Introduction
Fundamental units formed by interacting proteins
have been demonstrated to be responsible for implementing important
biological processes within cells (1). The identification of protein complexes
(modules or pathways) contributes to understanding the complex
formation and the higher level organization of cells, and the
dysregulation of protein complexes might be associated with the
development of disease. With the advance of microarray techniques,
a large number of physical interactions have been cataloged from
organisms, fueling the development of computational algorithms for
systematically mining protein complexes from the protein-protein
interaction (PPI) network. Despite the significant progress in the
aspect of extracting protein complexes from the PPI networks over
the last few years, computational methods are seriously constrained
by noise in current PPI data (2,3). Several
computational methods have been developed to predict modules from
networks relying on graph clustering, and clique finding, such as
MCODE (4), CMC (5) and STM (6).
In addition, the identification of dysregulated modules from
condition-specific networks will promote the process to reveal the
pathogenesis of disease. Recently, a systematic method for
uncovering dysregulated modules has been developed by integrating
PPI information and condition-specific gene expression profile
(7).
Chronic lymphocytic leukemia (CLL) is the most
common type of hematopoietic malignancies in adults (8). Nowadays, more and more patients are
diagnosed at early-stage, likely because of using routine blood
tests as well as the widespread availability of flow cytometry
(9–11). However, the treatment of CLL remains a
challenge (12). A previous study
demonstrated that TL1A could be considered as a negative regulator
of leukemic cell proliferation, that may influence the
physiopathology and clinical outcome of CLL at an early-stage
(13). Moreover, researchers
indicated that the inhibition of JAK was a potentially useful new
pharmacological approach to CLL treatment (14). However, the underlying mechanisms of
CLL have yet to be fully elucidated. Systematic tracking
dysregulated module behavior across specific conditions is
critical.
In this investigation, we proposed a straightforward
but systematic method to identify and compare the modules across
normal and CLLs through integrating PPI data and specific gene
expression profile of CLL. The results of this study will be
critical to reveal the molecular mechanism of this disease and
develop promising therapies.
Materials and methods
Data recruitment and
preprocessing
Here we selected the microarray expression profile
of E-GEOD-2466 (15) CLL from
ArrayExpress database to perform our analysis. The gene expression
profile numbered E-GEOD-2466 which was based on the platform of
A-AFFY-1 - Affymetrix GeneChip Human Genome U95Av2 [HG_U95Av2] and
A-AFFY-9 - Affymetrix GeneChip Human Genome U95A [HG_U95A] was
downloaded. E-GEOD-2466 comprised a total of 111 samples, including
100 genetically well-characterized B-CLL samples and 11 normal
control samples. Finally, we obtained the probe annotation data for
subsequent analysis.
With the goal of eliminating the influence of
non-specific hybridization, the expresso function from the Affy
package was used to obtain the probe data. Moreover, background
correction was conducted by means of robust multiarray average
(RMA) (16), quantiles (17) was employed to standardize the data,
MAS was utilized to implement perfect match (PM) and mismatch (MM)
correction (18), medianpolish
(16) was performed to summarize the
probe data, followed by the filtration of probe data by means of
featureFilter function. Ultimately, we obtained 20,102 genes from
the probe set.
Detection of differentially expressed
genes (DEGs)
The DEGs between the two groups were extracted
relying on limma package (19).
Empirical Bayes (eBayes) (20) as
well as a false discovery rate (FDR) (21) calibration of P-values were implemented
using ImFit function. The genes were extracted if these genes met
the following conditions: |logFC| ≥2, P<0.01.
PPI network construction
In this study, we downloaded all PPI data with
combine-score from the STRING database (22). The protein ID and gene symbols were
transformed and then the repeated ones were removed. We selected
the interaction relations with combined-score ≥0.65 to construct
the PPI sub-network.
Gene interactions re-weighted using Pearsons
correlation coefficient (PCC). Gene interactions of the PPI
sub-network of CLL and normal subjects were re-weighted by PCC. PCC
can evaluate the probability of two co-expressed genes, where value
ranged from −1 to +1 (23). Moreover,
the absolute value of PCC of each edge was defined as the value of
the interaction of the PPI sub-network. In addition, we defined PCC
of every gene pair as the weight value of the edge.
Uncovering modules from the
sub-network of PPI
Clique-merging, an approach used to identify the
modules, is similar to the previous algorithms for detecting
complexes from the PPI networks (2,5). Herein,
we used the clique-merging to extract the modules from PPI
sub-networks. In detail, we discovered all maximal cliques from the
network of the normal and disease conditions using fast depth-first
approach. Subsequently, the maximal cliques having nodes ≥5 were
selected. Last we arranged them in descending order according to
the score of a clique. This score was its weighted interaction
density (WID). According to the WID, we sorted these cliques.
A lot of maximal cliques may be overlapped. Hence,
these lapped cliques should be abandoned to decrease size or merged
into a bigger sub-graphs. In this study, the weighted
inter-connectivity between two cliques was employed to determine
whether these two lapped cliques were combined or not. Herein, t0
(a predefined overlap cut-off criteria) = 0.5 and tm (a predefined
m cut-off criteria) = 0.25 were selected to be the threshold in the
current study.
Identification of altered modules
The modules were respectively extracted from the PPI
networks of normal controls and CLL. For each module in the normal
and disease conditions, we calculated the modules correlation
density (MCD) based on the PCC of gene-gene interactions in PPI
network. MCD was measured as:
dcc(Si)=∑p,q∈SiPCC((p,q),N)(|Si|2).
The pairs of the disrupted or dysregulated modules
were evaluated through determining the module as a maximum weight
bipartite matching (24). A
similarity graph M = (Vm, Em) was constructed, where Vm = {S∪T},
and Em = ∪{(Si, Tj):J(Si,
Tj) ≥tJ, ΔCC(Si, Tj) ≥δ}, whereby
J(Si, Tj) =
|Si∩Tj|/|Si∪Tj| was the
Jaccard similarity (J) and ΔCC(Si, Tj) =
|dc(Si) -
dc(Ti)| was the differential correlation
density (ΔCC) between Si and Tj (7). In our study, the altered modules with tJ
≥2/3 as well as ΔCC ≥0.05 were considered as the disrupted
modules.
Gene composition of altered
modules
With the goal of better understanding the
differences of the altered modules in the two groups, we screened
out the missed and added genes. Significantly, the common part of
the missed as well as added genes was also identified. Moreover, in
order to obtain the relevant disease genes, we also took the
intersection of the DEGs and the intersection genes.
Results
Detection of DEGs
In our study, a total of 8,630 genes were screened
out for subsequent analysis. Using the threshold of |logFC| >2,
and a FDR <0.01, a total of 734 genes were identified to be
differentially expressed in CLL.
Disruptions in CLL PPI network
Under the combined-score ≥0.65, the PPI sub-networks
displayed equal numbers interactions (40,576) and nodes (6,068).
Their mean average scores were respectively 0.320 and 0.698 in the
normal and disease network. The result of PCC distribution in the
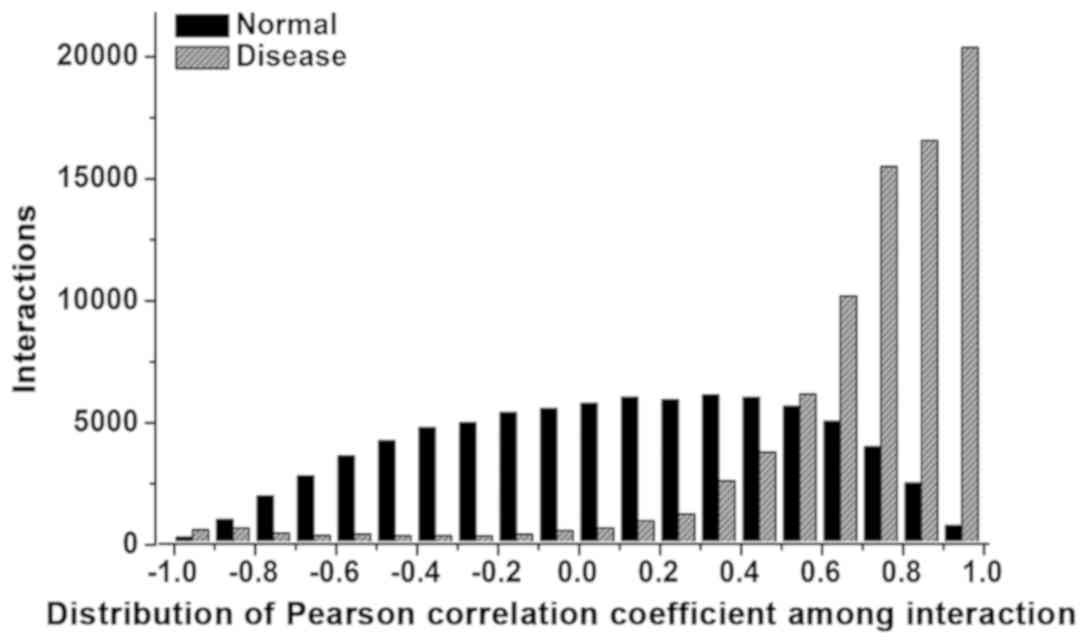
two networks is shown in Fig. 1.
Differences were observed in the PCC score distribution (−1 to 1)
of the two networks. Moreover, the count of the interactions in the
normal network was higher than that in the CLL network when the
weight distribution was ranging from −0.9 to −0.4.
Analysis of disruptions in CLL
modules
On the basis of the threshold of nodes ≥5, there
were 8,773 maximal cliques in the normal and CLL PPI sub-networks,
and then, these 8,773 cliques were utilized to conduct module
analysis. There were 1,805 and 703 modules from the PPI
sub-networks of normal and CLL, respectively (Table I). For the module of CLL, the average
module size was higher than that of module of normal. Moreover, for
CLL modules, it also showed an overall decrease in average
correlation comparing with the normal modules. The connection
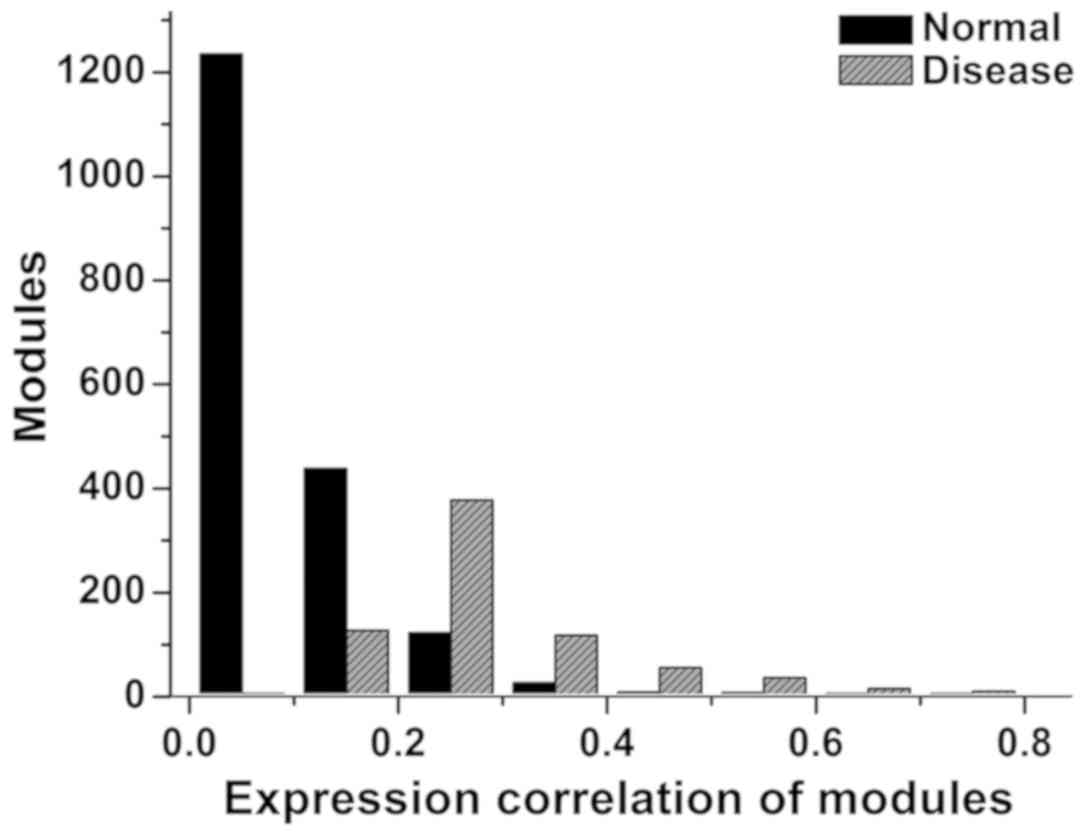
between the counts of modules and weighted density of modules is
shown in Fig. 2. The distribution of
modules in normal was higher under 0–0.2, while lower under 0.2–0.8
relative to that in disease condition.
 | Table I.Properties of normal and disease
modules. |
Table I.
Properties of normal and disease
modules.
|
|
|
| Correlation |
|---|
|
|
|
|
|
|---|
| Module set | No. of modules | Average module
size | Max | Avg | Min |
|---|
| Normal | 1,805 | 19.32798 | 0.5186083 | 0.06900261 | −0.1587027 |
| Disease |
703 | 43.46373 | 0.573716 | 0.0379185 | −0.1173073 |
Moreover, there were 646 disrupted
module pairs
Relative to the normal group, we found that all
altered modules were higher in the disease group based on the
module correlation density of the modules.
Gene composition of the altered
modules
There were 646 disrupted module pairs in total.
Among these module pairs, there were 145 missed genes and 353 added
genes in CLL modules relative to normal modules. Of note, 85 genes
were the common ones between the added and missed genes. In this
investigation, we obtained 9 common genes between the 85
intersection genes and the DEGs, of which were AKT1, UPF3A, SMG7,
RPN2, HSPB1, SPCS2, CDC16, COPS5 and PSMB10. Hence, we treated the
9 genes as the key genes involved in CLL.
Discussion
CLL is a variety of chronic lymphoproliferative
disorders (24). In our study, we
investigated gene profile E-GEOD-2466 and extracted the key genes
in CLL on the basis of systematically tracking the disrupted
modules of re-weighted PPI networks. In total, 9 key genes were
identified through comparing intersection genes in the altered
modules and DEGs.
In humans, RAC-α serine/threonine-protein kinase is
coded by AKT1 gene. AKT1 has been demonstrated to regulate a large
amount of biological processes, for example, cell proliferation,
growth, metabolism, survival, and angiogenesis. The AKT1 isoform
played a dominant role in the survival and chemoresistance of CLL
cells (25). AKT1 gene has been
inferred to be related to a variety of diseases. It was familiar in
cancer (26,27), tumors (28,29) and
insulin resistance (30). Another
study has also implicated that the expression of AKT1 was
associated with CLL (31,32).
Dolichyl-diphosphooligosaccharide-protein
glycosyltransferase subunit 2 is coded by RPN2 in humans. So far
there has not been much research on the correlation of RPN2 and a
disease, in addition to tumors. For example, it has been verified
that RPN2 silencing made breast cells hypersensitive to docetaxel
(33). Moreover, RPN2 has also
displayed a higher genomic alteration frequency in colorectal
cancer (34).
Heat-shock protein 27 (Hsp27) in humans is encoded
by HSPB1, which is a chaperone of the small heat-shock protein
group (35,36). As known, the common roles of small
heat-shock proteins are inhibition of apoptosis, thermotolerance,
and cell differentiation. HSPB1 was reported related to breast
cancer, shock, Alexander's disease and many kinds of tumors
(37–40). Dempsey et al (41) have explored that CLL patients
expressed significantly higher levels of Hsp27 in lymphocytes than
that expressed by lymphocytes from control subjects.
Taken together, we successfully identified a series
of key genes for understanding the potential mechanism of gene
expression of CLL on the basis of systematically uncovering the
altered modules based on the re-weighted PPI networks and DEGs.
These results have implications for understanding the mechanisms of
gene expression of CLL.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL and SFK conceived the study and drafted the
manuscript. ZW and XYZ acquired the data. CFL and SFK analyzed the
data and revised the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nguyen PV, Srihari S and Leong HW:
Identifying conserved protein complexes between species by
constructing interolog networks. BMC Bioinformatics. 14 Suppl
16:S82013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Srihari S and Leong HW: A survey of
computational methods for protein complex prediction from protein
interaction networks. J Bioinform Comput Biol. 11:12300022013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li X, Wu M, Kwoh CK and Ng SK:
Computational approaches for detecting protein complexes from
protein interaction networks: A survey. BMC Genomics. 11 Suppl
1:S32010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu G, Wong L and Chua HN: Complex
discovery from weighted PPI networks. Bioinformatics. 25:1891–1897.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hwang W, Cho YR, Zhang A and Ramanathan M:
A novel functional module detection algorithm for protein-protein
interaction networks. Algorithms Mol Biol. 1:242006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Srihari S and Ragan MA: Systematic
tracking of dysregulated modules identifies novel genes in cancer.
Bioinformatics. 29:1553–1561. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Byrd JC, Stilgenbauer S and Flinn IW:
Chronic lymphocytic leukemia. Hematology (Am Soc Hematol Educ
Program). 2004:163–183. 2004. View Article : Google Scholar
|
|
9
|
Abrisqueta P, Pereira A, Rozman C,
Aymerich M, Giné E, Moreno C, Muntañola A, Rozman M, Villamor N,
Hodgson K, et al: Improving survival in patients with chronic
lymphocytic leukemia (1980–2008): The Hospital Clinic of Barcelona
experience. Blood. 114:2044–2050. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rawstron AC, Böttcher S, Letestu R,
Villamor N, Fazi C, Kartsios H, de Tute RM, Shingles J, Ritgen M,
Moreno C, et al: European Research Initiative in CLL: Improving
efficiency and sensitivity: European Research Initiative in CLL
(ERIC) update on the international harmonised approach for flow
cytometric residual disease monitoring in CLL. Leukemia.
27:142–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
van den Broek EC, Kater AP, van de Schans
SA, Karim-Kos HE, Janssen-Heijnen ML, Peters WG, Nooijen PT,
Coebergh JW and Posthuma EF: Chronic lymphocytic leukaemia in the
Netherlands: Trends in incidence, treatment and survival,
1989–2008. Eur J Cancer. 48:889–895. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rozovski U, Hazan-Halevy I, Keating MJ and
Estrov Z: Personalized medicine in CLL: Current status and future
perspectives. Cancer Lett. 352:4–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cavallini C, Lovato O, Bertolaso A,
Zoratti E, Malpeli G, Mimiola E, Tinelli M, Aprili F, Tecchio C,
Perbellini O, et al: Expression and function of the TL1A/DR3 axis
in chronic lymphocytic leukemia. Oncotarget. 6:32061–32074. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Montresor A, Toffali L, Mirenda M, Rigo A,
Vinante F and Laudanna C: JAK2 tyrosine kinase mediates integrin
activation induced by CXCL12 in B-cell chronic lymphocytic
leukemia. Oncotarget. 6:34245–34257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haslinger C, Schweifer N, Stilgenbauer S,
Döhner H, Lichter P, Kraut N, Stratowa C and Abseher R: Microarray
gene expression profiling of B-cell chronic lymphocytic leukemia
subgroups defined by genomic aberrations and VH mutation status. J
Clin Oncol. 22:3937–3949. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Irizarry RA, Bolstad BM, Collin F, Cope
LM, Hobbs B and Speed TP: Summaries of Affymetrix GeneChip probe
level data. Nucleic Acids Res. 31:e15. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bolstad BM, Irizarry RA, Astrand M and
Speed TP: A comparison of normalization methods for high density
oligonucleotide array data based on variance and bias.
Bioinformatics. 19:185–193. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bolstad B: affy: Built-in processing
methods. https://www.bioconductor.org/packages/3.7/bioc/vignettes/affy/inst/doc/builtinMethods.pdf5–10.
2017
|
|
19
|
Smyth GK, Ritchie M, Thorne N, Wettenhall
J and Shi W: limma: Linear models for microarray data user's guide.
http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.361.8519&rep=rep1&type=pdf5–10.
2017
|
|
20
|
Datta S, Satten GA, Benos DJ, Xia J,
Heslin MJ and Datta S: An empirical bayes adjustment to increase
the sensitivity of detecting differentially expressed genes in
microarray experiments. Bioinformatics. 20:235–242. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Reiner A, Yekutieli D and Benjamini Y:
Identifying differentially expressed genes using false discovery
rate controlling procedures. Bioinformatics. 19:368–375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
von Mering C, Jensen LJ, Snel B, Hooper
SD, Krupp M, Foglierini M, Jouffre N, Huynen MA and Bork P: STRING:
Known and predicted protein-protein associations, integrated and
transferred across organisms. Nucleic Acids Res. 33:D433–D437.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hodge JC, Kim T-M, Dreyfuss JM,
Somasundaram P, Christacos NC, Rousselle M, Quade BJ, Park PJ,
Stewart EA and Morton CC: Expression profiling of uterine
leiomyomata cytogenetic subgroups reveals distinct signatures in
matched myometrium: Transcriptional profiling of the t(12;14) and
evidence in support of predisposing genetic heterogeneity. Hum Mol
Genet. 21:2312–2329. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kayar Y, Ekinci I, Bay I, Bayram Kayar N,
Hamdard J and Kazancıoğlu R: Acute renal failure due to leukaemic
infiltration in chronic lymphocytic leukaemia. Case Rep Med.
2015:4691362015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hofbauer SW, Krenn PW, Piñόn, Hofbauer J,
Pucher S, Asslaber D, Egle A, Hartmann TN and Greil R: The AKT1
isoform plays a dominant role in the survival and chemoresistance
of chronic lymphocytic leukaemia cells. Br J Haematol. 172:815–819.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carson JP, Kulik G and Weber MJ:
Antiapoptotic signaling in LNCaP prostate cancer cells: A survival
signaling pathway independent of phosphatidylinositol 3′-kinase and
Akt/protein kinase B. Cancer Res. 59:1449–1453. 1999.PubMed/NCBI
|
|
27
|
Kim D, Kim S, Koh H, Yoon SO, Chung AS,
Cho KS and Chung J: Akt/PKB promotes cancer cell invasion via
increased motility and metalloproteinase production. FASEB J.
15:1953–1962. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Goldstein M, Meller I and Orr-Urtreger A:
FGFR1 over-expression in primary rhabdomyosarcoma tumors is
associated with hypomethylation of a 5′ CpG island and abnormal
expression of the AKT1, NOG, and BMP4 genes. Genes Chromosomes
Cancer. 46:1028–1038. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ericson K, Gan C, Cheong I, Rago C,
Samuels Y, Velculescu VE, Kinzler KW, Huso DL, Vogelstein B and
Papadopoulos N: Genetic inactivation of AKT1, AKT2, and PDPK1 in
human colorectal cancer cells clarifies their roles in tumor growth
regulation. Proc Natl Acad Sci USA. 107:2598–2603. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cleasby ME, Reinten TA, Cooney GJ, James
DE and Kraegen EW: Functional studies of Akt isoform specificity in
skeletal muscle in vivo; maintained insulin sensitivity despite
reduced insulin receptor substrate-1 expression. Mol Endocrinol.
21:215–228. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhuang J, Hawkins SF, Glenn MA, Lin K,
Johnson GG, Carter A, Cawley JC and Pettitt AR: Akt is activated in
chronic lymphocytic leukemia cells and delivers a pro-survival
signal: The therapeutic potential of Akt inhibition. Haematologica.
95:110–118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
de Frias M, Iglesias-Serret D, Cosialls
AM, Coll-Mulet L, Santidrián AF, González-Gironès DM, de la Banda
E, Pons G and Gil J: Akt inhibitors induce apoptosis in chronic
lymphocytic leukemia cells. Haematologica. 94:1698–1707. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Honma K, Iwao-Koizumi K, Takeshita F,
Yamamoto Y, Yoshida T, Nishio K, Nagahara S, Kato K and Ochiya T:
RPN2 gene confers docetaxel resistance in breast cancer. Nat Med.
14:939–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang J, Yan B, Späth SS, Qun H, Cornelius
S, Guan D, Shao J, Hagiwara K, Van Waes C, Chen Z, et al:
Integrated transcriptional profiling and genomic analyses reveal
RPN2 and HMGB1 as promising biomarkers in colorectal cancer. Cell
Biosci. 5:532015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carper SW, Rocheleau TA and Storm FK: cDNA
sequence of a human heat shock protein HSP27. Nucleic Acids Res.
18:64571990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hunt CR, Goswami PC and Kozak CA:
Assignment of the mouse Hsp25 and Hsp105 genes to the distal region
of chromosome 5 by linkage analysis. Genomics. 45:462–463. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shi P, Wang M-M, Jiang L-Y, Liu H-T and
Sun J-Z: Paclitaxel-doxorubicin sequence is more effective in
breast cancer cells with heat shock protein 27 overexpression. Chin
Med J (Engl). 121:1975–1979. 2008.PubMed/NCBI
|
|
38
|
Shen G, Liang S, Xu Z, Zhou L, Xiao S, Xia
X, Li R, Liao Y, You C and Wei Y: Downregulated expression of HSP27
in human low-grade glioma tissues discovered by a quantitative
proteomic analysis. Proteome Sci. 8:172010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Iwaki T, Iwaki A, Tateishi J, Sakaki Y and
Goldman JE: Alpha B-crystallin and 27-kd heat shock protein are
regulated by stress conditions in the central nervous system and
accumulate in Rosenthal fibers. Am J Pathol. 143:487–495.
1993.PubMed/NCBI
|
|
40
|
Wu Y, Liu J, Zhang Z, Huang H, Shen J,
Zhang S, Jiang Y, Luo L and Yin Z: HSP27 regulates IL-1 stimulated
IKK activation through interacting with TRAF6 and affecting its
ubiquitination. Cell Signal. 21:143–150. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Dempsey NC, Leoni F, Ireland HE, Hoyle C
and Williams JH: Differential heat shock protein localization in
chronic lymphocytic leukemia. J Leukoc Biol. 87:467–476. 2010.
View Article : Google Scholar : PubMed/NCBI
|