48 h in response to TSA treatment at all
concentrations (1, 2, 3 and 4 µM; Fig.
1A). NF-κB activity was unchanged at 24 h following TSA
treatment, but was significantly enhanced at 48 h (Fig. 1B). TSA also increased the expression
of NF-κB downstream effector proteins (MMP-9, VEGF, XIAP and Bcl-2)
in Huh7/NF-κB-luc2 cells (Fig.
1C).
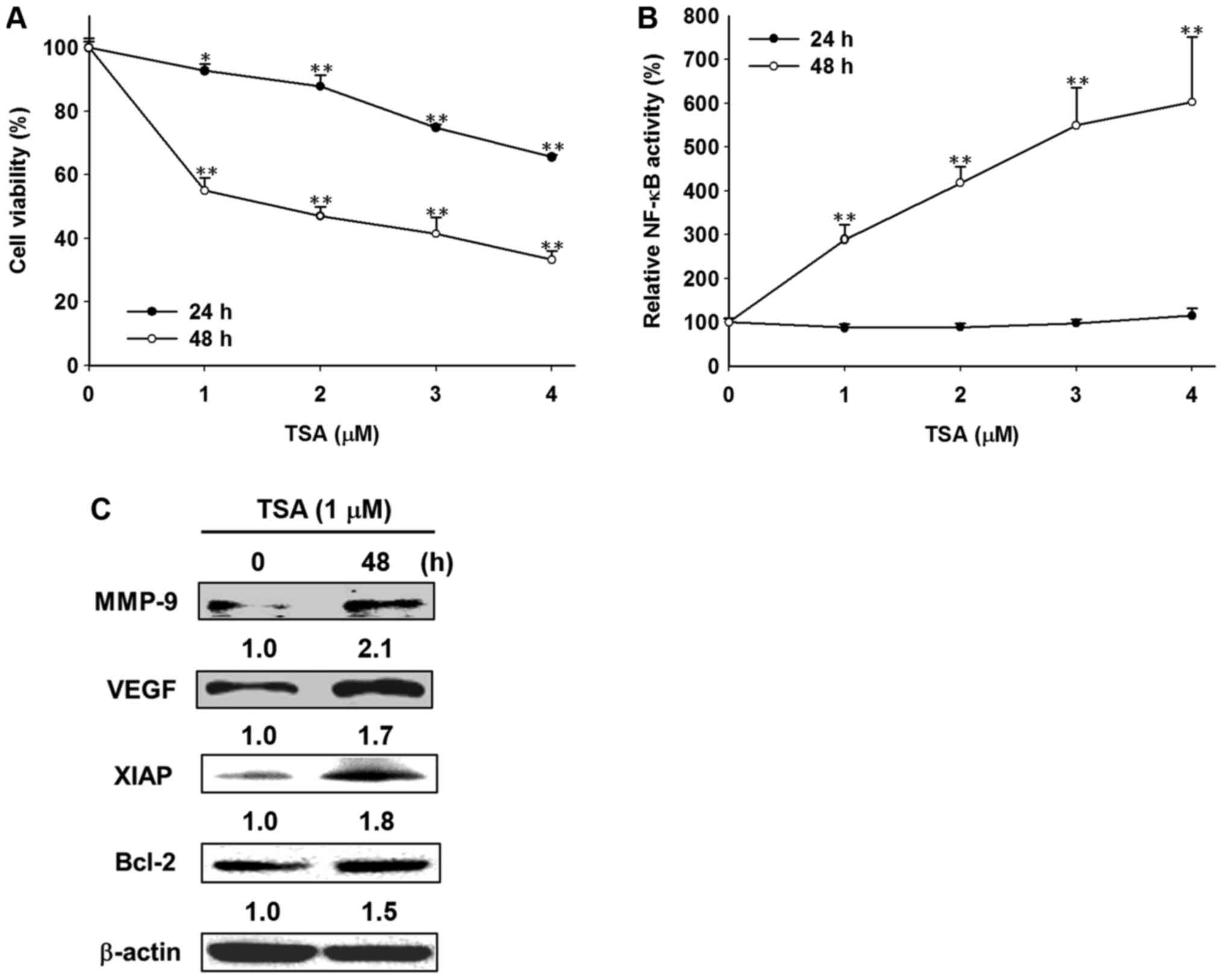 | Figure 1.Effects of TSA treatment on cell
viability, NF-κB activity and its downstream effector proteins in
Huh7/NF-κB-luc2 cells. (A) The cytotoxicity of TSA was
determined using an MTT assay. (B) NF-κB activity was determined by
bioluminescence imaging using NF-κB as the promoter and luciferase
gene as the reporter. (C) Cells were treated with TSA (1 µM) for 0
h (control) and 48 h, respectively. The expression of NF-κB
downstream effector proteins including VEGF, MMP-9, XIAP and Bcl-2
was assessed by western blotting. *P<0.05 and **P<0.01 vs.
control. TSA, trichostatin A; NF-κB, nuclear factor-κB; VEGF,
vascular endothelial growth factor; MMP-9, matrix
metalloproteinase-9; XIAP, X-linked inhibitor of apoptosis protein;
Bcl-2, B-cell lymphoma 2. |
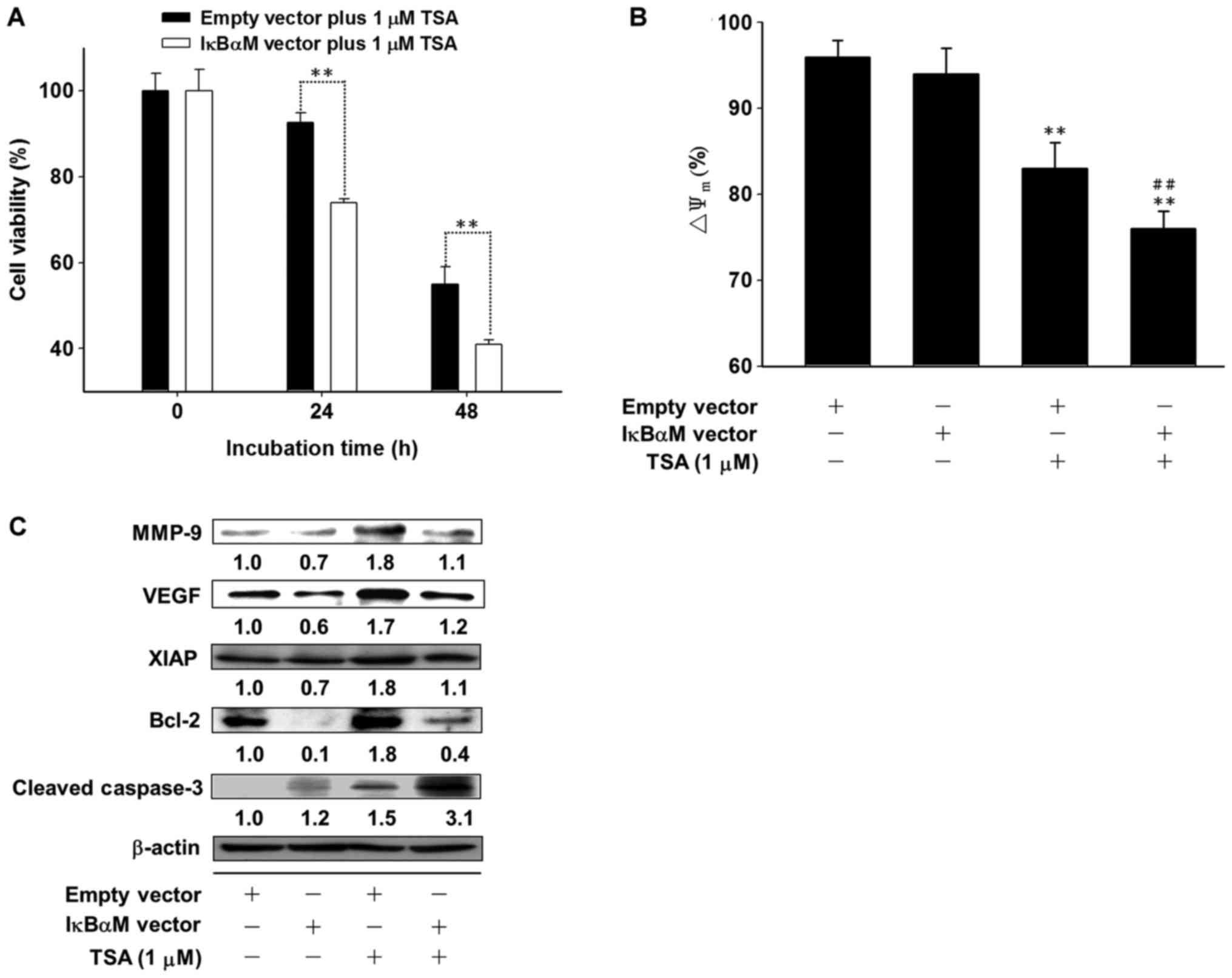
IκBαM vector enhances the antitumor
effect of TSA in Huh7/NF-κB-luc2 cells
An IκBαM vector was utilized as an NF-κB inhibitor
to analyze whether inhibition of NF-κB activation sensitized
Huh7/NF-κB-luc2 cells to TSA. It was observed that
TSA-induced decreases in cell viability and increased
ΔΨm loss were significantly enhanced in
IκBαM-transfected Huh7/NF-κB-luc2 cells compared with cells
transfected with empty vectors (Fig. 2A
and B). In addition, TSA-induced expression of NF-κB downstream
effector proteins was inhibited, whereas levels of cleaved
caspase-3 were increased by transfection of IκBαM into
Huh7/NF-κB-luc2 cells (Fig.
2C).
Sorafenib enhances the antitumor
effect of TSA in Huh7/NF-κB-luc2 via suppression of the ERK/NF-κB
signaling pathway
The three distinct treatment regimens of sorafenib
and TSA (TSA + sorafenib post-treatment, TSA + concurrent
sorafenib, and sorafenib pre-treatment + TSA, respectively) were
administered to verify that sorafenib influenced TSA-induced
cytotoxicity and NF-κB activity in HCC. Cell viability was
significantly decreased by sorafenib pre-treatment followed by TSA
when compared with the decreases resulting from the other treatment
schedules (concurrent and post-treatment) in Huh7/NF-κB-luc2
cells (Fig. 3A). Pre-treatment with
sorafenib followed by TSA significantly inhibited TSA-induced NF-κB
activity when compared with the inhibition observed following
concurrent sorafenib plus TSA or sorafenib post-treatment with TSA
in Huh7/NF-κB-luc2 cells (Fig.
3B). ERK, AKT, JNK and p38 inhibitors were used in the present
study to elucidate the downstream pathway/underlying molecular
mechanism by which sorafenib suppressed TSA-induced NF-κB activity
in Huh7/NF-κB-luc2 cells. The results revealed that
Huh7/NF-κB-luc2 cells treated with an ERK inhibitor
significantly decreased TSA-induced NF-κB activity (Fig. 3C, lane 9, compared with lane 7).
TSA-induced ERK phosphorylation was inhibited by sorafenib
pre-treatment in Huh7/NF-κB-luc2 cells (Fig. 3D, lane 4 compared, with lane 3).
TSA-induced expression of NF-κB downstream effector proteins,
including MMP-9, VEGF, XIAP, Bcl-2, were inhibited, whereas cleaved
caspase-3 was increased by sorafenib pre-treatment followed by TSA
in Huh7/NF-κB-luc2 cells (Fig.
3D, lane 4, compared with lane 3).
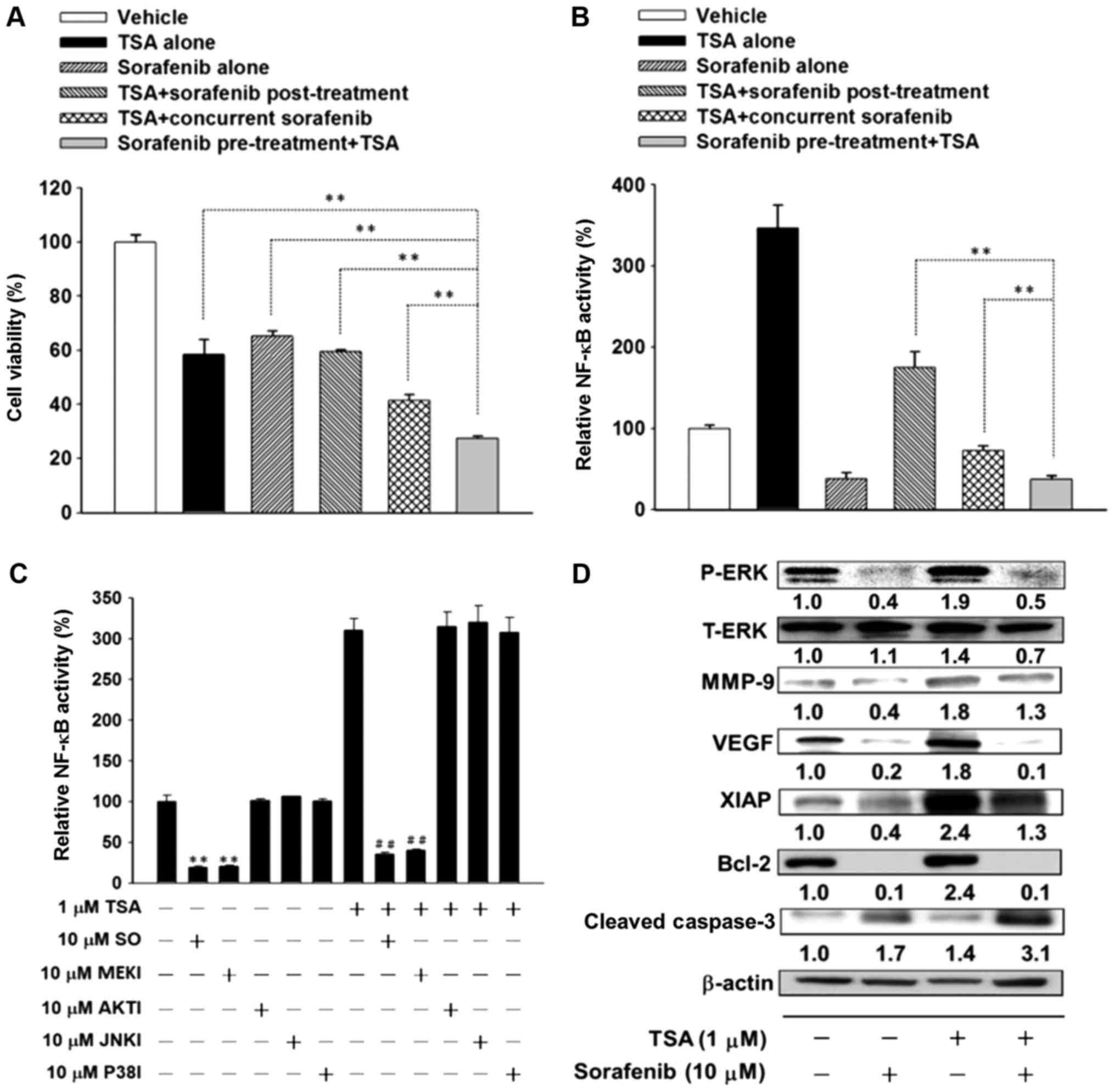 | Figure 3.Effects of combined sorafenib and TSA
treatment on cell viability and NF-κB signaling pathways in
Huh7/NF-κB-luc2 cells. (A) Cytotoxicity was determined with
an MTT assay **P<0.01 with comparisons indicated by lines. (B)
The expression of NF-κB was determined by bioluminescence imaging.
**P<0.01 with comparisons indicated by lines. (C) Cells were
pre-treated with MAPK inhibitors (10 µM; PD98059,
1L-6-hydroxymethyl-chiro-inositol-2-(R)-2-O-methyl-3-O-octadecyl-sn-glycerocarbonate,
SP600125 and SB203580 for 24 h followed by TSA (1 µM) for a further
24 h. The activation of NF-κB was determined by bioluminescence
imaging **P<0.01 vs. control, ##P<0.01 vs. TSA
alone. (D) Cells were treated with sorafenib (10 µM) for 48 h, TSA
(1 µM) for 48 h, and sorafenib (10 µM) for 24 h followed by TSA (1
µM) for 24 h. Western blots for total and phosphorylated ERKs,
NF-κB downstream effector proteins and cleaved caspase-3 are
presented. TSA, trichostatin A; NF-κB, nuclear factor-κB; MAPK,
mitogen-activated protein kinase; P-Phosphorylated; T-, total; ERK,
extracellular-signal-regulated kinase; MMP-9, matrix
metalloproteinase-9; VEGF, vascular endothelial growth factor;
XIAP, X-linked inhibitor of apoptosis protein; Bcl-2, B-cell
lymphoma 2; SO, sorafenib; MEKI, MAPK/ERK kinase inhibitor; AKTI,
protein kinase B inhibitor; JNKI, c-Jun N-terminal kinase
inhibitor; P38, p37 inhibitor. |
Discussion
Induction of NF-κB activity in cancer cells by TSA
may result in treatment resistance, whereas suppression of NF-κB
activation may enhance the anticancer effect of TSA in several
human cancer cells (9,14,25). The
results of the present study demonstrated that NF-κB activity in
Huh7/NF-κB-luc2 cells was induced after 48 h of TSA
treatment. Sorafenib, a multikinase inhibitor, has previously been
demonstrated to enhance the antitumor effects of SAHA in HCC in
vitro and in vivo; via Fas-associated death domain-like
interleukin-1β-converting enzyme suppression, cluster of
differentiation 95 activation and the inhibition of ERK/NF-κB
signaling pathways (10,21). However, it is unknown whether
sorafenib sensitizes HCC cells further, which are vulnerable to the
cytotoxicity effects of TSA and to elucidate the underlying
molecular mechanisms involved. In the present study, the IκBαM
vector used represented a positive control to verify the inhibition
of NF-κB activity on the anticancer effects of TSA in
Huh7/NF-κB-luc2 cells. Transfection of the IκBαM vector
significantly increased TSA-induced cytotoxicity, and significantly
decreased the ΔΨm, following TSA treatment with or without IκBαM
vector transfection compared with that of the sham group. The ΔΨm
of cells transfected with the IκBαM vector followed by TSA
treatment was significantly decreased compared with that treated
with TSA alone. Furthermore, expression of NF-κB downstream
effector proteins including MMP-9, VEGF, XIAP and Bcl-2 were
suppressed, whereas cleaved caspase-3 was increased, following TSA
treatment; the effect of which was enhanced when pretreated with
IκBαM vector transfection. The decreased ΔΨm and increased cleaved
caspase-3 levels represent early and late events in the apoptotic
processes, respectively (26,27). These results suggest that inhibition
of NF-κB activation and the expression of its downstream proteins
by pretreatment of sorafenib may increase TSA-induced apoptosis and
cell death. A previous study suggested that the radiosensitivity of
HCC cells may be modulated by sorafenib in a schedule-dependent
manner (28). Furthermore, concurrent
treatment with radiation and sorafenib and pre-treatment of
sorafenib were reported to improve the sensitizing enhancement
ratio for human SMMC-7721 and SK-HEP-1 HCC cell lines, respectively
(29). In another previous study,
pretreatment of sorafenib combined with radiation resulted in
improved therapeutic potential in three human HCC cell lines in
vitro and in vivo (30).
The basic strategy of combined chemotherapeutic drugs is to
increase the treatment success, reduce the dosage of either drug,
and ensure there is little or no increased toxicity to normal
tissue. It was previously reported that a combination of two
targeted drugs, Imatinib combined with Nilotinib or Dasatini, was
able to improve the therapeutics of chronic myeloid leukemia
(31). Our group have previously
demonstrated that sorafenib combined with SAHA increased the
therapeutic efficacy of HCC in vitro and in vivo,
which demonstrated that NF-κB activity was decreased 2-fold yet
increased 2-fold following treatment with SAHA (10 µM) for 24 and
48 h, respectively, therefore, the concurrent treatment which was
identified to increase the therapeutic efficacy was used in the
animal study (21). However, the
sequence effects of combined SAHA and sorafenib treatment were not
fully investigated in vitro. The combined enhanced antitumor
effect of sorafenib and TSA treatment may be dependent on a
sequence-dependent regime in HCC cells. Therefore, three
combinations of sorafenib and TSA treatments were investigated, to
identify the optimal treatment regime of HCC in vitro.
Notably, cell viability was not affected by TSA combined with
post-treatment of sorafenib compared with that of sorafenib
treatment alone. However, sorafenib pretreatment exhibited
increased cytotoxic effects compared with that of concurrent and
post-treatment combinations with TSA. To the best of our knowledge,
the present study is the first to demonstrate that combined
sorafenib and TSA, most specifically pre-treatment with sorafenib
followed by TSA, may prove to be a more effective therapeutic
treatment for HCC.
PD98059, a MEK inhibitor, has been demonstrated to
potentiate TSA-induced cell cycle arrest and apoptosis, and reverse
TSA-induced ERK/NF-κB activation in gastric cancer cells in
vitro (14). Similarly, sorafenib
has been revealed to enhance the therapeutic efficacy of SAHA
through the suppression of SAHA-induced ERK/NF-κB signaling
pathways in HCC, in vitro and in vivo (21). Notably, the results of the present
study revealed that TSA-induced NF-κB activity was inhibited by
sorafenib in Huh7/NF-κB-luc2 cells in a schedule-dependent
manner through the suppression of the MEK/ERK/NF-κB signaling
pathway.
Several previous studies have demonstrated that MAPK
family members (ERK, JNK, p38 and AKT) activate NF-κB and the
expression of its downstream effector proteins. In the present
study, MEK, AKT, JNK and p38 inhibitors were used to verify the
underlying molecular mechanisms of NF-κB downstream signaling
involved in the cytotoxicity of TSA in Huh7/NF-κB-luc2
cells. The results revealed that pre-treatment with sorafenib or
MEK inhibitor PD98059 significantly inhibited TSA-induced NF-κB
activity. The TSA-induced ERK phosphorylation and the expression of
NF-κB downstream effector proteins were inhibited by sorafenib
pre-treatment combined with TSA.
The results of the present study demonstrated that
the antitumor effects of combined sorafenib and TSA treatment in
human hepatocellular carcinoma Huh7/NF-κB-luc2 cells are
schedule-dependent. Sorafenib pre-treatment with TSA may be the
optimal combination with the highest therapeutic efficacy via the
suppression of the MEK/ERK/NF-κB signaling pathway. In conclusion,
this strategy may have potential benefits for the treatment of
advanced HCC in the clinic.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Biophotonics
and Molecular Imaging Research Center, Biomedical Engineering
Research and Development Center, and the Ministry of Education, Aim
for the Top University Plan, National Yang-Ming University, Taipei,
Taiwan (grant no. 106AC-BI1); and the National Yang-Ming University
Hospital, Yilan, Taiwan (grant no. RD2015-014).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JC, HC, FH and JH contributed to the conception and
design of the study. JC, HC, YL and FH performed the experiments
and wrote the first draft of the manuscript. JH organized the
research, contributed to the revision of the manuscript and gave
final approval of the version to be published. YC and WW made
substantial contributions to the analysis and interpretation of the
data and revised the manuscript critically for important
intellectual content. YC and WW agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved. Each author has participated
sufficiently in the work to take public responsibility for
appropriate portions of the content.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
All authors consent for the publication.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
TSA
|
trichostatin A
|
|
ERK
|
extracellular-signal-regulated
kinase
|
|
MMP-9
|
matrix metalloproteinase 9
|
|
VEGF
|
vascular endothelial growth factor
|
|
XIAP
|
X-linked inhibitor of apoptosis
protein
|
|
Bcl-2
|
B-cell lymphoma 2
|
|
ΔΨm
|
mitochondrial membrane potential
|
References
|
1
|
Pan LN, Lu J and Huang B: HDAC inhibitors:
A potential new category of anti-tumor agents. Cell Mol Immunol.
4:337–343. 2007.PubMed/NCBI
|
|
2
|
Mund C and Lyko F: Epigenetic cancer
therapy: Proof of concept and remaining challenges. Bioessays.
32:949–957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosato RR, Almenara JA and Grant S: The
histone deacetylase inhibitor MS-275 promotes differentiation or
apoptosis in human leukemia cells through a process regulated by
generation of reactive oxygen species and induction of
p21CIP1/WAF1. Cancer Res. 63:3637–3645. 2003.PubMed/NCBI
|
|
4
|
Baradari V, Höpfner M, Huether A, Schuppan
D and Scherubl H: Histone deacetylase inhibitor MS-275 alone or
combined with bortezomib or sorafenib exhibits strong
antiproliferative action in human cholangiocarcinoma cells. World J
Gastroenterol. 13:4458–4466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ngamphaiboon N, Dy GK, Ma WW, Zhao Y,
Reungwetwattana T, DePaolo D, Ding Y, Brady W, Fetterly G and Adjei
AA: A phase I study of the histone deacetylase (HDAC) inhibitor
entinostat, in combination with sorafenib in patients with advanced
solid tumors. Invest New Drugs. 33:225–232. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yuan H, Li AJ, Ma SL, Cui LJ, Wu B, Yin L
and Wu MC: Inhibition of autophagy significantly enhances
combination therapy with sorafenib and HDAC inhibitors
(vorinostatin) for human hepatoma cells. World J Gastroenterol.
20:4953–4962. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huang HL, Lee HY, Tsai AC, Peng CY, Lai
MJ, Wang JC, Pan SL, Teng CM and Liou JP: Anticancer activity of
MPT0E028, a novel potent histone deacetylase inhibitor, in human
colorectal cancer HCT116 cells in vitro and in vivo. PLoS One.
7:e436452012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen CH, Chen MC, Wang JC, Tsai AC, Chen
CS, Liou JP, Pan SL and Teng CM: Synergistic interaction between
the HDAC inhibitor, MPT0E028 and sorafenib in liver cancer cells in
vitro and in vivo. Clin Cancer Res. 20:1274–1287. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yao J, Duan L, Fan M and Wu X: NF-kappaB
signaling pathway is involved in growth inhibition, G2/M arrest and
apoptosis induced by trichostatin A in human tongue carcinoma
cells. Pharmaco Res. 54:406–413. 2006.
|
|
10
|
Zhang G, Park MA, Mitchell C, Hamed H,
Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, et al:
Vorinostat and sorafenib synergistically kill tumor cells via FLIP
suppression and CD95 activation. Clin Cancer Res. 14:5385–5399.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Duvic M and Vu J: Update on the treatment
of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biol
Targets Ther. 1:377–392. 2007.
|
|
12
|
Coradini D and Speranza A: Histone
deacetylase inhibitors for treatment of hepatocellular carcinoma.
Acta pharmacol Sin. 26:1025–1033. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan G, Graham K and Lanza-Jacoby S:
Curcumin enhances the anticancer effects of trichostatin a in
breast cancer cells. Mol Carcinogen. 52:404–411. 2013. View Article : Google Scholar
|
|
14
|
Yao J, Qian CJ, Ye B, Zhang X and Liang Y:
ERK inhibition enhances TSA-induced gastric cancer cell apoptosis
via NF-κB-dependent and Notch-independent mechanism. Life Sci.
91:186–193. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tai DI, Tsai SL, Chang YH, Huang SN, Chen
TC, Chang KS and Liaw YF: Constitutive activation of nuclear factor
kappaB in hepatocellular carcinoma. Cancer. 89:2274–2281. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li W, Tan D, Zenali MJ and Brown RE:
Constitutive activation of nuclear factor-kappa B (NF-κB) signaling
pathway in fibrolamellar hepatocellular carcinoma. Int J Clin Exp
Patho. 3:238–243. 2010.
|
|
17
|
Wang WH, Chiang IT, Liu YC, Hsu FT, Chen
HW, Chen CL, Lee YJ, Lin WJ and Hwang JJ: Simultaneous imaging of
temporal changes of NF-κB activity and viable tumor cells in
Huh7/NF-kappaB-tk-luc2/rfp tumor-bearing mice. In Vivo. 27:339–350.
2013.PubMed/NCBI
|
|
18
|
Pan CC, Kavanagh BD, Dawson LA, LI XA, Das
SK, Miften M and Tan RK: Radiation-associated liver injury. Int J
Radiat Oncol. 76 Suppl:S94–S100. 2010. View Article : Google Scholar
|
|
19
|
Liu YC, Chiang IT, Hsu FT and Hwang JJ:
Using NF-κB as a molecular target for theranostics in radiation
oncology research. Expert Rev Mol Diagn. 12:139–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dai Y, Guzman ML, Chen S, Wang L, Yeung
SK, Pei XY, Dent P, Jordan CT and Grant S: The NF (Nuclear
factor)-kappaB inhibitor parthenolide interacts with histone
deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in
human acute myeloid leukaemia cells. Brit J Haematol. 151:70–83.
2010. View Article : Google Scholar
|
|
21
|
Hsu FT, Liu YC, Chiang IT, Liu RS, Wang
HE, Lin WJ and Hwang JJ: Sorafenib increases efficacy of vorinostat
against human hepatocellular carcinoma through transduction
inhibition of vorinostat-induced ERK/NF-κB signaling. Int J Oncol.
45:177–188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chiang IT, Liu YC, Wang WH, Hsu FT, Chen
HW, Lin WJ, Chang WY and Hwang JJ: Sorafenib inhibits TPA-induced
MMP-9 and VEGF expression via suppression of ERK/NF-κB pathway in
hepatocellular carcinoma cells. In Vivo. 26:671–681.
2012.PubMed/NCBI
|
|
23
|
Kuo YC, Lin WC, Chiang IT, Chang YF, Chen
CW, Su SH, Chen CL and Hwang JJ: Sorafenib sensitizes human
colorectal carcinoma to radiation via suppression of NF-κB
expression in vitro and in vivo. Biomed Pharmacother. 66:12–20.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meidhof S, Brabletz S, Lehmann W, Preca
BT, Mock K, Ruh M, Schüler J, Berthold M, Weber A, Burk U, et al:
ZEB-1 associated drug resistance in cancer cells is reversed by
class I HDAC inhibitor mocetinostat. EMBO Mol Med. 7:831–847. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rundall BK, Denlinger CE and Jones DR:
Combined histone deacetylase and NF-kappaB inhibition sensitizes
non-small cell lung cancer to cell death. Surgery. 136:416–425.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Domingo-Domenech J, Pippa R, Tapia M,
Gascon P, Bachs O and Bosch M: Inactivation of NF-kappaB by
proteasome inhibition contributes to increased apoptosis induced by
histone deacetylase inhibitors in human breast cancer cells. Breast
Cancer Res Tr. 112:53–62. 2008. View Article : Google Scholar
|
|
27
|
Ly JD, Grubb DR and Lawen A: The
mitochondrial membrane potential (deltapsi(m)) in apoptosis; an
update. Apoptosis. 8:115–128. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li Q, Hu Y, Xi M, He L, Zhao L and Liu M:
Sorafenib modulates the radiosensitivity of hepatocellular
carcinoma cells in vitro in a schedule-dependent manner. BMC
Cancer. 12:4852012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu W, Gu K, Yu Z, Yuan D, He M, Ma N, Lai
S, Zhao J, Ren Z, Zhang X, et al: Sorafenib potentiates irradiation
effect in hepatocellular carcinoma in vitro and in vivo. Cancer
Lett. 329:109–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J CH, Chuang HY, Hsu FT, Chen YC,
Chien YC and Hwang JJ: Sorafenib pretreatment enhances radiotherapy
through targeting MEK/ERK/NF-κB pathway in human hepatocellular
carcinoma-bearing mouse model. Oncotarget. 2016.(in press).
|
|
31
|
Komarova NL, Katouli AA and Wodarz D:
Combination of two but not three current targeted drugs can improve
therapy of chronic myeloid leukemia. PLoS One. 4:e44232019.
View Article : Google Scholar
|