Introduction
Taxol, which is a type of microtubule-stabilizing
antitumor drug, has a broad antineoplastic spectrum and induces
cell apoptosis in various types of human cancer (1). Taxol exerts its anticancer effects via
several mechanisms: Inhibition of microtubule assembly and protein
isoprenylation, cell cycle arrest at the G2/M phase, and
stimulation of cell apoptosis and DNA fragmentation (2,3). Taxol has
been demonstrated to be efficacious for castration-resistant
prostate cancer in previous studies (4,5). However,
the majority of patients with prostate cancer gradually acquired
drug resistance to Taxol following a series of repeated treatments,
which may seriously affect their prognosis (1,6). Thus,
there is an increasing requirement to develop novel agents which
could enhance chemosensitivity to Taxol in patients with prostate
cancer.
As an important signaling molecule, antioxidant and
toxicant, nitric oxide (NO) is involved in multiple pathological
and physiological processes. JS-K
(C13H16N6O8; Chemical
Abstracts Service no. 205432-12-8), a glutathione
transferase-activated nitric oxide-donor prodrug, is reported to
promote high intracellular levels of NO and result in cytotoxicity
to human prostate cancer cells (7–10). Our
previous study demonstrated that JS-K was able to increase the
anticancer effects of doxorubicin, which is a type of anthracycline
and potent antitumor drug, in human renal carcinoma cells (11). In the present study, the effect of
JS-K on the chemosensitivity of human prostate cancer cells to
Taxol was investigated. The results of the present study
demonstrated that JS-K increased the cytotoxic effects of Taxol on
prostate cancer cells. The results of the present study also
demonstrated the function of the accumulation of reactive oxygen
species (ROS) in apoptosis in prostate cancer cells induced by the
combination of JS-K and Taxol. These results revealed the essential
functions and mechanisms of JS-K on Taxol-induced apoptosis in
prostate cancer cells.
Materials and methods
Drugs and reagents
Taxol and the nitric oxide prodrug JS-K were
purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA),
and dissolved in 100% dimethylsulfoxide (DMSO) as stock solutions
(Taxol, 10 mM; JS-K, 5 mM). The final concentration of DMSO did not
exceed 0.1% in any experiment. N-acetylcysteine (NAC) and oxidized
glutathione (GSSG) were obtained from Beyotime Institute of
Biotechnology (Haimen, China) and dissolved in PBS to
concentrations of 100 mM (NAC) and 5 mM (GSSG). Antibodies against
BRI1-associated receptor kinase 1 (Bak, cat no. 3814),
Bcl-2-associated X protein (Bax, cat no. 2772), B-cell lymphoma
(Bcl-2, cat no. 2876), cleaved caspase-3 (cat no. 9661), caspase-7
(cat no. 9494) and caspase-9 (cat no. 9502) were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA) and an antibody
against GAPDH was purchased from Abcam (Cambridge, MA, USA) to be
used as a loading control. Horseradish peroxidase (HRP) goat
anti-rabbit and anti-mouse IgG secondary antibodies were purchased
from EarthOx Life Sciences (Millbrae, CA, USA).
Cell lines and cell culture
Human prostate cancer 22RV1 and PC-3 cell lines were
purchased from Guangzhou RiboBio Co., Ltd. (Guangzhou, China).
22RV1 cells were cultured in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and PC-3 cells were cultured in
high glucose Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified atmosphere
containing 5% CO2. All culture media were supplemented
with 10% (v/v) fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 100 U/ml penicillin and 100 U/ml streptomycin.
Cell viability assay
The viability of prostate cancer cells was detected
using Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies,
Inc., Kumamoto, Japan) according to the manufacturer's protocol.
Briefly, 22RV1 and PC-3 cells were seeded in 96-well plates (NEST
Biotechnology Co., Ltd, Jiangsu, China) at a density of
5×103 cells/well and then cultured for 24 h. Cells were
pretreated with/without JS-K (1 µM) for 24 h and then exposed to
Taxol (2 µM) for another 24 h, the culture medium was removed and
replaced with 100 µl medium containing CCK-8 reagent and then
incubated at 37°C for 2 h. Absorbance was recorded at a wavelength
of 450 nm in a 96-well plate reader (EnSpire 2300 Multilabel
Reader; PerkinElmer, Inc., Waltham, MA, USA). Half-maximal
inhibitory concentrations (IC50) were calculated by
probit method using SPSS software (version 18.0; SPSS, Inc.,
Chicago, IL, USA).
Colony formation assay
Cells were pretreated with/without JS-K (1 µM) for 6
h and then exposed to Taxol (2 µM) for another 6 h. Viable cells
were harvested and seeded into 6-well plates (NEST Biotechnology
Co., Ltd.) at a density of 1.5×103 cells/well. Cells
were cultured for 2 weeks for assessment of colony formation.
Colony formation was analyzed by staining the cells with a Crystal
Violet Staining Solution (Beyotime Institute of Biotechnology).
Briefly, cells were fixed with 4% paraformaldehyde for 30 min at
room temperature. Cells were stained with crystal violet staining
solution (10 mg/ml) for 30 min at room temperature and then washed
with ultrapure water. The macroscopic clones (>1 mm) were
photographed with digital camera.
Apoptosis assay
Apoptotic cells were quantified using a fluorescein
isothiocyanate (FITC) Annexin V Apoptosis Detection kit (BD
Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer's protocol. Briefly, following pretreatment
with/without JS-K (1 µM) for 24 h and then exposure to Taxol (2 µM)
for another 24 h,s the prostate cancer cells were collected, washed
with PBS and resuspended in binding buffer. Cells were incubated
with annexin V-FITC and propidium iodide for 15 min at room
temperature in the dark, prior to flow cytometric analysis. The
stained cells were analyzed by flow cytometry within 1 h.
Measurement of intracellular ROS
To determine the accumulation of intracellular ROS
in prostate cancer cells, a ROS assay kit (Beyotime Institute of
Biotechnology) was used. Briefly, following pretreatment
with/without JS-K (1 µM) for 24 h and then exposure to Taxol (2 µM)
for an additional 24 h, cells were collected and resuspended with
serum-free medium containing dihydrofluorescein diacetate (10 µM).
Following incubation at 37°C for 20 min in the dark, the cells were
harvested and analyzed using flow cytometry with excitation at 488
nm and emission at 525 nm.
Measurement of mitochondrial membrane
potential
22RV1 and PC-3 cells were seeded into 6-well plates
at a density of 3×105 cells/well and cultured for 24 h,
then were pretreatment with/without JS-K (1 µM) for 6 h and then
exposed to Taxol (2 µM) for an additional 6 h. The JC-1
Mitochondrial Membrane Potential assay kit (Beyotime Institute of
Biotechnology) was used according to the manufacturer's protocol.
The cells were analyzed using flow cytometry.
Glutathione assay
Cells were plated in 6-well plates and pretreated
with/without JS-K (1 µM) for 6 h and then exposed to Taxol (2 µM)
for an additional 6 h. The cells were then harvested and lysed by
two successive rounds of freezing and thawing. The supernatant was
collected by centrifuging at 10,000 × g at 4°C for 10 min and
analyzed for reduced glutathione (GSH) and GSSG levels using a GSH
and GSSG assay kit (Beyotime Institute of Biotechnology) according
to the manufacturer's protocol.
Measurement of adenosine triphosphate
(ATP) levels
Intracellular ATP levels were measured using an ATP
assay kit (Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Briefly, prostate cancer cells were
pretreated with/without JS-K (1 µM) for 6 h and then exposed to
Taxol (2 µM) for another 6 h and incubated in 200 µl lysis buffer
at 4°C for 5 min with gentle shaking. The supernatant of lysis
buffer was then collected by centrifugation at 12,000 × g at 4°C
for 10 min. Firefly luciferase activity was detected using a Sirius
L luminometer (Titertek-Berthold, Pforzheim, Germany)
Preferably.
Caspase-Glo 3/7 and Caspase-Glo 9
assays
To examine cell apoptosis following treatment with
JS-K and Taxol, caspase-3/7 and caspase-9 activities were analyzed
using Caspase-Glo 3/7 and Caspase-Glo 9 assays (Promega
Corporation, Madison, WI, USA) according to the manufacturer's
protocol. Briefly, cells were seeded into 96-well plates (NEST
Biotechnology Co., Ltd.) and pretreated with/without JS-K (1 µM)
for 24 h and then exposed to Taxol (2 µM) for another 24 h An equal
volume of Caspase-Glo 3/7 or Caspase-Glo 9 reagent was added to
each well, and the cells were incubated for 1 h at room temperature
in the dark. The luminescence was measured using a Sirius L
luminometer (Titertek-Berthold, Bad Wildbad, Germany).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from 22RV1 and PC-3 cells
using an E.Z.N.A. Total RNA Kit I (Omega Bio-tek, Inc. Norcross,
GA) and the mRNAs were reversely transcribed into stable cDNA with
a PrimeScript RT reagent Kit with gDNA Eraser (Takara Bio, Inc.,
Dalian, China). RT-qPCR analysis was performed by using the SYBR
Premix Ex Taq II kit (TaKaRa). The thermocycling conditions
included: 95°C for 1 min, followed by 40 cycles of amplification at
95°C for 5 sec and 60°C for 30 sec. The levels of mRNAs were
expressed as 2−ΔΔCq (12).
The sequences of forward and reverse primers for each gene are
listed in Table I.
 | Table I.Sequences for target gene primers. |
Table I.
Sequences for target gene primers.
| Gene | Primer sequence
(5′-3′) | Tm, °C |
|---|
| GAPDH | TGCACCACCAACTGCTTAG
(F) | 60.07 |
|
|
AGTAGAGGCAGGGATGATGTTC (R) | 59.72 |
| Bak | CCCAACCCATTCACTACAGG
(F) | 59.80 |
|
| CCCACTTAGAACCCTCCAGA
(R) | 59.80 |
| Bcl-2 | CTTTGAGTTCGGTGGGGTCA
(F) | 59.80 |
|
| GGGCCGTACAGTTCCACAAA
(R) | 59.80 |
| Bax | AAGCTGAGCGAGTGTCTCAAG
(F) | 60.00 |
|
|
CAAAGTAGAAAAGGGCGACAAC (R) | 58.20 |
Western blot analysis
Cells were lysed with radioimmunoprecipitation
buffer (Beyotime Institute of Biotechnology) supplemented with 1 mM
phenylmethylsulfonyl fluoride (Beyotime Institute of Biotechnology)
and proteins were extracted by centrifugation at 10,000 × g at 4°C
for 15 min. Total protein was determined by a BCA Protein assay kit
(Beyotime Institute of Biotechnology). Proteins (40 µg/lane) were
loaded onto SDS-PAGE gels (12%) then transferred onto
polyvinylidene difluoride membranes (Merck KGaA, Darmstadt,
Germany), which were blocked with 5% non-fat milk in Tris-buffered
saline and 1% Tween 20 (TBS-T) and incubated with the
aforementioned primary antibodies (1:1,000) in diluent (5% BSA in
TBS-T) overnight at 4°C. Following six washes with TBS-T for 5 min
each, the membranes were probed with HRP-conjugated goat
anti-rabbit or anti-mouse IgG secondary antibody (#E030120,
#E030110; EarthOx Life Sciences, Millbrae, CA, USA; dilution
1:10,000 in TBS-T containing 5% BSA) for 2 h at room
temperature.
Statistical analysis
All experiments were performed in triplicate. The
results are presented as the mean ± standard deviation. All data
were analyzed using one-way analysis of variance using SPSS
software (version 18.0; SPSS, Inc., Chicago, IL, USA). Differences
between treatments were assessed using a Fisher's least significant
difference test. P<0.05 was considered to indicate a
statistically significant difference.
Results
JS-K promotes Taxol-induced
suppression of proliferation and apoptosis of prostate cells
Prostate cancer cells were exposed to Taxol, JS-K or
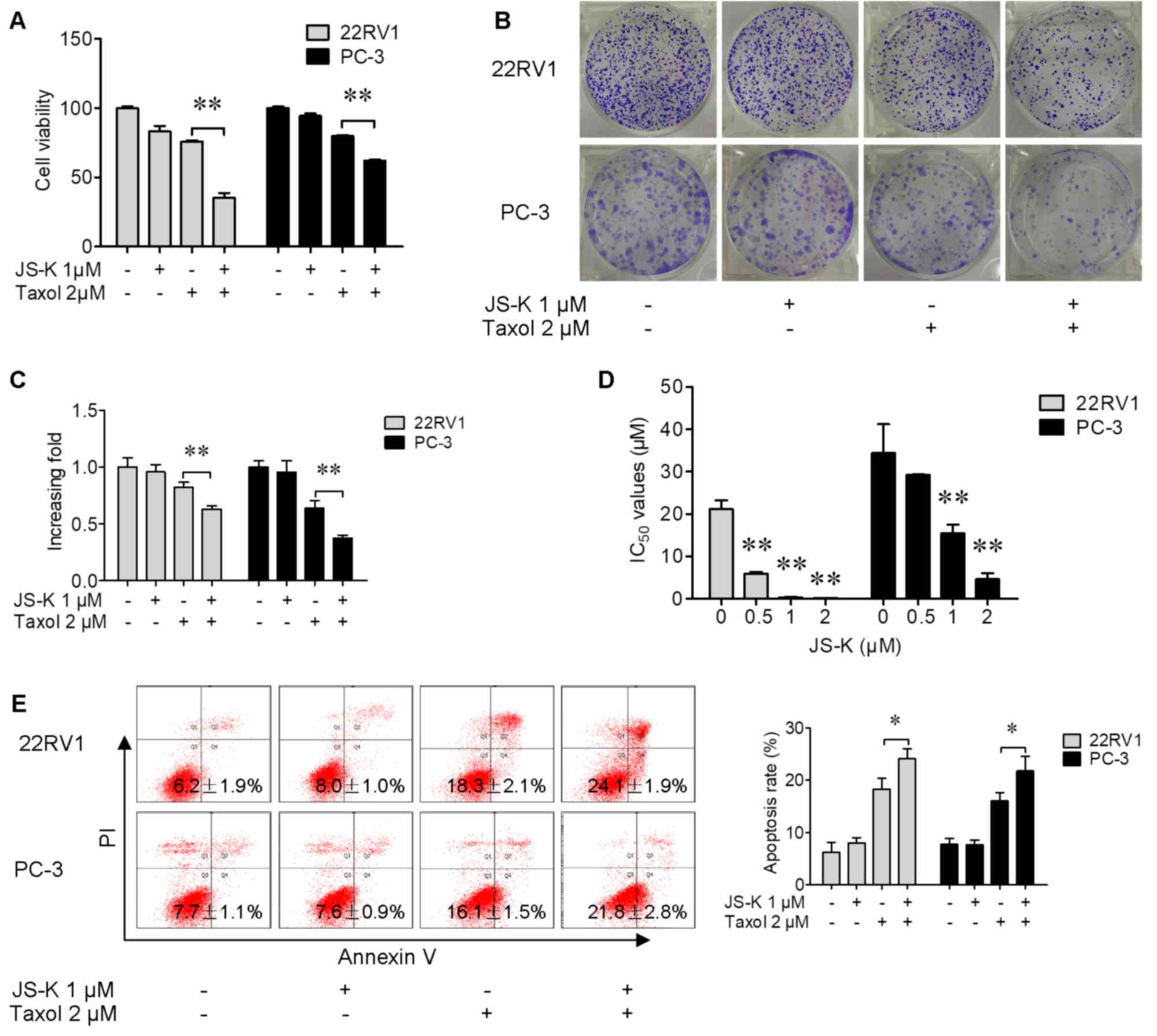
their combination for 24 h. A CCK-8 assay (Fig. 1A) and a colony formation assay
(Fig. 1B and C) were performed to
evaluate the effects of Taxol and JS-K on prostate cancer cells,
and the data indicated that the number of viable cells was
significantly decreased by treatment with Taxol and JS-K together
compared with Taxol alone. The half-maximal inhibitory
concentrations of Taxol were significantly decreased when cells
were pretreated with JS-K (1 µM) for 24 h (Fig. 1D). These data suggested that JS-K was
able to significantly promote Taxol-induced suppression of prostate
cancer cell proliferation. The apoptosis-inducing effect of Taxol,
JS-K and their combination was investigated. It was revealed that
JS-K significantly increased apoptosis (Fig. 1E) in Taxol-treated prostate cancer
cells. These results indicated that JS-K significantly increased
Taxol-induced suppression of proliferation and apoptosis of
prostate cancer cells.
JS-K increases Taxol-induced ROS
production and decreases the Taxol-induced decrease in the GSH/GSSG
ratio in prostate cancer cells
The effects of JS-K on Taxol-induced ROS production
were assessed; it was revealed that ROS production was
significantly increased in prostate cancer cells that were treated
with Taxol and JS-K together (Fig.
2A). The levels of GSH and GSSG were investigated; it was
revealed that the GSH/GSSG ratio was significantly decreased in
cells treated with Taxol and JS-K together compared with Taxol
alone (Fig. 2B).
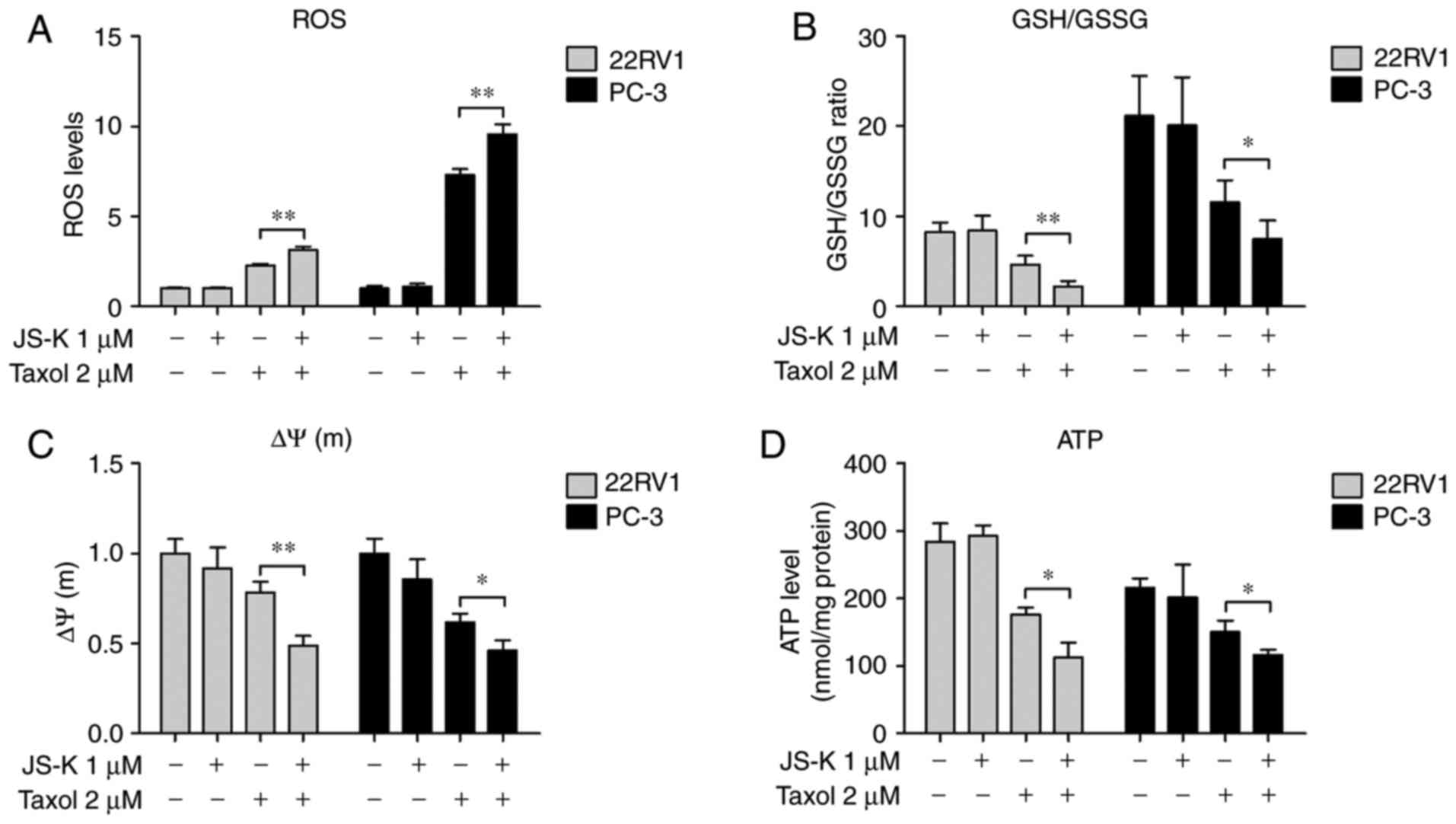 | Figure 2.Synergistic effects of JS-K and Taxol
on ROS production, GSH/GSSG ratio, mitochondrial membrane potential
and ATP levels in prostate cancer cells. Prostate cancer cells were
treated with JS-K and Taxol for 6 h and (A) ROS generation, (B)
GSH/GSSG ratio, (C) mitochondrial membrane potential and (D) ATP
levels were determined. The data are presented as the mean ±
standard deviation for three independent experiments. *P<0.05,
**P<0.01. GSH, reduced glutathione; GSSG, oxidized glutathione;
ATP, adenosine triphosphate; ROS, reactive oxygen species; ΔΨ(m),
mitochondrial membrane potential. |
JS-K exacerbates the Taxol-induced
decrease in mitochondrial membrane potential and ATP levels in
prostate cancer cells
The Taxol and JS-K-induced increase in total ROS and
decrease in GSH/GSSG ratio (Fig. 2A and
B) may contribute to increased mitochondrial dysfunction and
lead to mitochondria-mediated apoptosis. To investigate the
dysfunction in mitochondrion, the mitochondrial membrane potential
and intracellular levels of ATP in prostate cancer cells treated
with JS-K and Taxol was determined. It was revealed that the
mitochondrial membrane potential (Fig.
2C) and intracellular levels of ATP (Fig. 2D) was significantly decreased in cells
treated with Taxol and JS-K together compared with Taxol alone.
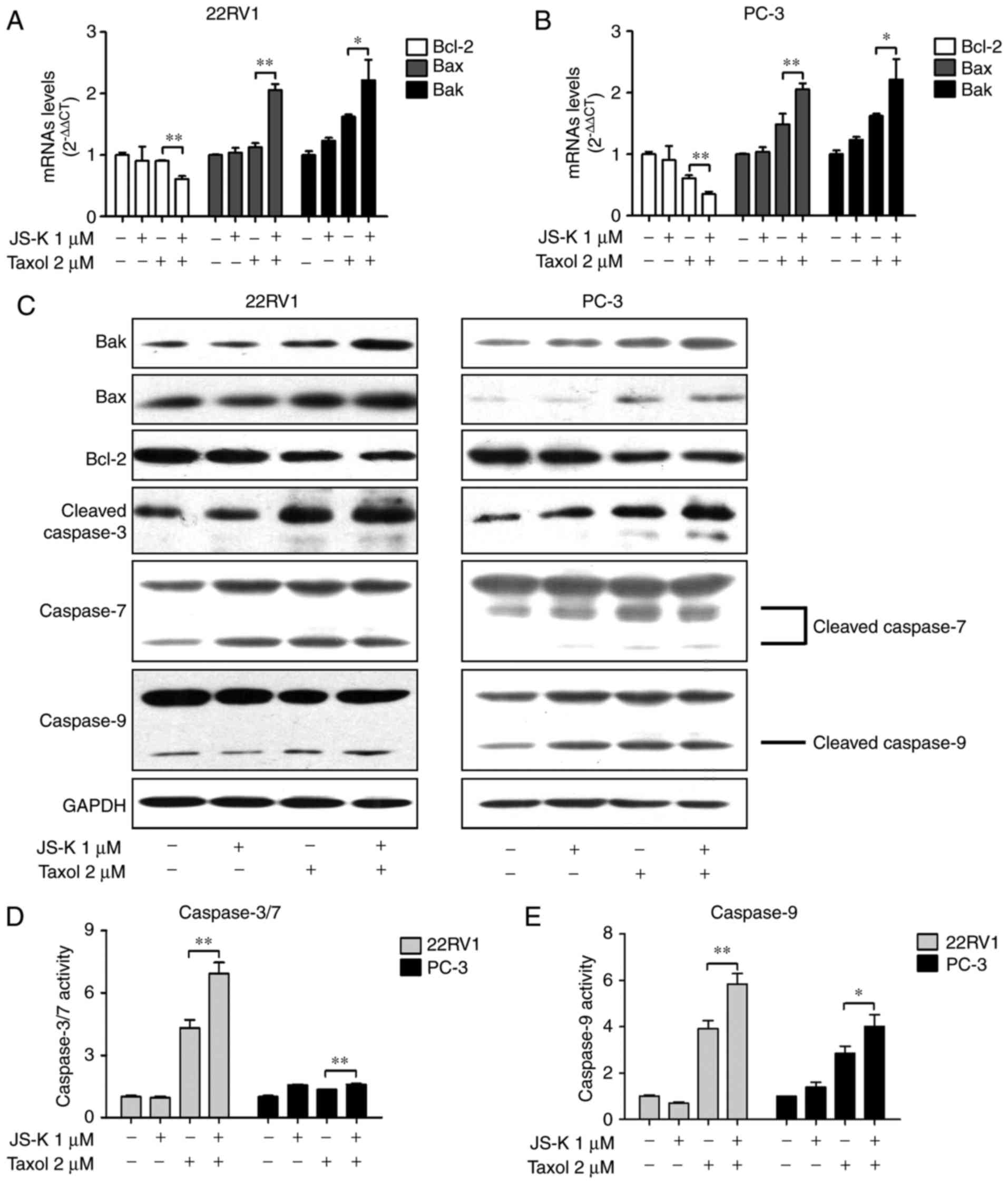
JS-K and Taxol synergistically
regulates the apoptosis-associated signaling pathway
In the present study, prostate cancer cells were
treated with Taxol, JS-K or their combination for 24 h. mRNA
(Fig. 3A and B) and protein (Fig. 3C and Table
II) levels of Bax and Bak were upregulated in response to the
combination of Taxol and JS-K, whereas Bcl-2 levels were
downregulated. The levels and activities of caspase-3/7/9 were
investigated; increased levels of cleaved caspase-3, cleaved
caspase-7 and cleaved caspase-9 (Fig.
3C) and increased activity of caspase-3/7 (Fig. 3D) and caspase-9 (Fig. 3E) in cells treated with Taxol and JS-K
together compared with the cells treated with Taxol alone was
revealed.
| Table II.Expression levels of related proteins
following treatment with JS-K and Taxol. |
Table II.
Expression levels of related proteins
following treatment with JS-K and Taxol.
|
| Expression levels of
22RV1 | Expression levels of
PC-3 |
|---|
|
|
|
|
|---|
| Treatment | Control | JS-K | Taxol | JS-K+Taxol | Control | JS-K | Taxol | JS-K+Taxol |
|---|
| Bak | 1.00 | 1.06 | 1.68 | 3.74 | 1.00 | 1.23 | 2.04 | 3.12 |
| Bax | 1.00 | 1.01 | 1.26 | 1.82 | 1.00 | 1.11 | 4.56 | 6.44 |
| Bcl-2 | 1.00 | 0.85 | 0.62 | 0.62 | 1.00 | 0.81 | 0.65 | 0.79 |
| Cl-caspase-3 | 1.00 | 1.10 | 1.72 | 2.46 | 1.00 | 1.37 | 2.21 | 3.42 |
| Caspase-7 (35
kDa) | 1.00 | 1.68 | 157 | 1.81 | 1.00 | 0.92 | 1.02 | 1.21 |
| Caspase-7 (30
kDa) | − | − | − | − | 1.00 | 1.32 | 2.26 | 2.06 |
| Caspase-7 (20
kDa) | 1.00 | 2.56 | 2.76 | 2.76 | 1.00 | 4.35 | 13.04 | 21.74 |
| Caspase-9 (47
kDa) | 1.00 | 0.89 | 0.70 | 0.84 | 1.00 | 1.40 | 1.57 | 1.83 |
| Caspase-9 (37
kDa) | 1.00 | 0.89 | 1.00 | 1.53 | 1.00 | 2.04 | 2.54 | 2.71 |
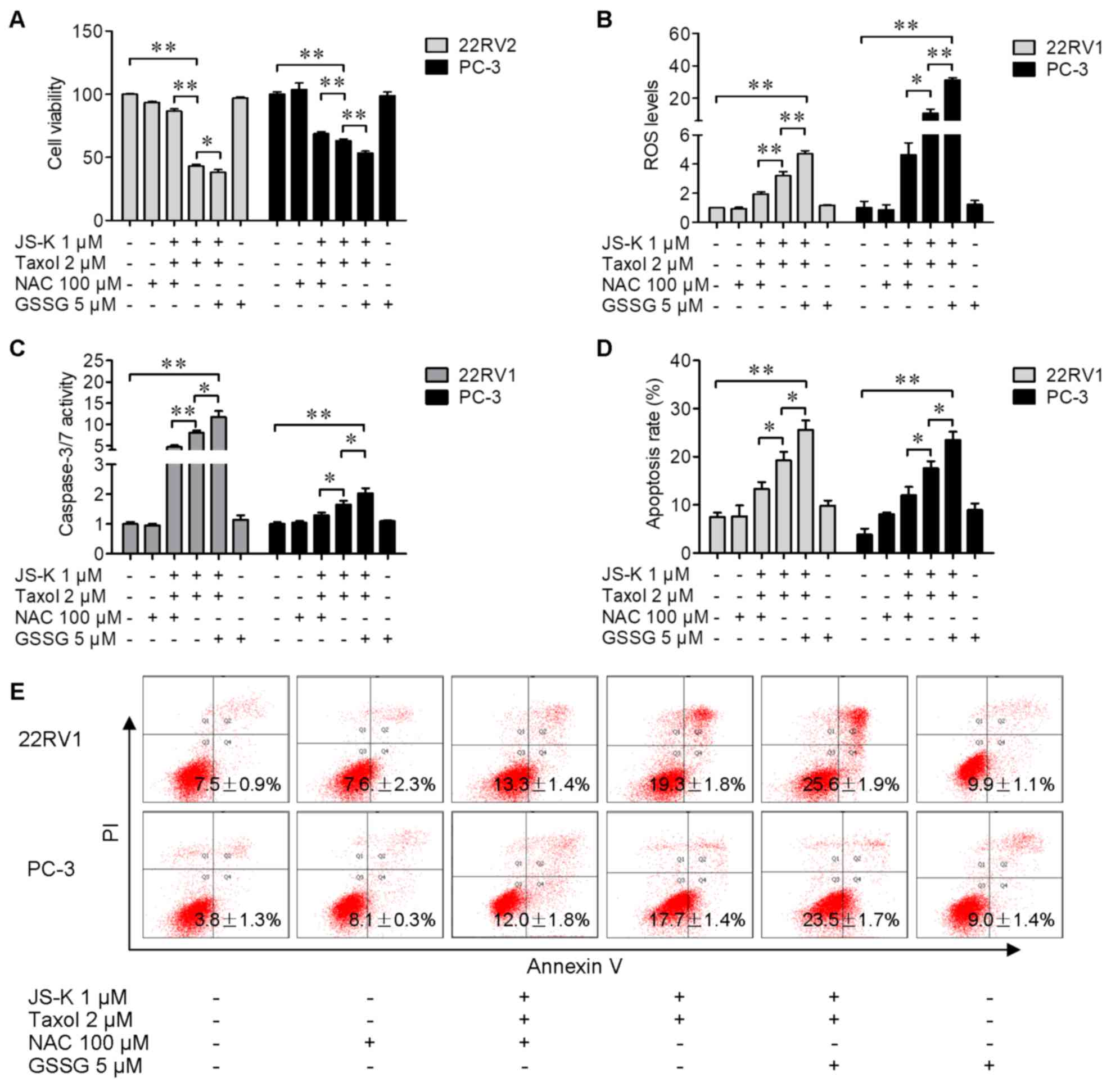
NAC reverses JS-K-induced anticancer
activity of Taxol and GSSG exacerbates the effects of JS-K
To investigate the effects of ROS on Taxol and
JS-K-induced prostate cancer cell proliferation suppression and
apoptosis, prostate cancer cells were treated with Taxol and JS-K
in the presence or absence of the antioxidant NAC (100 µM) or
pro-oxidant GSSG (5 µM) for 24 h. Cells treated with JS-K and Taxol
together was demonstrated to decrease the number of viable cells
(Fig. 4A) and increase ROS levels
(Fig. 4B), caspase 3/7 activity
(Fig. 4C) and apoptosis (Fig. 4D and E) in prostate cancer cells;
whereas NAC (100 µM) reversed the JS-K-induced increase in
chemosensitivity of cells to Taxol (Fig.
4A-D). Furthermore, GSSG (5 µM) exacerbated the JS-K-induced
increase in chemosensitivity of cells to Taxol (Fig. 4A-D). These results indicated the
functions of ROS in JS-K-sensitized Taxol exerting anticancer
activity in human prostate cancer cells.
Discussion
Taxol is recognized as an effective anticancer drug
for treating castration-resistant prostate cancer and has been
clinically challenged by its acquisition of drug resistance
following repeated treatments (1,4,6). In the present study, it was indicated
that JS-K increased Taxol-induced proliferation suppression and
apoptosis in prostate cancer cells. Taxol was identified to
increase ROS generation in cancer cells (13). Our previous study demonstrated that
JS-K promotes apoptosis in prostate cancer cells via induction of
intracellular ROS accumulation (10),
and the results of the present study were consistent with these
results.
Apoptosis is a major process of programmed cell
death, which involves two distinct apoptosis pathways including
mitochondrial and death receptor pathways (14,15).
Abnormal ROS generation is a critical event during
mitochondria-associated apoptosis. As vital signaling molecules,
ROS regulate multiple intracellular signal transduction pathways
that are involved in various cellular processes including cell
proliferation, cell cycle progression, invasion, migration and
apoptosis in cancer (16,17). Our previous studies have demonstrated
that JS-K, an important glutathione transferase-activated NO donor
prodrug, was able to stimulate intercellular accumulation of ROS
which resulted in cytotoxicity and apoptosis in cancer cells
derived from human urogenital tumors (10,18). In
the present study, it was demonstrated that JS-K markedly increased
intracellular ROS generation in Taxol-treated prostate cancer
cells. It was also revealed that administration of the prooxidant
GSSG exacerbated the inducing effect of JS-K on Taxol-induced
apoptosis. However, the inducing effect of JS-K was inhibited in
the presence of the antioxidant NAC. These data suggested that
upregulated ROS generation may be a mechanism by which JS-K
increases the anticancer activity of Taxol in prostate cancer
cells.
GSH, as well as its oxidized form GSSG, are crucial
elements involved in mitochondrial dysfunction, redox balance and
apoptosis (19,20). The physiological balance of GSH and
GSSG is used to indicate the general cellular oxidation/reduction
microenvironments and the redox balance (21). The results of the present study have
demonstrated that JS-K significantly decreased the GSH/GSSG ratio,
which resulted in a redox imbalance. Apoptosis induced by
mitochondrial dysfunction is a fundamental apoptotic pathway
accompanied by a series of events including decreases in
mitochondrial membrane potential and ATP production, and activation
of caspase-9 (15,22,23). As an
energy source in cellular metabolism, ATP production would be
influenced by a decline in mitochondrial membrane potential during
cellular apoptosis (24). The results
of the present study demonstrated that JS-K significantly promoted
Taxol-induced mitochondria-associated apoptosis, and exacerbated
the decrease in mitochondrial membrane potential and ATP levels,
and exacerbated the activation of caspase-9. In addition, Bcl-2
family members are important regulators of apoptosis, and
dysregulation of Bcl-2 family members occurs in a number of
diseases and stress-induced apoptosis (25,26). As
members of the Bcl-2 family, Bak, Bax and Bcl-2 serves crucial
functions in mitochondrial stress-induced cellular apoptosis, and
the appropriate balance of Bcl-2 and Bax is vital for cellular
survival (27). In the present study,
JS-K significantly exacerbated the Taxol-induced increase in
expression levels of Bax and Bak proteins, and the decrease in
expression of Bcl-2 protein. These results suggested that JS-K
promoted Taxol-induced apoptosis through a mitochondria-mediated
pathway.
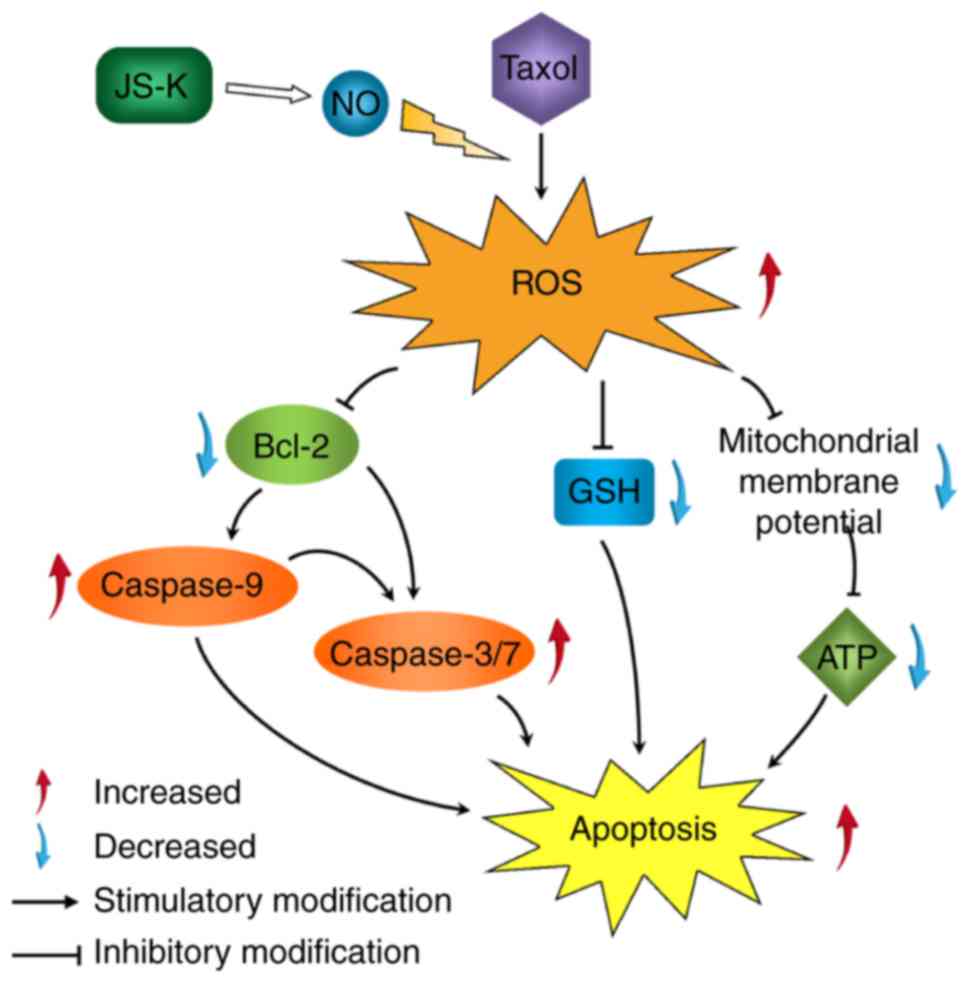
In conclusion, the results of the present study
indicated that the addition of JS-K increased the anticancer effect
of Taxol on prostate cancer cells. It was revealed that the effects
of combined treatment with JS-K and Taxol were reversed by an
antioxidant and exacerbated by a pro-oxidant. It was hypothesized
that ROS activation, induced by the combination of JS-K and Taxol,
induced apoptosis in prostate cancer cells (Fig. 5). Further investigation is required to
identify the roles and mechanisms underlying the combination of
JS-K and Taxol.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Funds (grant no. 81272833) of China.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
MNQ and JJL designed the experiments. MNQ, SZ, LZK,
HCT and XZ performed the experiments. MNQ and JJL were involved in
data and statistical analyses. MNQ and JJL wrote the article and
prepared figures. JJL provided guidance and the financial support.
All authors reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Weaver BA: How Taxol/paclitaxel kills
cancer cells. Mol Biol Cell. 25:2677–2681. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rowinsky EK, Cazenave LA and Donehower RC:
Taxol: A novel investigational antimicrotubule agent. J Natl Cancer
Inst. 82:1247–1259. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Danesi R, Figg WD, Reed E and Myers CE:
Paclitaxel (taxol) inhibits protein isoprenylation and induces
apoptosis in PC-3 human prostate cancer cells. Mol Pharmacol.
47:1106–1111. 1995.PubMed/NCBI
|
|
4
|
Obasaju C and Hudes GR: Paclitaxel and
docetaxel in prostate cancer. Hematol Oncol Clin North Am.
15:525–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kopczyńska E: Role of microRNAs in the
resistance of prostate cancer to docetaxel and paclitaxel. Contemp
Oncol (Pozn). 19:423–427. 2015.PubMed/NCBI
|
|
6
|
Sobue S, Mizutani N, Aoyama Y, Kawamoto Y,
Suzuki M, Nozawa Y, Ichihara M and Murate T: Mechanism of
paclitaxel resistance in a human prostate cancer cell line, PC3-PR,
and its sensitization by cabazitaxel. Biochem Biophys Res Commun.
479:808–813. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grimm EA, Sikora AG and Ekmekcioglu S:
Molecular pathways: Inflammation-associated nitric-oxide production
as a cancer-supporting redox mechanism and a potential therapeutic
target. Clin Cancer Res. 19:5557–5563. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Laschak M, Spindler KD, Schrader AJ,
Hessenauer A, Streicher W, Schrader M and Cronauer MV: JS-K, a
glutathione/glutathione S-transferase-activated nitric oxide
releasing prodrug inhibits androgen receptor and WNT-signaling in
prostate cancer cells. BMC Cancer. 12:1302012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tan G, Qiu M, Chen L, Zhang S, Ke L and
Liu J: JS-K, a nitric oxide pro-drug, regulates growth and
apoptosis through the ubiquitin-proteasome pathway in prostate
cancer cells. BMC Cancer. 17:3762017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu M, Chen L, Tan G, Ke L, Zhang S, Chen
H and Liu J: JS-K promotes apoptosis by inducing ROS production in
human prostate cancer cells. Oncol Lett. 13:1137–1142. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qiu M, Ke L, Zhang S, Zeng X, Fang Z and
Liu J: JS-K, a GST-activated nitric oxide donor prodrug, enhances
chemo-sensitivity in renal carcinoma cells and prevents cardiac
myocytes toxicity induced by Doxorubicin. Cancer Chemother
Pharmacol. 80:275–286. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang SQ, Wang C, Chang LM, Zhou KR, Wang
JW, Ke Y, Yang DX, Shi HG, Wang R, Shi XL, et al: Geridonin and
paclitaxel act synergistically to inhibit the proliferation of
gastric cancer cells through ROS-mediated regulation of the
PTEN/PI3K/Akt pathway. Oncotarget. 7:72990–73002. 2016.PubMed/NCBI
|
|
14
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu WS: The signaling mechanism of ROS in
tumor progression. Cancer Metastasis Rev. 25:695–705. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qiu M, Chen L, Tan G, Ke L, Zhang S, Chen
H and Liu J: A reactive oxygen species activation mechanism
contributes to JS-K-induced apoptosis in human bladder cancer
cells. Sci Rep. 5:151042015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nie F, Zhang X, Qi Q, Yang L, Yang Y, Liu
W, Lu N, Wu Z, You Q and Guo Q: Reactive oxygen species
accumulation contributes to gambogic acid-induced apoptosis in
human hepatoma SMMC-7721 cells. Toxicology. 260:60–67. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song X, Xie L, Wang X, Zeng Q, Chen TC,
Wang W and Song X: Temozolomide-perillyl alcohol conjugate induced
reactive oxygen species accumulation contributes to its
cytotoxicity against non-small cell lung cancer. Sci Rep.
6:227622016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brigelius-Flohé R and Maiorino M:
Glutathione peroxidases. Biochim Biophys Acta. 1830:3289–3303.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Giampazolias E and Tait SW: Mitochondria
and the hallmarks of cancer. FEBS J. 283:803–814. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allan LA and Clarke PR: Apoptosis and
autophagy: Regulation of caspase-9 by phosphorylation. FEBS J.
276:6063–6073. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cosentino K and García-Sáez AJ:
Mitochondrial alterations in apoptosis. Chem Phys Lipids.
181:62–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kvansakul M and Hinds MG: The Bcl-2
family: Structures, interactions and targets for drug discovery.
Apoptosis. 20:136–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Siddiqui WA, Ahad A and Ahsan H: The
mystery of BCL2 family: Bcl-2 proteins and apoptosis: An update.
Arch Toxicol. 89:289–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tasyriq M, Najmuldeen IA, In LL, Mohamad
K, Awang K and Hasima N: 7α-hydroxy-β-sitosterol from chisocheton
tomentosus induces apoptosis via dysregulation of cellular
Bax/Bcl-2 ratio and cell cycle arrest by downregulating ERK1/2
activation. Evid Based Complement Alternat Med. 2012:7653162012.
View Article : Google Scholar : PubMed/NCBI
|