Introduction
Head and neck squamous cell carcinoma (HNSCC)
encompasses a heterogeneous group of tumors with aggressive nature
and is the fifth most common carcinoma worldwide (1,2). HNSCC
accounts for ~4% of all malignancies worldwide and 5% of all cancer
mortalities (3).
The choice of cancer treatment depends on the site
of the primary tumor, the stage of the disease, treatment
toxicities and the expected oncological and functional outcomes.
One approach to improve treatment efficacy is to add novel
molecular targeted agents to the classical treatment regimens
(4).
Monoclonal antibodies targeting the epidermal growth
factor receptor have shown clinical benefits in palliative and
curative settings (5). However, only
a minority of patients presenting with current or metastatic HNSCC
exhibited tumor regression with these agents, and the majority of
patients develop acquired tumor resistance following several months
of treatments (4). Therefore in order
to develop agents that target novel proteins, the identification of
novel genes in HNSCC is required.
In the present study, the mutated genes in HNSCC
were screened by searching the Cancer Genome Atlas (TCGA). The TCGA
research network has profiled and analyzed large numbers of human
tumors in order to identify molecular aberrations at DNA, RNA,
protein and epigenetic levels (6–13). It was
identified that there were many genes with amplification and
deletion in HNSCC (Table I),
including C9orf53. C9orf53 is a protein-coding gene. C9orf53 (also
named CDKN2A-AS1) is associated with Alzheimer's disease (14). The present study focused on the role
of C9orf53 in HNSCC, and it was found that the main type of C9orf53
mutation in HNSCC is deletion. Importantly, it was observed that
deletions in C9orf53 are associated with lower patient survival
rates. In the cell experiments, the role of C9orf53 in HNSCC was
investigated in vitro by overexpressing or inhibiting
C9orf53 expression. In conclusion, the present study revealed the
role of C9orf53 in HNSCC, and the study may provide a novel
therapeutic target for future investigation.
 | Table I.Expression of genes that are deleted
or amplified in HNSCC. |
Table I.
Expression of genes that are deleted
or amplified in HNSCC.
| Genes | P-value |
|---|
| Amplification |
|
| FADD | P<0.001 |
| USP13 | P<0.001 |
| DCUN1D1 | P<0.001 |
| EGFR | P<0.001 |
| CCND1 | P<0.001 |
| FGF3 | P<0.001 |
| MIR548K | P<0.001 |
| PPFIA1 | P<0.001 |
| POU5F1B | P<0.001 |
| CASC8 | P<0.001 |
| CCAT1 |
4.82×10−22 |
| FGFR1 |
1.15×10−18 |
| RGP1 |
1.24×10−13 |
| LINC00393 |
1.34×10−11 |
| MYRFL |
5.81×10−11 |
| MLANA | 0.147 |
| CDK6 | 0.259 |
| SAMD9L | 0.259 |
| SNAI2 |
9.67×10−9 |
| NFIB |
2.38×10−8 |
| E2F1 |
9.36×10−6 |
| ANKRD39 |
5.09×10−5 |
| LRRC14B |
7.44×10−5 |
| ERBB2 |
1.16×10−4 |
| TINAG |
2.37×10−4 |
| CD44 |
5.72×10−4 |
| LINC00452 |
2.99×10−3 |
| PTP4A1 | 0.037 |
| Deletion |
|
| C9ORF53 | P<0.001 |
| ZNF532 | P<0.001 |
| CDKN2A | P<0.001 |
| RN7SL5P |
1.42×10−39 |
| RNY3P4 |
3.13×10−23 |
| STK11 |
1.77×10−19 |
| FAM72C |
4.76×10−19 |
| PDE4D |
6.60×10−18 |
| RB1 |
4.14×10−17 |
| PARD3 |
1.80×10−14 |
| LINC00971 | 0.101 |
| KIAA0825 |
1.25×10−9 |
| MIR3182 |
4.80×10−9 |
| CBWD3 |
2.74×10−7 |
| PTPRG |
1.29×10−6 |
| GPN2 |
1.62×10−6 |
| ZNF750 |
2.67×10−6 |
| KCNIP4 |
1.51×10−5 |
| RNA5SP431 |
1.61×10−5 |
| RAB24 |
1.09×10−4 |
| C9ORF163 |
1.24×10−3 |
| LINC00645 |
7.31×10−3 |
| LRRN1 | 0.0239 |
Materials and methods
Frequency of gene alteration and
patient survival analysis for cancer
The data on the frequency of C9orf53 alteration,
mRNA level analysis and patient survival were obtained from TCGA
via cBioportal for Cancer Genomics (http://www.cbioportal.org/public-portal/index.do)
(15,16).
Patients
Surgical specimens from 19 patients with HNSCC (age
range: 36–75 years; mean age: 54.3±7.4 years; Sex ratio: Male to
Female 1.7:1) and matched normal tissues adjacent to tumors were
obtained postoperatively in June, 2009 from the Department of Head
and Neck Surgery, Changhai Hospital, Second Military Medical
University (Shanghai, China). All patients gave signed, informed
consent for their tissues to be used for scientific research.
Ethics approval for the present study was obtained from Changhai
Hospital, Second Military Medical University (Shanghai, China). All
diagnoses were based on pathological and/or cytological findings.
The histological features of the specimens were evaluated by senior
pathologists according to the World Health Organization
classification criteria (17). The
tissues were obtained prior to chemotherapy and radiotherapy. The
samples were immediately frozen and stored at −80°C prior to
reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) assay.
Cell culture
293, SCC-5 and SCC-9 cells were obtained from the
Cell Bank of Chinese Academy of Science (Shanghai, China). 293,
SCC-5 and SCC-9 cells were cultured in 37°C, 5%CO2, and
in Dulbecco's modified Eagle's medium (Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum
(Hyclone; GE Healthcare Life Sciences), 2 mM L-glutamine and 100
µg/ml penicillin/streptomycin (Sangon Biotech Co., Ltd., Shanghai,
China) as described in previous studies (18–20).
RNA extraction and RT-qPCR
RNA was extracted with Trizol (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. The cDNA synthesis and RT-qPCR were
subsequently performed using the Qiagen system as described
previously (21). RT-qPCR analysis
was performed using standard protocols on Applied Biosystem 7500 HT
sequence Detection system. The relative mRNA levels of C9orf53 were
normalized to levels of the housekeeping gene GAPDH and calculated
using the 2−ΔΔCq method (22). The primers used are as follows: GAPDH
forward, 5′-CCATGTTCGTCATGGG-TGTGAACCA-3′ and reverse,
5′-GCCAGTAGAGGCAGGGATGATGTTG-3′ and C9orf53 forward,
5′-AAGAATTCGGCACGAGGGTT-3′ and reverse,
5′-CTCTGCCACAGTGGGATTGT-3′.
MTT assay
For MTT assay, 5×103 cells per well were seeded in
triplicate in a 96-well plate with complete growth medium. The
cells were counted over 5 days using the MTT assay (Promega
Corporation, Madison, WI, USA) as described previously (19,20). The
data was measured using the Microtiter plate reader (Promega
Corporation) at 570 nm.
Transfection of C9orf53 overexpression
plasmid and small-interfering (si)RNA
C9orf53 overexpression plasmid (pcDNA3.1-C9orf53;),
C9orf53-siRNA (5′-GTGTGATTTCGTAAACAGATA-3′) and control-siRNA were
designed and constructed by Sangon Biotech Company (Shanghai,
China). Untransfected cells were used as a blank control.
Transfections were performed using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. SCC-5 and SCC-9 cells were seeded into
24-well plates at a density of 5×104 cells/well and were
allowed to culture overnight. DNA Plasmid (500 ng) with
Lipofectamine® 2000 and siRNA (600 ng) with
Lipofectamine® 2000 were mixed for transfection. Then
DNA-Lipofectamine® 2000 complexes were added into wells
at 37°C for 24 h prior to subsequent experimentation.
Apoptosis assay
SCC-5 and SCC-9 cells were labeled with Annexin
V-fluorescein isothiocyanate and propidium iodide (PI) using the
apoptosis detecting kit (Invitrogen; Thermo Fisher Scientific,
Inc.) following the manufacturer's instructions. Then samples were
analyzed by fluorescence-activated cell sorting (FACS) assay with a
flow cytometer (BD FACSVerse™ flow cytometer, with BD FACSuite™
software v1.0.6, BD Biosciences, San Jose, CA, USA) (23).
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments. The difference between
groups was analyzed using two-tailed, paired Student's t-test when
only two groups were compared. The Wilcoxon matched-pairs signed
rank test was used to determine if there was a statistically
significant difference in the expression of C9orf53 between matched
pairs. The difference between groups was analyzed using one-way
ANOVA with post hoc contrasts by Student-Newman-Keuls test, when
three or more than three groups were compared. Correlation analysis
was performed using two-tailed Person's correlation coefficient
analysis. Patient survival was determined by Kaplan-Meier analysis
(with log-rank test). Statistical analyses were performed using
SPSS software (version 17.0; IBM Corp., Armonk, NY, USA). P<0.05
was considered significantly different.
Results
Screening of novel genes amplified or
deleted in HNSCC
To identify the potential target genes of HNSCC, the
genes that are amplified or deleted in HNSCC were screened. It was
observed that there were a number of genes amplified or deleted in
HNSCC (Table I). To date, to the best
of our knowledge, there is no study on the role of C9orf53 in
HNSCC. Therefore, in the present study, the role of C9orf53 in
HNSCC was investigated. Initially, mutations in C9orf53 were
investigated in various types of cancer and mutations were
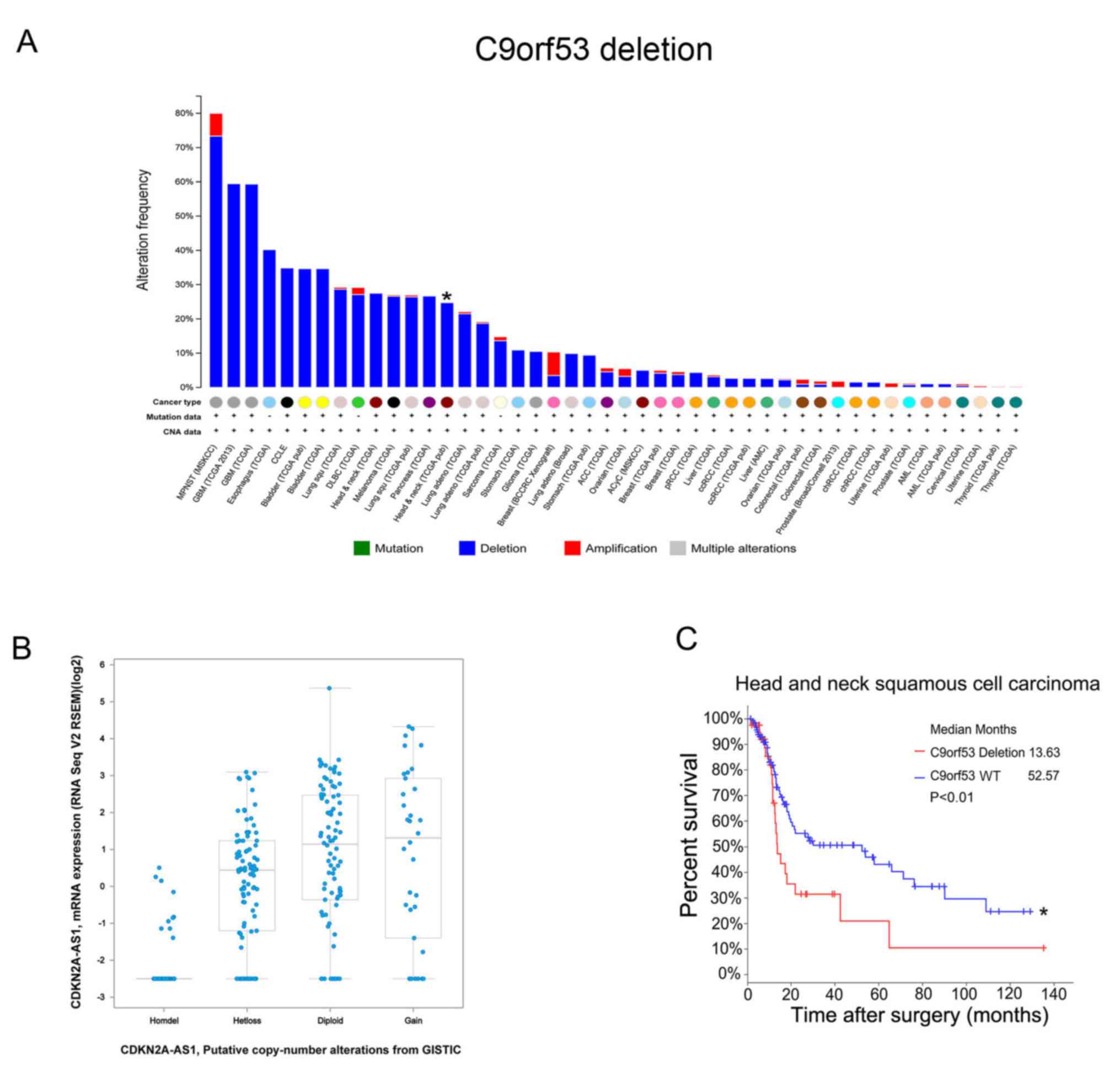
identified in 302 HNSCC tissue samples. There were 27.5% HNSCC
tissues with C9orf53 deletions (Fig.
1A). Next, it was observed that a low expression of C9orf53
mRNA was associated with C9orf53 deletions (Fig. 1B). Importantly, these data indicated
that C9orf53 deletion was associated with a decreased survival rate
(Fig. 1C).
Reduced expression of C9orf53 in HNSCC
tissues compared with normal tissues
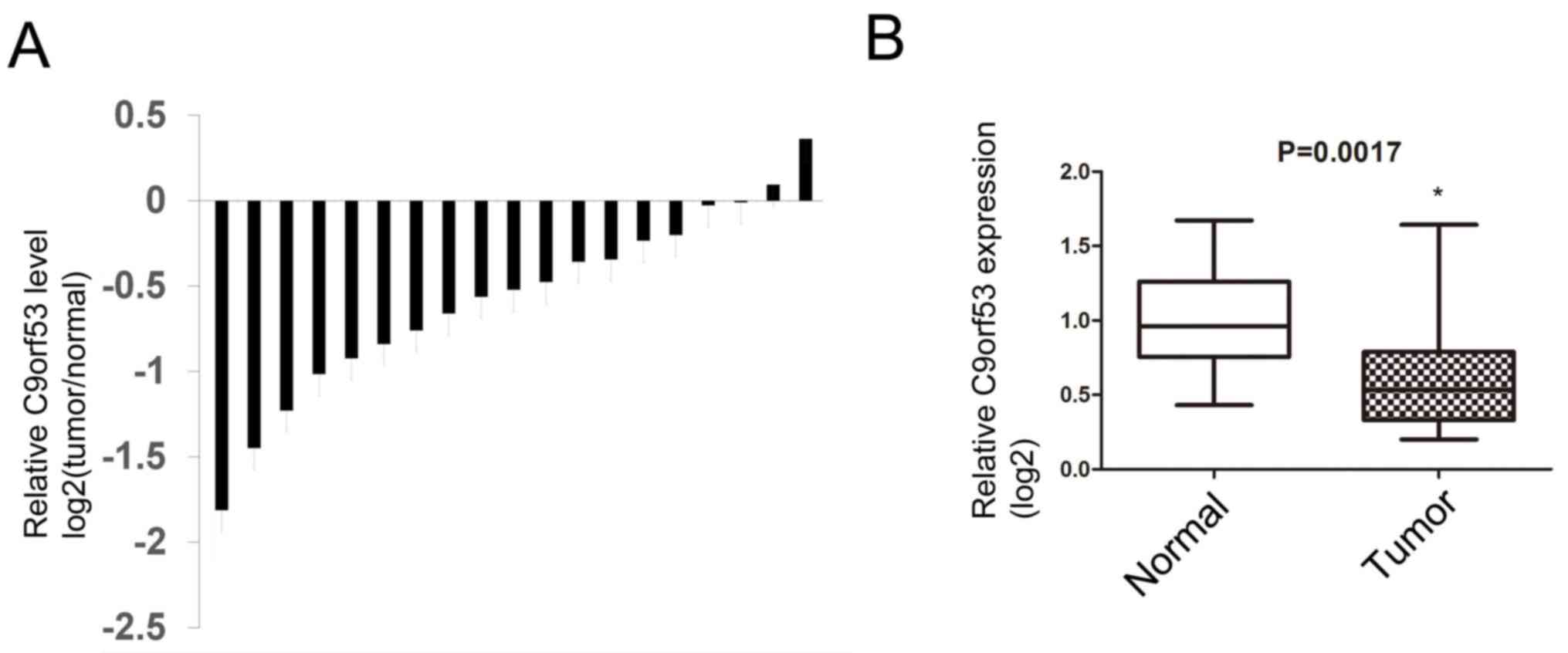
Next, 19 surgical specimens and matched
tumor-adjacent normal tissues were obtained from patients with
HNSCC. The levels of C9orf53 mRNA were assayed by RT-qPCR. It was
identified that 17 of the 19 HNSCC tissues exhibited lower C9orf53
expression compared with matched normal tissues that were adjacent
to tumors (Fig. 2A). Furthermore, the
median C9orf53 expression in HNSCC tissues was lower compared with
normal tissues that were adjacent to tumors, and the standard
deviation of C9orf53 expression in HNSCC was higher compared with
normal tissues (Fig. 2B).
Reduced expression of C9orf53 promotes
proliferation and overexpression of C9orf53-induced apoptosis
Next, the role of C9orf53 in vitro was
investigated. The levels of C9orf53 in HNSCC cell lines (SCC-5 and
SCC-9) were analyzed, and 293 cells were used as a control. It was
observed that the levels of C9orf53 in SCC-5 and SCC-9 were lower
compared with 293 cells (Fig. 3A). As
SCC-5 cells exhibited lower C9orf53 levels compared with SCC-9
cells, C9orf53 was overexpressed in SCC-5 cells by transfection
with pcDNA3.1-C9orf53. C9orf53 expression was suppressed in SCC-9
cells by transfection with si-C9orf53. The levels of C9orf53 were
determined by RT-qPCR, 48 h following transfection. It was observed
that the level of C9orf53 was successfully increased following
transfection of pcDNA3.1-C9orf53 in SCC-5 cells and that C9orf53
expression was successfully inhibited following transfection with
si-C9orf53 in SCC-9 cells (Fig. 3B).
As transfection with si-C9orf53-1 resulted in the greatest decrease
in C9orf53 expression (Fig. 3B),
si-C9orf53-1 was selected for subsequent experiments. Following the
overexpression or inhibition of C9orf53, proliferation was examined
by MTT assay. It was found that overexpressing C9orf53 inhibited
proliferation, and suppressing C9orf53 promoted proliferation
(Fig. 3C). Next, the cell apoptosis
was examined by FACS assay. The overexpression of C9orf53 increased
the rate of cell apoptosis (Fig.
3D).
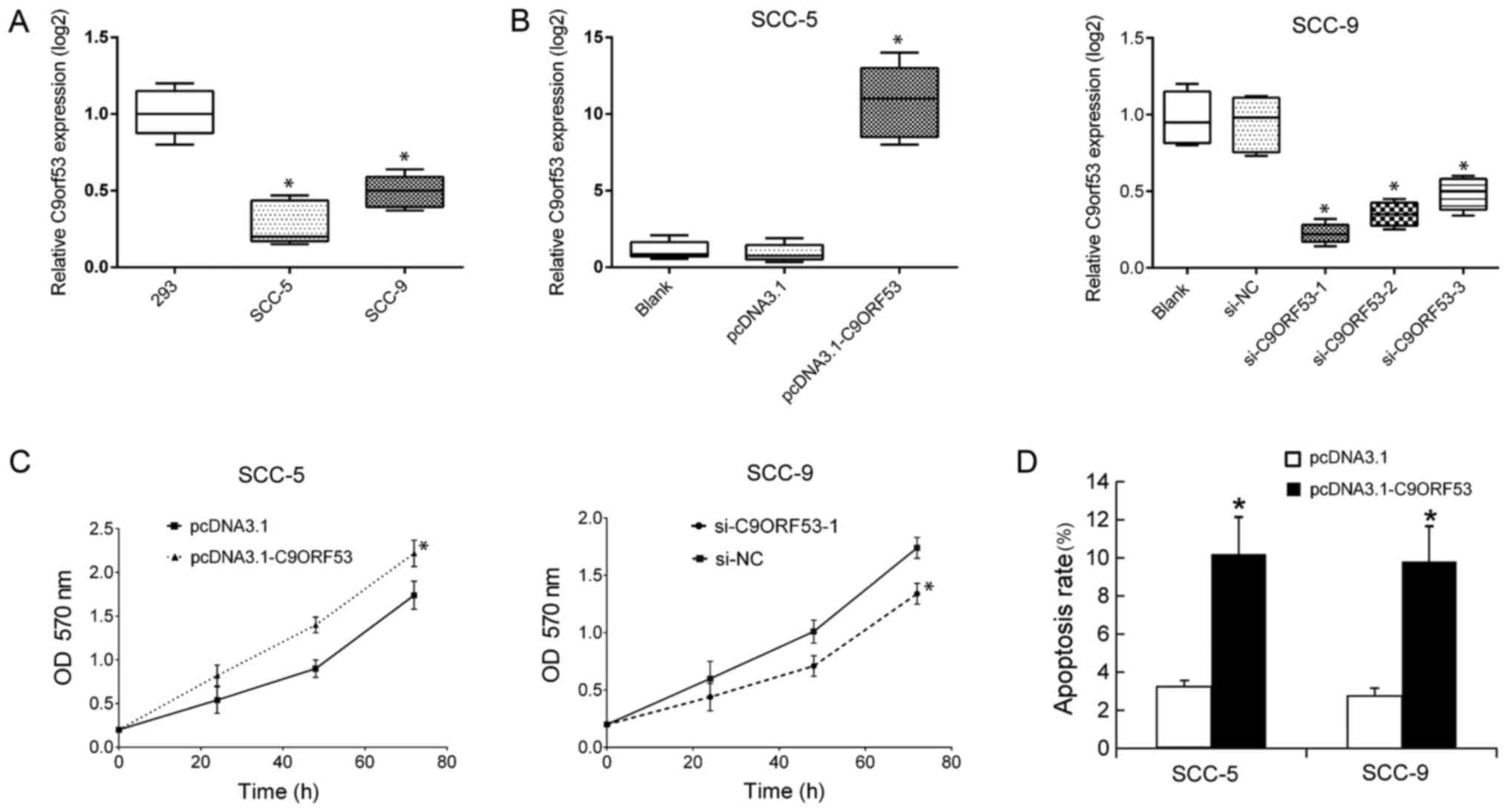 | Figure 3.Suppression of C9orf53 promotes
proliferation and induces apoptosis. The levels of C9orf53 in 293,
SCC-5 and SCC-9 cells were assayed by RT-qPCR. (A) Data on C9orf53
expression are displayed in box plots. *P<0.05 vs. 293 cells.
(B) The levels of C9orf53 in SCC-5 cells were assayed by RT-qPCR 48
h following transfection of pcDNA3.1-C9orf53. All data on C9orf53
expression are displayed in box plots. *P<0.05 pcDNA3.1-C9orf53
vs. control. (C) Following the transfection of pcDNA3.1-C9orf53 or
siC9orf53-1, cell proliferation was analyzed by MTT assay at the
indicated time points. (D) Cell apoptosis was analyzed by FACS, 48
h following the transfection of pcDNA3.1-C9orf53 or siC9orf53-1.
All experiments were repeated four times. The differences between
two groups were analyzed using two-tailed Student's t-test, and the
differences between groups were analyzed using one-way ANOVA when
three or more groups were compared. *P<0.05 vs.
pcDNA3.1-transfected cells. C9ORF53, CDKN2A antisense RNA 1; FACS,
fluorescence-activated cell sorting analysis; NC, negative control;
OD, optical density; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; siRNA, small-interfering RNA. |
Discussion
In the present study, the role of C9orf53 in HNSCC
was studied. It was identified that C9orf53 was deleted in HNSCC,
and the levels of C9orf53 in HNSCC tissues were lower in tumor
tissues compared with matched normal tissues that were adjacent to
tumor tissues, Notably, the deletion of C9orf53 and a lower
expression level of C9orf53 were associated with the rate of
patient survival. Subsequently, the role of C9orf53 was confirmed
by overexpressing and suppressing C9orf53 expression in
vitro. The suppression of C9orf53 was able to promote
proliferation and induce apoptosis and apoptosis. To the best of
our best knowledge, this might be the first report of the role of
C9orf53 in cancer.
The data from the present study indicated that the
effects of C9orf53 on proliferation and apoptosis were relative
minor. However, the difference in survival between patients with
C9orf53 deletion and wild-type C9orf53 was significant. It is
surprising that these minor influences were able to cause an effect
on patient survival. It was hypothesized that although the effects
of C9orf53 on cell proliferation and apoptosis were rather minor,
the cells proliferated exponentially in vivo. Therefore,
minor differences in C9orf53 were able to cause a long-lasting
effect on patient survival (130 months).
Recently, a comprehensive landscape of somatic
genomic alteration of HNSCC was provided by TCGA (24). It was indicated that
human-papillomavirus-associated tumors are dominated by helical
domain mutations of the oncogene
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α.
Smoking-related HNSCCs demonstrated near universal loss-of-function
tumor protein p53 mutations and CDKN2A inactivation. Whether the
role of C9orf53 in HNSCC is associated with human papillomavirus
infection or smoking will be investigated in further studies.
In conclusion, the present study investigated the
role of C9orf53 in HNSCC. The data indicated that the deletion and
decreased level of C9orf53 might promote the growth of HNSCC cells.
The present study might provide a novel therapeutic target for
further investigations.
Acknowledgements
The authors would like to thank Dr. Chaoxiong Zhang
(West China School of Public Health and Healthy Food Evaluation
Research Center, Sichuan University, Chengdu, China) for assistance
with the discussion.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81271353) and PLA
Research Project (grant no. 2011XL015) and ‘The 12th Five-Year
Plan’ for Medicsal Science Development of PLA Research Project
(grant no. CWS11J300), the Key Project on the Integration of
Industry, Education, and Research and Medicine of Science and
Technology Commission of Shanghai Municipality (grant nos.
12DZ1940503 and 13DZ1942704).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
GW, HJ and YL collected patient data and performed
cell experiments. YW and XC performed transfection and apoptosis
analysis. YZ and DW contributed to study design and manuscript
writing.
Ethics approval and consent to
participate
The present study was approved by Ethics Committee
of Second Military Medical University (Shanghai, China).
Patient consent for publication
All patients gave informed consent for the use of
their tissues and publication of the data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu CJ, Lin SC, Chen YJ, Chang KM and
Chang KW: Array-comparative genomic hybridization to detect
genomewide changes in microdissected primary and metastatic oral
squamous cell carcinomas. Mol Carcinog. 45:721–731. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shieh TM, Lin SC, Liu CJ, Chang SS, Ku TH
and Chang KW: Association of expression aberrances and genetic
polymorphisms of lysyl oxidase with areca-associated oral
tumorigenesis. Clin Cancer Res. 13:4378–4385. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmitz S, Ang KK, Vermorken J, Haddad R,
Suarez C, Wolf GT, Hamoir M and Machiels JP: Targeted therapies for
squamous cell carcinoma of the head and neck: Current knowledge and
future directions. Cancer Treat Rev. 40:390–404. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Allegra CJ, Jessup JM, Somerfield MR,
Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF and
Schilsky RL: American society of clinical oncology provisional
clinical opinion: Testing for KRAS gene mutations in patients with
metastatic colorectal carcinoma to predict response to
anti-epidermal growth factor receptor monoclonal antibody therapy.
J Clin Oncol. 27:2091–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, .
Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA,
Ellrott K, Shmulevich I, Sander C and Stuart JM: The cancer genome
atlas pan-cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Verhaak RG, Hoadley KA, Purdom E, Wang V,
Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al:
Integrated genomic analysis identifies clinically relevant subtypes
of glioblastoma characterized by abnormalities in PDGFRA, IDH1,
EGFR, and NF1. Cancer Cell. 17:98–110. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network:
Integrated genomic analyses of ovarian carcinoma. Nature.
474:609–615. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cancer Genome Atlas Network: Comprehensive
molecular characterization of human colon and rectal cancer.
Nature. 487:330–337. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the catalogue of somatic
mutations in cancer. Nucleic Acids Res. 29:D945–D950. 2011.
View Article : Google Scholar
|
|
12
|
Collins FS and Barker AD: Mapping the
cancer genome. Scientific American. 296:50–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stratton MR, Campbell PJ and Futreal PA:
The cancer genome. Nature. 458:719–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Emanuele E, Lista S, Ghidoni R, Binetti G,
Cereda C, Benussi L, Maletta R, Bruni AC and Politi P: Chromosome
9p21.3 genotype is associated with vascular dementia and
Alzheimer's disease. Neurobiol Aging. 32:1231–1235. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: an open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barnes L, Eveson JW, Reichart P and
Sidransky D: Pathology and genetics of head and neck tumours. IARC.
2005.
|
|
18
|
Wu N, Liu C, Bai C, Han YP, Cho WC and Li
Q: Over-expression of deubiquitinating enzyme USP14 in lung
adenocarcinoma promotes proliferation through the accumulation of
beta-catenin. Int J Mol Sci. 14:10749–10760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu N, Zhang C, Bai C, Han YP and Li Q:
miR-4782-3p Inhibited non-small cell lung cancer growth via USP14.
Cell Physiol Biochem. 33:457–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song B, Zhang C, Li G, Jin G and Liu C:
MiR-940 inhibited pancreatic ductal adenocarcinoma growth by
targeting MyD88. Cell Physiol Biochem. 35:1167–1177. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hou J, Lin L, Zhou W, Wang Z, Ding G, Dong
Q, Qin L, Wu X, Zheng Y, Yang Y, et al: Identification of miRNomes
in human liver and hepatocellular carcinoma reveals miR-199a/b-3p
as therapeutic target for hepatocellular carcinoma. Cancer Cell.
19:232–243. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu J, Wen M, Huang Y, He X, Wang Y, Wu Q,
Li Z, Castellanos-Martin A, Abad M, Cruz-Hernandez JJ, et al:
C2ORF40 suppresses breast cancer cell proliferation and invasion
through modulating expression of M phase cell cycle genes.
Epigenetics. 8:571–583. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cancer Genome Atlas Network: Comprehensive
genomic characterization of head and neck squamous cell carcinomas.
Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|