Introduction
The prevalence of colorectal cancer (CRC) is
increasing, and CRC has one of the highest cancer morbidities
reported worldwide. Over 47,000 CRC-related deaths occurred in
Japan in 2012; in terms of site-specific cancer mortality, these
figures corresponded to the third highest in men and the highest in
women (1). CRC without lymph node
metastasis can be largely cured by surgical resection alone
(2), but the prognosis becomes poor
once CRC has progressed to lymph node metastasis or distant organ
metastasis. With lymph node metastasis, CRC is more likely to recur
than CRC without lymph node metastasis (3), and distant metastases greatly reduce the
5-year survival rate. The liver is the most common metastatic organ
(4,5),
regardless of whether metastasis occurs in a synchronous or
metachronous manner, and liver metastasis is a major factor
affecting the prognosis of CRC. For CRC treatment strategies, the
prevention of liver metastasis is one of the most important issues.
Thus, multimodality therapies have been rigorously established,
including surgical resection (6),
radiation (7,8), chemotherapy (EORTC trial) (9), and molecular target therapies (PRIME
trial, FIRE-3 trial) (10,11).
Various genetic changes have been reported as
factors involved in the therapeutic effect of anti-epidermal growth
factor receptor (EGFR) monoclonal antibody, including K-ras genomic
mutation (12,13) and EGFR genomic amplification (14,15). These
factors have already been applied in clinical practice, but
treatment failure continues to occur frequently. Recently, the
phosphatase of regenerating liver-3 (PRL-3)-induced activation of
EGFR was reported to be primarily attributable to the
transcriptional downregulation of protein tyrosine phosphatase 1B
(PTP1B), an inhibitory phosphatase for EGFR, and oncogene addiction
to EGFR, which enhances tumour sensitivity in anti-EGFR cancer
therapy in patients with CRC (16).
Accordingly, the PRL-3 gene status has attracted attention as a new
therapeutic biomarker for CRC.
A number of genetic and epigenetic abnormalities are
related to colorectal tumour progression (17). Normal colorectal mucosa changes into
adenoma, accompanied by APC gene mutations (18), and subsequent progression to atypical
adenoma is related to K-ras gene mutations (19). p53 gene mutations finally cause
malignant transformation (20).
Moreover, there have been critical reports that 8q chromosome
amplification is associated with the acquisition of metastatic
potential (21). This locus includes
many oncogene candidates, such as PRL-3 and c-myc. PRL-3 and c-myc
genes exhibit gene amplification accompanied by gene overexpression
in CRC (21), and an association with
the progression of CRC or vascular invasion has been reported
(22). PRL-3 is a member of the
protein tyrosine phosphatase family, the members of which play
important roles in signalling pathways. We recently reported that
PRL-3 genomic amplification and overexpression were confirmed in
gastric cancer, oesophageal squamous cell cancer, and CRC (23–26), and
we revealed a relationship between these changes and cancer
invasion and a poor prognosis. Especially in terms of CRC, PRL-3
genomic gains (defined larger as over two-fold) were significantly
increased in liver metastases (26).
In the current study, the genomic copy numbers of
PRL-3, c-myc and EGFR were simultaneously investigated in both
primary CRC and corresponding liver metastases to clarify the
clinical relevance of these oncogenes during metastatic
formation.
Patients and methods
Patients
A total of 35 patients with histologically confirmed
liver metastases (synchronous, n=11 and metachronous, n=24) of CRC
underwent surgical resection of the liver and primary tumours at
the Department of Surgery, Kitasato University School of Medicine
(Sagamihara, Japan), between 1993 and 2007. Nakayama et al
previously reported 44 cases in this series (26), but 9 of those cases were excluded
because additional exploration was impossible due to a lack of
available DNA from the tumour tissues. The 35 cases in the present
series were composed of stage I (n=3), stage IIA (n=7), stage IIB
(n=1), stage IIIB (n=9), stage IIIC (n=2), stage IVA (n=9), and
stage IVB (n=4) disease at the time of the diagnosis of the primary
CRC.
Clinicopathological factors
The included clinicopathological factors were age,
sex, tumour portion, tumour size, 7th UICC pT factor, 7th UICC pN
factor, ly factor (lymphatic permeation), v factor (vascular
permeation), synchronous or metachronous liver metastasis, and 7th
UICC stage, in addition to the genomic copy statuses of the PRL-3,
c-myc and EGFR genes.
DNA extraction from the tissues
Tissue sections from the tumours (primary CRC or the
corresponding liver metastases from CRC) and corresponding normal
mucosa obtained at least 5 cm from the tumour edge were precisely
dissected on haematoxylin and eosin-stained slides; genomic DNA was
subsequently extracted using a QIAamp® DNA FFPE kit
(Qiagen GmbH, Hilden, Germany).
PRL-3, c-myc, and EGFR genomic gain
statuses in primary CRC and liver metastases
Quantitative polymerase chain reaction (qPCR) was
performed to quantify each genomic amplification in triplicate
samples using iQ™ Supermix and the iCycler iQ™ Real-Time PCR
Detection system (both Bio-Rad Laboratories, Hercules, CA, USA). To
normalise each gene copy number per cell, β-actin was used as an
endogenous reference, as previously described (23,24). The
ΔCt values were calculated as Ct (PRL-3)-Ct (β-actin) for each
sample. The copy number relative to the corresponding normal tissue
was determined as 2−ΔΔCt, where ΔΔCt=ΔCt (tumour)-ΔCt
(corresponding normal tissues). The PCR conditions and sequences
for the β-actin/PRL-3 primers and probes have been previously
described (23) (β-actin: Forward,
5′-tggtgtttgtctctctgactaggtg-3′; reverse,
5′-ctaagtgtgctggggtcttgg-3′; probe, 5′-tggctcgtgtgacaaggccatg-3′,
PRL-3: 5′-aaagattggcgagaacagca-3′; reverse,
5′-atcccagacacacaccgaac-3′; probe,
5′-tggtgtttgtctctctgactaggtg-3′). The EGFR primers and probe were
based on those reported by Moroni et al (15) (forward, 5′-gaattcggatgcagagcttc-3′;
reverse, 5′-gacatgctgcggtgttttc-3′; probe,
5′-ctctgtttcagggcatgaactact-3′). The c-myc primers and probe were
prepared using Primer 3 software (forward,
5′-agcccactggtcctcaagag-3′; reverse, 5′-cttggcagcaggatagtccttc-3′;
probe, 5′-tctccacacatcagcacaactacgcagc-3′). DNA ratios for the
tumour (primary CRC or liver metastasis) tissues (T) relative to
the corresponding normal tissues (N) (T/N ratio) that were equal to
or greater than 2-fold were defined as positive genomic gains.
Immunohistochemical study
Liver metastases from CRC were immunohistochemically
stained for PRL-3, c-myc, EGFR, and E-cadherin using the following
respective antibodies: anti-human/-mouse/-rat PRL-3 antibody Clone
334407, Human c-Myc Antibody Monoclonal Mouse IgG1 Clone #9E10 cat.
no. MAB3696 (both R&D Systems Inc., Minneapolis, MN, USA), Anti
EGFR UltraMAB (R) UM570070 (OriGene Technologies Inc., Rockville,
MD, USA), and G-10:sc-8426 diluted 1:200 (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA).
FFPE tissue blocks of cases 24 and 26 were cut into
thin sections (4 µm thick) that were then deparaffinized with
xylene and dehydrated through a stepwise series of ethanol. For
antigen activation, samples were immersed in pH 6 citrate buffer
and boiled in a microwave for 15 min. The sections were then
incubated in 3% aqueous hydrogen peroxide for 15 min to inactivate
endogenous peroxidases. The sections were incubated with each of
the above primary antibodies overnight at 4°C. The secondary
antibody reaction was performed using the Histofine Simple Stain
MAX-PO (MULTI) kit (Nichirei, Tokyo, Japan) according to the
manufacturer's protocol. Color was developed by incubating with
ImmPACT DAB (Vector Laboratories, Inc., Burlingame, CA, USA) for 5
min. Mayer's Hematoxylin Solution was used to stain nuclei. The
slides were observed with an optical microscope at magnification,
400.
Statistical analysis
Continuous variables were evaluated using a paired
Student t-test, and categorical variables were evaluated using the
Fisher exact test or the Chi square test, as appropriate. P<0.05
was considered to indicate a statistically significant difference.
All the calculations were performed using JMP® 10
software (SAS Institute Inc., Cary, NC, USA).
Results
Genomic gains in PRL-3, c-myc and EGFR
in primary CRC and corresponding liver metastases as evaluated
using Q-PCR
In this study, Q-PCR protocols for the c-myc gene
and the EGFR gene were initially developed to assess the genomic
copy number, and these protocols were used in addition to the
previously reported protocol for the PRL-3 gene (26). Representative genomic quantifications
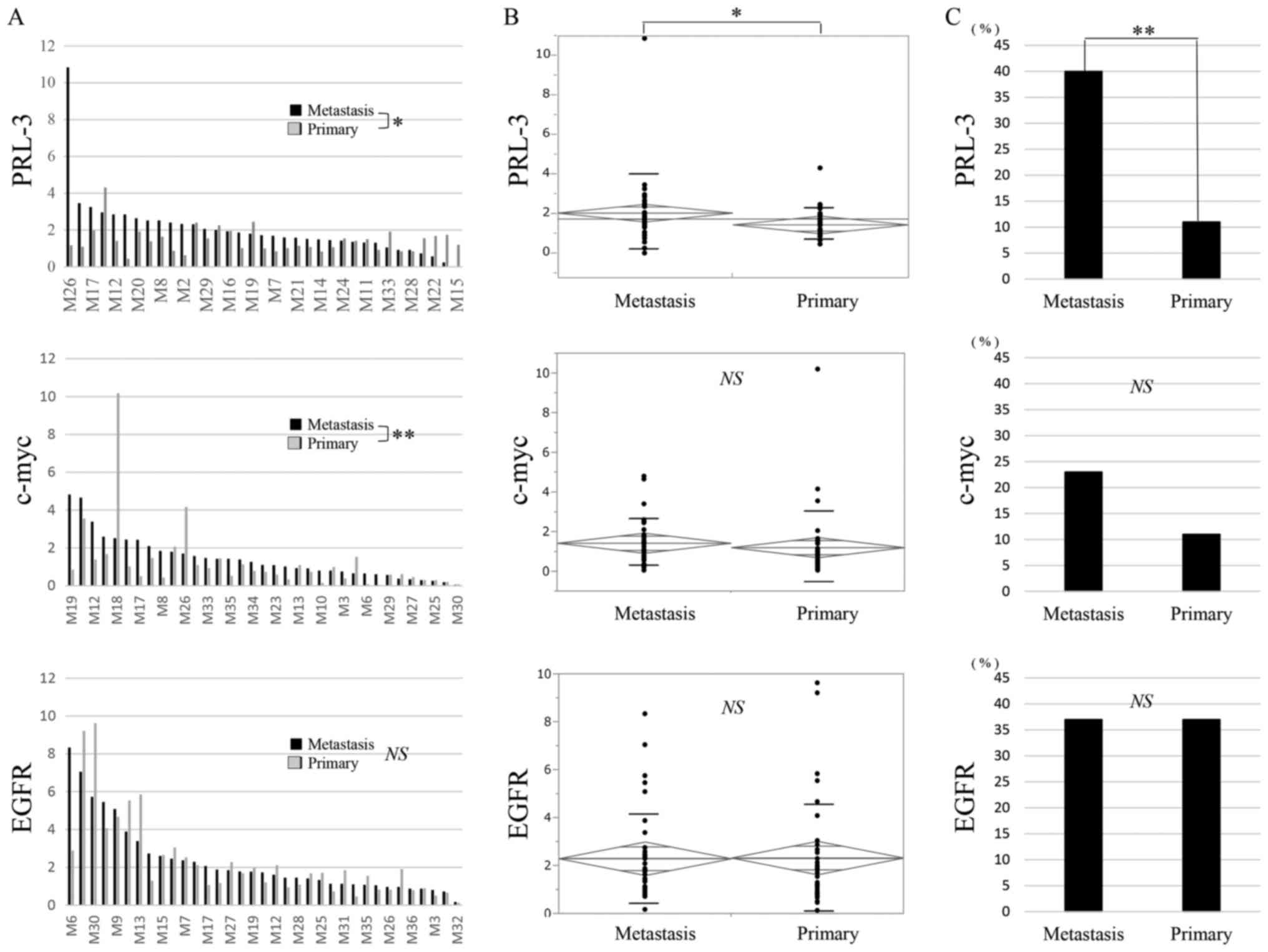
for the c-myc gene and the EGFR gene are shown in Fig. 1A and B, respectively. In Fig. 1A, c-myc genomic gains (2.1-fold for
both) were recognised only in the liver metastases, compared with
the corresponding primary tumours (Primary) and the corresponding
normal tissues (Normal). In Fig. 1B,
EGFR genomic gains (5.7-fold and 9.6-fold, respectively) were
recognised in both the liver metastases and the corresponding
primary tumours (Primary), compared with the corresponding normal
tissues (Normal).
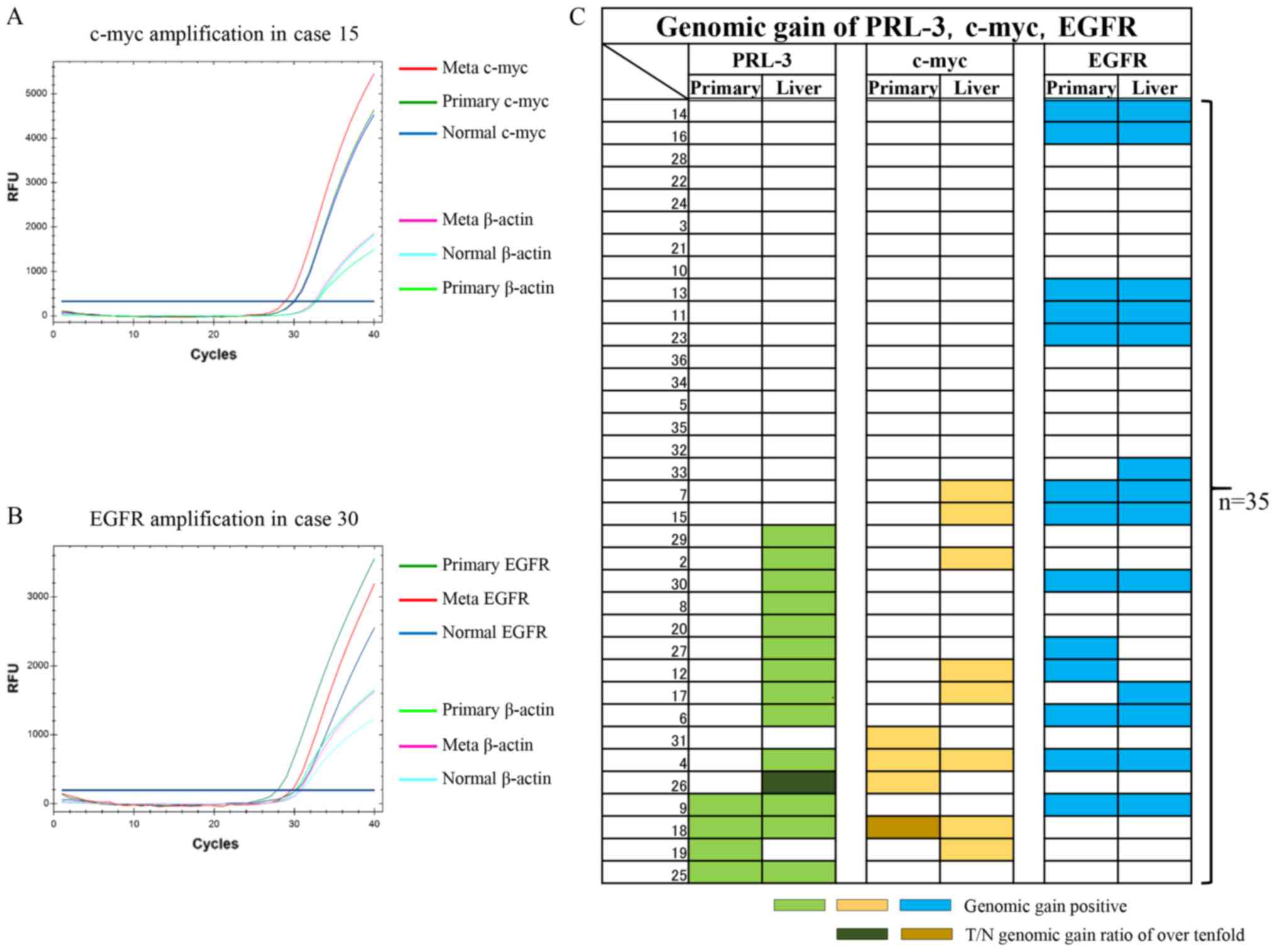 | Figure 1.qPCR analysis examining genomic gains
in c-myc and EGFR genes as well as gains in the PRL-3 gene in 35
primary CRC and corresponding liver metastases. (A) Representative
case with a c-myc gain. The β-actin gene was used as an internal
control for the DNA. Note that the β-actin levels of the Meta,
Primary and Normal tissues were almost the same, unlike the levels
of the c-myc gene. (B) Representative case with an EGFR gain. The
β-actin gene was used as an internal control for the DNA. Note that
the β-actin levels of the Meta, Primary and Normal tissue were
almost the same, unlike the levels of the EGFR gene. (C) Panel
showing the genomic gain statuses of the PRL-3 (light green), c-myc
(yellow) and EGFR (blue) genes using coloured bars. The two
dark-coloured bars show T/N genomic gain ratios of over 10-fold.
qPCR, quantitative polymerase chain reaction analysis; CRC,
colorectal cancer; EGFR, epidermal growth factor receptor; PRL-3,
phosphatase of regenerating liver-3; RFU, relative fluorescence
unit. |
The genomic gain statuses of c-myc and EGFR were
then compared with the PRL-3 genomic gain status in the 35
previously reported primary CRC and corresponding liver metastases
(Fig. 1C) (26). In the primary CRC tumours, genomic
gains in PRL-3, c-myc, and EGFR were seen in 4 (11%), 4 (11%), and
13 (37%) cases, respectively. Among the 3 genes, EGFR genomic gains
were the most frequent, followed by PCR-3 and c-myc, in the primary
CRC tissues. Genomic gains in the 3 genes were mutually exclusive
in the primary CRC tissues. As a result, a genomic gain in any of
the 3 genes was observed in 18 cases (51%).
In the corresponding liver metastases, genomic gains
in PRL-3, c-myc, and EGFR were seen in 14 (40%), 8 (23%), and 13
(37%) cases, respectively. A genomic gain in any of the 3 genes was
observed in 23 cases (66%). Among the 3 genes, a genomic gain in
PRL-3 was the most frequent, followed by EGFR and c-myc. A genomic
gain in c-myc was partially redundant with PRL-3 in the liver
metastases. Interestingly, genomic gains in the EGFR gene were
consistent between both primary CRC and liver metastases
(P=0.0000008).
T/N ratios of PRL-3, c-myc and EGFR
genetic copies between primary CRC and corresponding liver
metastases
The T/N ratios of the PRL-3 gene are shown for
primary CRC and the corresponding liver metastases in Fig. 2A. The T/N ratio ranged from 0.4 to 4.3
(mean, 1.4) in the primary CRC and from 0 to 10.8 (mean, 2.0) in
the corresponding liver metastases. A significant difference in the
T/N ratios of the PRL-3 gene was observed between the primary CRC
and the corresponding liver metastases when compared using a t-test
(Fig. 2A; P=0.03) and an analysis of
variance (ANOVA) (Fig. 2B; P=0.03). A
genomic gain in PRL-3 was more frequently seen in the liver
metastases than in the corresponding primary CRC (Fig. 2C; P=0.004).
The T/N ratios of the c-myc gene are also shown for
primary CRC and the corresponding liver metastases in Fig. 2A. The T/N ratio ranged from 0.05 to
10.2 (mean, 1.2) in the primary CRC and from 0.05 to 4.8 (mean,
1.4) in the corresponding liver metastases. A significant
difference in the T/N ratios of the c-myc gene was observed between
the primary CRC and the corresponding liver metastases when
compared using a Wilcoxon signed-rank sum analysis (Fig. 2A; P=0.009), but a significant
difference was not seen using an ANOVA (Fig. 2B). A genomic gain in c-myc was more
frequently seen in the liver metastases than in the corresponding
primary CRC (Fig. 2C).
The T/N ratios of the EGFR gene are also shown for
primary CRC and the corresponding liver metastases in Fig. 2A. The T/N ratio ranged from 0.1 to 9.6
(mean, 2.3) in the primary CRC and from 0.2 to 8.3 (mean, 2.3) in
the corresponding liver metastases. A significant difference in the
T/N ratio of the EGFR gene was not observed between the primary CRC
and the corresponding liver metastases when compared using a
Wilcoxon analysis (Fig. 2A) or an
ANOVA (Fig. 2B). The genomic gain in
EGFR was equal between the primary CRC and the corresponding liver
metastases (Fig. 2C).
Clinicopathological analysis of PRL-3,
c-myc and EGFR genomic gains in primary CRC tissues and
corresponding liver metastases
We then investigated the associations between PRL-3,
c-myc, and EGFR genomic gains and clinicopathological factors in
the 35 primary CRC. In the primary CRC tissues, genomic gains in
the PRL-3 and c-myc genes were not correlated with any factors,
while that of EGFR was significantly associated with tumour size
(Table I, P=0.04). In the liver
metastases, the c-myc genomic status was significantly associated
with the v factor (P<0.01), and the EGFR genomic status was
significantly correlated with tumour size (P=0.04).
 | Table I.Univariate relations of
clinicopathological factors to the three genomic gain in liver
metastasis of CRC. |
Table I.
Univariate relations of
clinicopathological factors to the three genomic gain in liver
metastasis of CRC.
|
|
| PRL-3 genomic gain
(%) | c-myc genomic gain
(%) | EGFR genomic gain
(%) |
|---|
|
|
|
|
|
|
|---|
|
| Number (%) | Primary |
| Liver |
| Primary |
| Liver |
| Primary |
| Liver |
|
|---|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|---|
| Patient
variable | n=35 | (−) n=31 | (+) n=4 | P-value | (−) n=21 | (+) n=14 | P-value | (−) n=31 | (+) n=4 | P-value | (−) n=27 | (+) n=8 | P-value | (−) n=22 | (+) n=13 | P-value | (−) n=22 | (+) n=13 | P-value |
|---|
| Age, years median
(range) | 62 (37–82) |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
≥60 | 19 (54) | 16 (46) | 3 (9) |
| 13 (37) | 6 (17) |
| 16 (46) | 3 (9) |
| 14 (40) | 5 (14) |
| 14 (40) | 5 (14) |
| 13 (37) | 6 (17) |
|
|
>60 | 16 (46) | 15 (43) | 1 (3) |
| 8 (23) | 8 (23) |
| 15 (43) | 1 (3) |
| 13 (37) | 3 (9) |
| 8 (23) | 8 (23) |
| 9 (26) | 7 (20) |
|
| Sex |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
|
|
|
| NS |
|
Male | 24 (69) | 22 (63) | 2 (6) |
| 14 (37) | 10 (29) |
| 22 (63) | 2 (6) |
| 18 (51) | 6 (17) |
| 14 (40) | 10 (29) |
| 15 (43) | 9 (26) |
|
|
Female | 11 (34) | 9 (26) | 2 (6) |
| 7 (20) | 4 (11) |
| 9 (26) | 2 (6) |
| 9 (26) | 2 (6) |
| 8 (23) | 3 (9) |
| 7 (20) | 4 (11) |
|
| Tumour size, cm
(mean ± SD) | 4.72 (±1.97) |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| 0.04 |
|
| 0.04 |
|
≥4.7 | 19 (54) | 17 (49) | 2 (6) |
| 12 (34) | 7 (20) |
| 18 (51) | 1 (3) |
| 14 (40) | 5 (14) |
| 9 (26) | 10 (28) |
| 9 (26) | 10 (28) |
|
|
>4.7 | 16 (46) | 14 (40) | 2 (6) |
| 9 (26) | 7 (20) |
| 13 (37) | 3 (9) |
| 13 (37) | 3 (9) |
| 13 (37) | 3 (9) |
| 13 (37) | 3 (9) |
|
| Tumour
position |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
Colon | 28 (80) | 25 (71) | 3 (9) |
| 16 (46) | 12 (34) |
| 22 (63) | 6 (17) |
| 24 (69) | 4 (11) |
| 17 (29) | 11 (31) |
| 18 (51) | 10 (29) |
|
|
Rectum | 7 (20) | 6 (17) | 1 (3) |
| 5 (14) | 2 (6) |
| 5 (14) | 2 (6) |
| 7 (20) | 0 |
| 5 (14) | 2 (6) |
| 4 (11) | 3 (9) |
|
| N factor |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
| N0 | 12 (34) | 11 (31) | 1 (3) |
| 7 (20) | 5 (14) |
| 9 (26) | 3 (9) |
| 8 (23) | 4 (11) |
| 7 (20) | 5 (14) |
| 8 (23) | 4 (11) |
|
| N1 | 13 (37) | 11 (31) | 2 (6) |
| 9 (26) | 4 (11) |
| 12 (34) | 1 (3) |
| 10 (29) | 3 (9) |
| 8 (23) | 4 (11) |
| 9 (26) | 4 (11) |
|
| N2 | 10 (29) | 9 (26) | 1 (3) |
| 5 (14) | 5 (14) |
| 10 (29) | 0 |
| 9 (26) | 1 (3) |
| 6 (17) | 4 (11) |
| 5 (14) | 5 (14) |
|
| ly factor |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
ly0 | 2 (6) | 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
|
|
ly1 | 12 (34) | 12 (34) | 0 |
| 8 (23) | 4 (11) |
| 9 (26) | 3 (9) |
| 9 (26) | 3 (9) |
| 6 (17) | 6 (17) |
| 6 (17) | 6 (17) |
|
|
ly2 | 13 (37) | 8 (23) | 4 (11) |
| 6 (17) | 6 (17) |
| 11 (31) | 1 (3) |
| 9 (26) | 3 (9) |
| 7 (20) | 5 (14) |
| 7 (20) | 5 (14) |
|
|
ly3 | 8 (23) | 8 (23) | 0 |
| 4 (11) | 4 (11) |
| 8 (23) | 0 |
| 6 (17) | 2 (6) |
| 6 (17) | 2 (6) |
| 6 (17) | 2 (6) |
|
| v factor |
|
|
| NS |
|
| NS |
|
| NS |
|
| <0.01 |
|
| NS |
|
| NS |
| v0 | 3 (9) | 3 (9) | 0 |
| 3 (9) | 0 |
| 2 (6) | 1 (3) |
| 3 (9) | 0 |
| 3 (9) | 0 |
| 3 (9) | 0 |
|
| v1 | 10 (29) | 9 (26) | 1 (3) |
| 7 (20) | 3 (9) |
| 10 (29) | 0 |
| 9 (26) | 1 (3) |
| 7 (20) | 3 (9) |
| 9 (26) | 4 (11) |
|
| v2 | 13 (37) | 11 (31) | 2 (6) |
| 7 (20) | 6 (17) |
| 12 (34) | 1 (3) |
| 11 (31) | 2 (6) |
| 6 (17) | 7 (20) |
| 7 (20) | 6 (17) |
|
| v3 | 9 (26) | 8 (23) | 1 (3) |
| 4 (11) | 5 (14) |
| 7 (20) | 2 (6) |
| 4 (11) | 5 (14) |
| 6 (17) | 3 (9) |
| 6 (17) | 3 (9) |
|
| 7th UICC stage |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
| I | 3 (9) | 3 (9) | 0 |
| 1 (3) | 2 (6) |
| 2 (6) | 1 (3) |
| 2 (6) | 1 (3) |
| 3 (9) | 0 |
| 3 (9) | 0 |
|
|
IIA | 7 (20) | 7 (20) | 0 |
| 5 (14) | 2 (6) |
| 7 (20) | 0 |
| 5 (14) | 2 (6) |
| 2 (6) | 5 (14) |
| 3 (9) | 4 (11) |
|
|
IIB | 1 (3) | 0 | 1 (3) |
| 0 | 1 (3) |
| 0 | 1 (3) |
| 0 | 1 (3) |
| 1 (3) | 0 |
| 1 (3) | 0 |
|
|
IIC | 0 | 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
|
|
IIIA | 0 | 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
| 0 | 0 |
|
|
IIIB | 9 (26) | 6 (17) | 3 (9) |
| 3 (9) | 6 (17) |
| 8 (23) | 1 (3) |
| 6 (17) | 3 (9) |
| 5 (14) | 4 (11) |
| 6 (17) | 3 (9) |
|
|
IIIC | 2 (6) | 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 2 (6) | 0 |
| 1 (3) | 1 (3) |
|
|
IVA | 9 (26) | 9 (26) | 0 |
| 7 (20) | 2 (6) |
| 8 (23) | 1 (3) |
| 8 (23) | 1 (3) |
| 6 (17) | 3 (9) |
| 5 (14) | 4 (11) |
|
|
IVB | 4 (11) | 4 (11) | 0 |
| 3 (9) | 1 (3) |
| 4 (11) | 0 |
| 4 (11) | 0 |
| 3 (9) | 1 (3) |
| 3 (9) | 1 (3) |
|
| Liver
metastatic |
|
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
| NS |
|
Simultaneous | 11 (31) | 11 (31) | 0 |
| 8 (23) | 3 (9) |
| 10 (29) | 1 (3) |
| 10 (29) | 1 (3) |
| 8 (23) | 3 (9) |
| 7 (20) | 4 (11) |
|
|
Metachronous | 24 (69) | 20 (57) | 4 (11) |
| 13 (37) | 11 (31) |
| 21 (88) | 3 (9) |
| 21 (88) | 7 (20) |
| 14 (40) | 10 (29) |
| 15 (43) | 9 (26) |
|
Based on the clinical data for the liver metastases,
the genomic gains in PRL-3 and EGFR were relatively independent,
while that of c-myc was redundant with that of PRL-3 in the liver
metastases. The former observation reflects the central role of the
PRL-3/EGFR pathway in tumour progression through PRL-3-induced EGFR
oncogene addiction (16), while the
latter may reflect the proximity of c-myc to the PRL-3 genomic
locus (21).
Immunohistochemical experiments for
liver metastasis
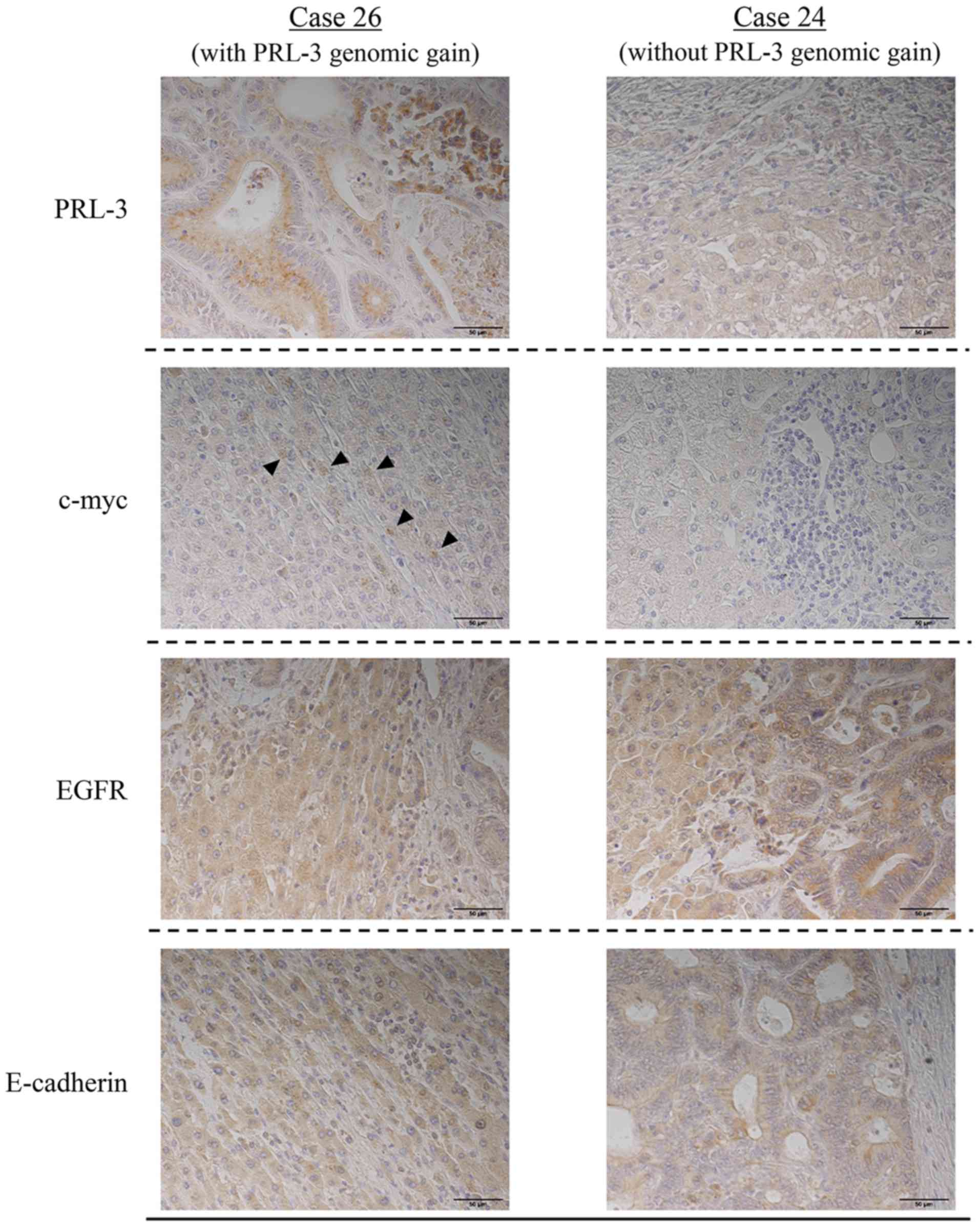
Cases 26 and 24, which were representative of the
presence and absence of a PRL-3 genomic gain, respectively, were
used to investigate changes in the protein expression of c-myc or
EGFR according to PRL-3 genomic gain (Fig. 3). Neither of these cases exhibited a
c-myc or an EGFR genomic gain. PRL-3 protein was observed in case
26 but not in case 24, suggesting a correlation with its gene
amplification. The expression of EGFR protein was observed in both
case 26 and 24, while c-myc protein was only expressed in case
26.
Moreover, the expression of E-cadherin protein was
examined in liver metastases to clarify the relationship between
EMT markers and amplification of the PRL-3 gene. E-cadherin protein
was observed in both cases 26 and 24, suggesting that the
expressions of PRL-3 protein and E-cadherin protein were not
correlated.
Discussion
Gene copy number changes are major aberrations that
occur mostly upstream of cancer cells, and their potential role as
molecular targets in cancer therapy has been reported (27). Nakayama et al (26) showed, for the first time, that a
genomic gain in PRL-3 is associated with lymph node metastasis and
liver metastasis in CRC. In the current study, Q-PCR analyses for
the c-myc gene and the EGFR gene were additionally performed using
both liver metastases and the corresponding primary tumours, and
the results were compared with the genomic gain status of PRL-3 to
investigate the relationship among the 3 genes during liver
metastasis from CRC.
In our previous experiment, the genomic gain in
PRL-3 in a primary tumour was associated with the liver metastasis
of CRC, progression, a poor tumour grade, and a poor prognosis
(26); in the present study, however,
no significant correlations between a genomic gain in PRL-3 and
clinicopathological factors were observed except for liver
metastasis because of the relatively small number of samples that
were tested. Nevertheless, the genomic copy numbers of PRL-3 in the
liver metastases were significantly higher than those in the
corresponding primary tumours (P=0.004) (Fig. 2C). Because of this result, we
additionally examined the genomic gains in the c-myc and EGFR genes
in the same patients.
Both PRL-3 and c-myc are located adjacently on
chromosome 8q24. Buffart et al (21) reported that the gene amplification of
PRL-3 and c-myc in advanced CRC was observed more frequently than
that in early-stage CRC, but in their heat map, the copy number
ratio of these two genes did not necessarily coincide in the same
patient. Moreover, Saha et al (28) showed that gene amplification of c-myc
was absent in cases with PRL-3 amplified on chromosome 8q24.3, and
Zimmerman et al (29)
confirmed an increase in c-myc protein in a PRL-3-knockout mouse
model. Hence, these two genes are not necessarily synchronised for
genomic amplification even though they are located on the same
chromosome, indicating a complementary role in liver metastasis.
Our results supported this hypothesis, since PRL-3 and c-myc
genomic gene gains were both present in the primary tumour tissue
in only once case (Fig. 1C). On the
other hand, the gains in these two genes were often redundant in
liver metastases. In terms of gene amplification, the relationship
between the PRL-3 and c-myc genes was considered to be
complementary prior to liver metastasis and synergistic thereafter.
Gains in both the PRL-3 and the c-myc genes might occur together at
the time of liver metastasis, while genomic gains in these two
genes might occur separately in primary CRC. The PRL-3 genomic gain
is likely to contribute more to the liver metastatic capacity than
a gain in c-myc because 1 case with liver metastasis from a stage I
primary cancer exhibited the highest genomic gain in PRL-3, but the
genomic gain in c-myc was relatively low in the corresponding liver
metastasis (Fig. 1C).
In this study, the genomic gain in EGFR was
correlated with the tumour size (P=0.04) in both the liver
metastases and the corresponding primary tumours (Table I). EGFR gene activation is known to be
important for tumour progression, and EGFR-targeting therapies have
been rigorously established for CRC and non-small cell lung cancer
in clinical practice. Overexpression of the EGFR gene is frequently
recognised in CRC (25–80%) and is associated with aggressive
disease and a poor prognosis (30,31). In
our study, a genomic gain in EGFR was not associated with the
acquisition of liver metastatic potential but was useful as a
predictor of liver metastasis, since it was consistent between the
liver metastases and the corresponding primary tumours
(P=0.0000008).
In a combination analysis of the 3 genes, their
positive gains were exclusive of each other in primary CRC. The
PRL-3 gene exerts various functions, such as proliferation and the
growth and inhibition of apoptosis signals, through the activation
of EGFR and c-myc. In the present study, 23 cases (66%) exhibited
gains in any of the 3 genes; moreover, the genomic gains in c-myc
and EGFR in liver metastases were significantly associated with
vascular invasion factor and tumour growth, respectively. The
PRL-3, c-myc, and EGFR genes are likely to have complementary roles
in the proliferation of liver metastases in CRC. In liver
metastasis, a PRL-3 genomic gain is correlated with its own protein
expression, and c-myc protein was induced by the PRL-3 protein or a
genomic gain (Fig. 3). PRL-3 protein
might increase the expression of c-myc protein in liver metastasis
independently of the EGFR protein. PRL-3-targeted therapy, which
targets a point furthest upstream in the signalling cascade, may be
effective against CRC with liver metastasis as a new treatment to
improve prognosis in patients with a PRL-3 genomic gain.
Anti-EGFR monoclonal antibodies have been used in
patients with unresectable advanced or recurrent CRC who are
negative for RAS mutations. The K-ras gene wild type is seen in
60–70% of CRC with metastasis; however, the actual response rate to
anti-EGFR monoclonal antibody was reported to be 10–40% (32). Moreover, some reports have indicated
that only about 10% of patients with CRC and metastasis who are
resistant to chemotherapy responded to treatment with P-mab or
cetuximab (33,34). Therefore, it is necessary to predict
responders to anti-EGFR monoclonal antibody therapy. Balin-Gauthier
et al (35) reported that a
response to cetuximab was correlated with the level of
phosphorylated EGFR but not with the level of EGFR expression. On
the other hand, another report indicated that the response to
anti-EGFR treatment is influenced by the EGFR amplification status
(15). Al-Aidaroos et al
(16) showed that in mice, PRL-3
activates EGFR leading to EGFR oncogene addiction and is a
molecular marker that is indicative of a response to anti-EGFR
monoclonal drugs. Intriguingly, there have been reports describing
the effects of monoclonal antibodies against EGFR, if the EGFR gene
is amplified, even in the presence of a K-ras exon 2 mutation
(16). Whether the expression of
PRL-3 or EGFR has a greater effect on anti-EGFR treatments in CRC
cell lines remains unclear. However, considering that PRL-3 is
located upstream of EGFR, the PRL-3 gene could be a superior
therapeutic target than EGFR in anti-EGFR monoclonal antibody
therapy. In a preliminary investigation performed at our hospital,
anti-EGFR antibody treatment tended to have a good therapeutic
effect (partial response) in cases with a high expression of PRL-3
(data not shown), similar to a recent report that actually
demonstrated this effect in a prospective cohort study (16).
Limitations
First, the number of target cancer tissues was
limited, and the tissues had been archived. Several effects arising
from the long-term preservation of tissues used for DNA extraction
and tissue immunostaining are conceivable. In the future, we would
like to perform additional studies using a larger number of freshly
obtained CRC tissues and corresponding liver metastases. Second,
all the experiments were performed in vitro. Since in
vivo experiments are necessary for the development of clinical
applications of PRL-3 as a biomarker, we plan to perform
prospective research involving such investigations in the near
future.
Finally, we summarised the PRL-3/EGFR/K-ras pathway
based on previous reports (Fig. 4).
Numerous reports have indicated that EGFR is involved in cell
proliferation and survival through individual critical pathways,
such as MAPK, PI3K/Akt, and JAK/STAT, as well as those of feedback
mechanisms (36,37). PRL-3 has been reported to be involved
in the activation of EGFR through the inhibition of PTP1B (12), the promotion of cell motility and
invasion related to Src and Csk (38,39), and
the up-regulation of the PI3K/Akt pathway through the
down-regulation of phosphatase and tensin homologue deleted
chromosome 10 (PTEN), which are inhibitors of the PI3K pathway for
the dephosphorylation of PI3K products (40).
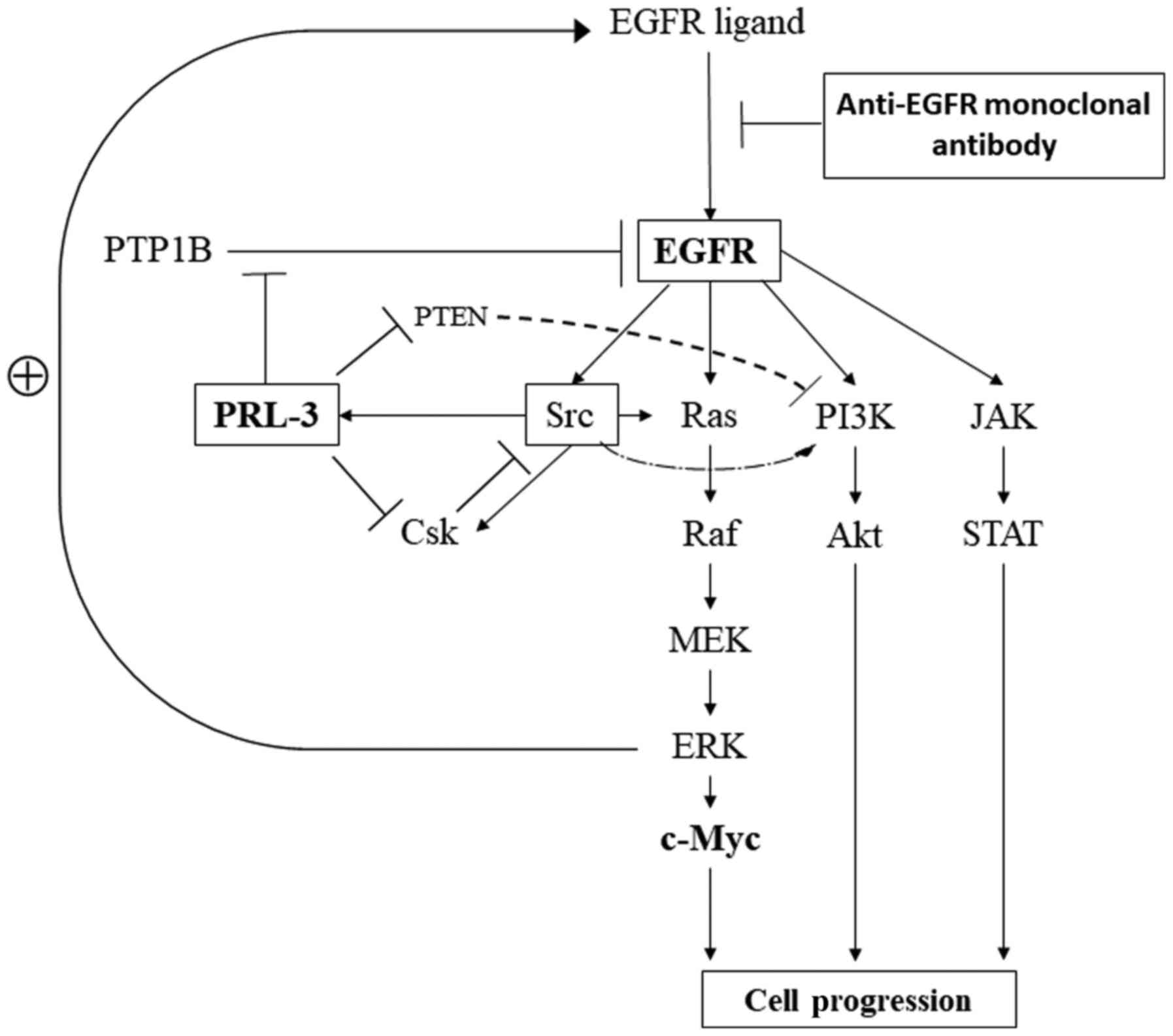 | Figure 4.Signalling pathways related to
PRL-3/EGFR/c-myc genes in cancer cells. EGFR, epidermal growth
factor receptor; PTP1B, protein tyrosine phosphatase 1B; PTEN,
phosphatase and tensin homolog; PRL-3, phosphatase of regenerating
liver-3; PI3K, phospho-inositide 3 kinase; JAK, Janus kinase; Csk,
C-terminal Src kinase; STAT, signal transducers and activators of
transcription; MEK, mitogen-activated protein kinase kinase; ERK,
extracellular signal-regulated kinase; EGFR, epidermal growth
factor receptor. |
Genomic gain aberrations in the copy numbers of
PRL-3, c-myc and EGFR are frequently observed in liver metastases
from CRC. These results may be beneficial for the treatment of CRC
patients with liver metastasis and may be useful for the
identification of patients who are likely to respond well to
anticancer therapies in the near future.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analysed during this study are
included in the published article.
Authors' contributions
Conception and design: TT, KYa and MW undertook
study conception and design and TT, TK, KYo, SI, NN, HKaw, HKat, TS
and TN were responsible for the acquisition and analysis and
interpretation of data. TT and KYa drafted the article and revised
it critically for important intellectual content. TT, KYa, TK, KYo,
SI, NN, HKaw, HKat, TS, TN and MW gave final approval for this
version to be published.
Ethics approval and consent to
participate
This study was conducted in accordance with the
Declaration of Helsinki and was approved by the Research Ethics
Committee of Kitasato University School of Medicine. All patients
agreed to the use of their samples in scientific research. And,
written informed consents for pathological investigation were
obtained from all patients.
Patient consent for publication
Informed consent for publication was obtained from
all patients.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Watanabe T, Itabashi M, Shimada Y, Tanaka
S, Ito Y, Ajioka Y, Hamaguchi T, Hyodo I, Igarashi M, Ishida H, et
al: Japanese Society for Cancer of the Colon and Rectum (JSCCR)
guidelines 2014 for treatment of colorectal cancer. Int J Clin
Oncol. 20:207–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Katoh H, Yamashita K, Wang G, Sato T,
Nakamura T and Watanabe M: Anastomotic leakage contributes to the
risk for systemic recurrence in stage II colorectal cancer. J
Gastrointest Surg. 15:120–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Katoh H, Yamashita K, Wang G, Sato T,
Nakamura T and Watanabe M: Prognostic significance of preoperative
bowel obstruction in stage III colorectal cancer. Ann Surg Oncol.
18:2432–2441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yamashita K and Watanabe M: Clinical
significance of tumor markers and an emerging perspective on
colorectal cancer. Cancer Sci. 100:195–199. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Katoh H, Yamashita K, Sato T, Ozawa H,
Nakamura T and Watanabe M: Prognostic significance of peritoneal
tumour cells identified at surgery for colorectal cancer. Br J
Surg. 96:769–777. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kato T, Yasui K, Hirai T, Kanemitsu Y,
Mori T, Sugihara K, Mochizuki H and Yamamoto J: Therapeutic results
for hepatic metastasis of colorectal cancer with special reference
to effectiveness of hepatectomy: Analysis of prognostic factors for
763 cases recorded at 18 institutions. Dis Colon Rectum. 46 10
Suppl:S22–S31. 2003.PubMed/NCBI
|
|
7
|
Nakamura T, Yamashita K, Sato T, Ema A,
Naito M and Watanabe M: Neoadjuvant chemoradiation therapy using
concurrent S-1 and irinotecan in rectal cancer: Impact on long-term
clinical outcomes and prognostic factors. Int J Radiat Oncol Biol
Phys. 89:547–555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wong SL, Mangu PB, Choti MA, Crocenzi TS,
Dodd GD III, Dorfman GS, Eng C, Fong Y, Giusti AF, Lu D, et al:
American Society of Clinical Oncology 2009 clinical evidence review
on radiofrequency ablation of hepatic metastases from colorectal
cancer. J Clin Oncol. 28:493–508. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nordlinger B, Sorbye H, Glimelius B,
Poston GJ, Schlag PM, Rougier P, Bechstein WO, Primrose JN, Walpole
ET, Finch-Jones M, et al: Perioperative chemotherapy with FOLFOX4
and surgery versus surgery alone for resectable liver metastases
from colorectal cancer (EORTC Intergroup trial 40983): A randomised
controlled trial. Lancet. 371:1007–1016. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Douillard JY, Siena S, Cassidy J,
Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham
D, Jassem J, et al: Final results from PRIME: Randomized phase III
study of panitumumab with FOLFOX4 for first-line treatment of
metastatic colorectal cancer. Ann Oncol. 25:1346–1355. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Heinemann V, von Weikersthal LF, Decker T,
Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmuller
C, Kahl C, et al: FOLFIRI plus cetuximab versus FOLFIRI plus
bevacizumab as first-line treatment for patients with metastatic
colorectal cancer (FIRE-3): A randomised, open-label, phase 3
trial. Lancet Oncol. 15:1065–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diaz LA Jr, Williams RT, Wu J, Kinde I,
Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al:
The molecular evolution of acquired resistance to targeted EGFR
blockade in colorectal cancers. Nature. 486:537–540. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Amado RG, Wolf M, Peeters M, Van Cutsem E,
Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, et
al: Wild-type KRAS is required for panitumumab efficacy in patients
with metastatic colorectal cancer. J Clin Oncol. 26:1626–1634.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Moroni M, Veronese S, Benvenuti S,
Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M,
Siena S and Bardelli A: Gene copy number for epidermal growth
factor receptor (EGFR) and clinical response to antiEGFR treatment
in colorectal cancer: A cohort study. Lancet Oncol. 6:279–286.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Al-Aidaroos AQ, Yuen HF, Guo K, Zhang SD,
Chung TH, Chng WJ and Zeng Q: Metastasis-associated PRL-3 induces
EGFR activation and addiction in cancer cells. J Clin Invest.
123:3459–3471. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vogelstein B and Kinzler KW: The path to
cancer-three strikes and you're out. N Engl J Med. 373:1895–1898.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ashton-Rickardt PG, Dunlop MG, Nakamura Y,
Morris RG, Purdie CA, Steel CM, Evans HJ, Bird CC and Wyllie AH:
High frequency of APC loss in sporadic colorectal carcinoma due to
breaks clustered in 5q21-22. Oncogene. 4:1169–1174. 1989.PubMed/NCBI
|
|
19
|
Vogelstein B, Fearon ER, Hamilton SR, Kern
SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos
JL: Genetic alterations during colorectal-tumor development. N Engl
J Med. 319:525–532. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Linsalata M, Notarnicola M, Caruso MG, Di
Leo A, Guerra V and Russo F: Polyamine biosynthesis in relation to
K-ras and P-53 mutations in colorectal carcinoma. Scand J
Gastroenterol. 39:470–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Buffart TE, Coffa J, Hermsen MA, Carvalho
B, van der Sijp JR, Ylstra B, Pals G, Schouten JP and Meijer GA:
DNA copy number changes at 8q11-24 in metastasized colorectal
cancer. Cell Oncol. 27:57–65. 2005.PubMed/NCBI
|
|
22
|
Sugimachi K, Niida A, Yamamoto K,
Shimamura T, Imoto S, Iinuma H, Shinden Y, Eguchi H, Sudo T,
Watanabe M, et al: Allelic imbalance at an 8q24 oncogenic SNP is
involved in activating MYC in human colorectal cancer. Ann Surg
Oncol. 21 Suppl 4:S515–S521. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ooki A, Yamashita K, Kikuchi S, Sakuramoto
S, Katada N and Watanabe M: Phosphatase of regenerating liver-3 as
a convergent therapeutic target for lymph node metastasis in
esophageal squamous cell carcinoma. Int J Cancer. 127:543–554.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ooki A, Yamashita K, Kikuchi S, Sakuramoto
S, Katada N, Waraya M, Kawamata H, Nishimiya H, Nakamura K and
Watanabe M: Therapeutic potential of PRL-3 targeting and clinical
significance of PRL-3 genomic amplification in gastric cancer. BMC
Cancer. 11:1222011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hatate K, Yamashita K, Hirai K, Kumamoto
H, Sato T, Ozawa H, Nakamura T, Onozato W, Kokuba Y, Ihara A and
Watanabe M: Liver metastasis of colorectal cancer by
protein-tyrosine phosphatase type 4A, 3 (PRL-3) is mediated through
lymph node metastasis and elevated serum tumor markers such as CEA
and CA19-9. Oncol Rep. 20:737–743. 2008.PubMed/NCBI
|
|
26
|
Nakayama N, Yamashita K, Tanaka T,
Kawamata H, Ooki A, Sato T, Nakamura T and Watanabe M: Genomic gain
of the PRL-3 gene may represent poor prognosis of primary
colorectal cancer, and associate with liver metastasis. Clin Exp
Metastasis. 33:3–13. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saha S, Bardelli A, Buckhaults P,
Velculescu VE, Rago C, St Croix B, Romans KE, Choti MA, Lengauer C,
Kinzler KW and Vogelstein B: A phosphatase associated with
metastasis of colorectal cancer. Science. 294:1343–1346. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zimmerman MW, Homanics GE and Lazo JS:
Targeted deletion of the metastasis-associated phosphatase Ptp4a3
(PRL-3) suppresses murine colon cancer. PLoS One. 8:e583002013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee JC, Wang ST, Chow NH and Yang HB:
Investigation of the prognostic value of coexpressed erbB family
members for the survival of colorectal cancer patients after
curative surgery. Eur J Cancer. 38:1065–1071. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Porebska I, Harlozińska A and Bojarowski
T: Expression of the tyrosine kinase activity growth factor
receptors (EGFR, ERB B2, ERB B3) in colorectal adenocarcinomas and
adenomas. Tumour Biol. 21:105–115. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Allegra CJ, Jessup JM, Somerfield MR,
Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF and
Schilsky RL: American Society of Clinical Oncology provisional
clinical opinion: Testing for KRAS gene mutations in patients with
metastatic colorectal carcinoma to predict response to
anti-epidermal growth factor receptor monoclonal antibody therapy.
J Clin Oncol. 27:2091–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saltz LB, Meropol NJ, Loehrer PJ Sr,
Needle MN, Kopit J and Mayer RJ: Phase II trial of cetuximab in
patients with refractory colorectal cancer that expresses the
epidermal growth factor receptor. J Clin Oncol. 22:1201–1208. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cunningham D, Humblet Y, Siena S, Khayat
D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype
C, et al: Cetuximab monotherapy and cetuximab plus irinotecan in
irinotecan-refractory metastatic colorectal cancer. N Engl J Med.
351:337–345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Balin-Gauthier D, Delord JP, Rochaix P,
Mallard V, Thomas F, Hennebelle I, Bugat R, Canal P and Allal C: In
vivo and in vitro antitumor activity of oxaliplatin in combination
with cetuximab in human colorectal tumor cell lines expressing
different level of EGFR. Cancer Chemother Pharmacol. 57:709–718.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Fiordalisi JJ, Dewar BJ, Graves LM,
Madigan JP and Cox AD: Src-mediated phosphorylation of the tyrosine
phosphatase PRL-3 is required for PRL-3 promotion of Rho
activation, motility and invasion. PLoS One. 8:e643092013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liang F, Liang J, Wang WQ, Sun JP, Udho E
and Zhang ZY: PRL3 promotes cell invasion and proliferation by
down-regulation of Csk leading to Src activation. J Biol Chem.
282:5413–5419. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang H, Quah SY, Dong JM, Manser E, Tang
JP and Zeng Q: PRL-3 down-regulates PTEN expression and signals
through PI3K to promote epithelial-mesenchymal transition. Cancer
Res. 67:2922–2926. 2007. View Article : Google Scholar : PubMed/NCBI
|