Introduction
Colorectal cancer (CRC) is one of the most common
malignant heterogeneous tumours in the world. Almost 70% of
colorectal tumours develop from an adenocarcinoma sequence and the
remaining develop via a serrated neoplasia pathway named for the
pattern of crypts in the precursor polyps (1). In general, the molecular pathways that
lead to colorectal carcinogenesis are distinguished by three basic
pathways: Chromosomal instability (CIN), the CpG pathway of the
methylation phenotype (CIMP) and microsatellite instability (MSI)
(2). Hereditary non-polyposis
colorectal carcinoma (HNPCC), or Lynch syndrome (LS), develops
through the MSI pathway. LS is an autosomal dominant disease
characterised by DNA dysfunction, namely in mismatch repair (MMR)
genes. Mutations in these genes lead to errors in microsatellites.
LS diagnosis was first postulated using clinical Amsterdam I and II
criteria. In 2006, there was a revision of the Bethesda guidelines,
which included clinical as well as morphological features of
tumours. Recent studies showed that LS may be early onset or occur
in people over 50 years of age. Screening criteria among patients
with CRC have been expanded. Multiple guidelines recommend tumour
screening through MMR protein expression or MSI. Tumour genomic
profiling using next-generation sequencing (NGS) panels to define
the spectrum of mutations in an individual's tumour is becoming
increasingly widespread (3).
Hereditary tumour groups carry germline mutations in the MMR genes:
MLH1, MSH2, PMS1, PMS2, MSH3, MSH6 and EPCAM. MMR
genes repair DNA damage that occurs during replication; the MMR
system is responsible for replacing mismatched nucleotides
(4).
MSI positivity in sporadic cancers is usually the
result of epigenetic inactivation. The most common epigenetic
change is CpG island hypermethylation in the MLH1 promoter
region. Methylation leads to loss of function of tumour suppressor
genes, MMR genes and genes responsible for regulation of cell
growth and division (5). In CRC, the
V600E mutation in BRAF, a gene that codes a protein in the
RAS/RAF/MAPK pathway, suggests a sporadic origin for the disease.
The V600E mutation is not usually found in hereditary forms of
cancer and therefore allows the selection of patients diagnosed
with sporadic carcinomas who are positive for MSI (6).
Screening for MSI status in CRC patient tissues,
together with BRAF mutation detection and identification of
hypermethylation status, represent an algorithm to stratify CRC
patients. Currently, NGS is one of the most promising tools used to
detect germline mutations. The Miseq system (Illumina, USA) offers
an NGS workflow based on target amplification. Targeted DNA
enrichment methods allow even higher genome throughput at a reduced
cost per sample (7). NGS allows
massive parallel sequencing that affords maximal tumour genomic
assessment (8).
In the present study, we observed CRC patients with
the aim of identifying suspected LS patients who were carriers of
MMR mutations. For future analysis, we would like to apply
molecular methods to diagnosed patients older than 50 years.
Materials and methods
Specimens
We obtained 300 DNA samples from Slovak patients
with CRC in collaboration with the Department of Molecular Biology
(Jessenius Faculty of Medicine in Martin), Department of Pathologic
Anatomy (Jessenius Faculty of Medicine in Martin) and University
Hospital in Martin. Our analyses were performed on DNA derived from
microdissected formalin-fixed-paraffin-embedded (FFPE) tumour
tissue, and DNA was extracted with the Black Prep FFPE DNA kit
(Analytik Jena AG, Jena, Germany), according to manufacturer's
protocol.
All tumours were examined with immunohistochemistry
(IHC) for the expression of MMR proteins. IHC analysis was
performed using 4 µm tissue sections from FFPE blocks. IHC results
were diagnosed at the Department of Pathologic Anatomy (Jessenius
Faculty of Medicine in Martin).
In the present study, we acquired only 5 blood
samples from suspected LS patients according to our criteria. DNA
was isolated from whole blood using the DNeasy Blood and Tissue kit
(Qiagen GmbH, Hilden, Germany). DNA concentration was measured on a
Qubit® 2.0 Fluorometer (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Samples were diluted to
working solutions at a concentration of 1–2 ng/µl and stored at
−20°C.
All patients were informed about the study and
provided written informed consent. The present study was a part of
projects that were approved by the Ethical Committee at Jessenius
Faculty of Medicine in Martin.
MSI polymerase chain reaction (PCR)
and fragment analyses
To determine the tumour MSI, MSI PCR was performed
using the MSI Analysis System v1.2 kit (Promega Corporation,
Madison, WI, USA) with a GeneAmp PCR System 9700 instrument
(Applied Biosystems; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. MSI analysis based on PCR is
considered the gold standard approach to detect microsatellite
status, and it is sensitive for identifying LS. MSI testing is a
method-for-function detection which cannot be replaced under
certain conditions, such as MSI tumours with intact IHC expression
(non-functional protein expression). The MSI assay was performed
using a pentaplex PCR with 5 quasimonomorphic mononucleotide
markers (NR-27, NR-21, NR-24, BAT-25 and BAT-26) and 2
pentanucleotide markers (PENTA C and PENTA D) (9–11). The
MSI Analysis System was optimised to amplify 1–2 ng/µl of genomic
DNA in a 10 µl reaction volume. For a positive amplification
control, we used high molecular weight K562 (Promega Corporation).
After PCR, 1 µl of amplified product was added to 9.5 µl Hi-Di™
Formamide (Applied Biosystems; Thermo Fisher Scientific, Inc.) and
0.5 µl Internal Lane Standard 600 (ILS 600; Promega Corporation).
Denatured samples were loaded on the ABI 3500 (Applied Biosystems;
Thermo Fisher Scientific, Inc.). The results were evaluated by
GeneMapper software (Applied Biosystems; Thermo Fisher Scientific,
Inc.) by comparing the patient's sample with the K562 control
and/or non-tumour tissue. Samples with only one altered
microsatellite marker were classified as MSI-low (MSI-L). Samples
with ≥2 altered markers were classified as MSI-high (MSI-H).
Samples with no altered markers were classified as microsatellite
stable (MSS).
Sanger sequencing for BRAF
c.1799T>A (V600E)
The presence of the V600E mutation was monitored by
Sanger sequencing using an ABI 3500 (Applied Biosystems; Thermo
Fisher Scientific, Inc.). PCR products for Sanger sequencing were
amplified with the primers BRAF Ex15F
(5′-TCATAATGCTTGCTCTGATAGGA-3′) and BRAF Ex15R
(5′-GGCCAAAATTTAATCAGTGGA-3′). PCR products were evaluated by
electrophoresis on a 1.75% agarose gel and purified with the
NucleoSpin Gel and PCR Clean-Up kit (Machery-Nagel GmbH, Düren,
Germany). Sequencing PCR was prepared using the BigDye Terminator
v1.1 Cycle Sequencing kit (Thermo Fisher Scientific, Inc.). PCR
condition optimisation was described previously (12,13).
SigmaSpin Post-Reaction Clean-Up (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) was used for the second purification and in
this analysis according to the information provided by the
manufacturer. Results were evaluated using Chromas Pro software
(Technelysium Pty Ltd., South Brisbane, QLD, Australia). RKO cell
lines that carried the V600E mutation were used as positive control
and DNA from a healthy individual was the negative control.
Bisulfite treatment
The EpiTect Bisulfite kit (Qiagen GmbH) was used for
bisulfite treatment of 1 ng/µl-2 µg/µl of genomic DNA in RNase-free
water in a 20 µl volume, according to the manufacturer's
instructions. The modified DNA was eluted to a final concentration
of 40 ng/µl. EpiTect Controls DNAs (Qiagen GmbH), which included
fully methylated or unmethylated DNA, were used as controls. The
samples were stored at −20°C until further analysis.
Nested methylation-specific PCR
(MS-PCR)
To detect the MLH1 methylation status, we
used nested two-step accession for increased sensitivity (14), with modifications according to
Lasabová et al (15). MS-PCR
was performed using a GeneAmp PCR System 9700 and amplified with
specific primers to distinguish methylated (M) from unmethylated
(U) DNA according to Herman et al (16). The sequences of our modified external
(MLH1ExF/R) and internal (MLH1UF/R and MLH1MF/R) primer sets and
the size of PCR products are presented in Table I. PCR conditions for the nested PCR
step with external primers were as follows: 95°C for 8 min, then 20
cycles of 95°C for 30 sec, 62°C for 30 sec and 72°C for 30 sec and
finally 72°C for 8 min. The PCR products of this amplification were
diluted 1:500 and we prepared the second PCR using internal primers
for methylated or unmethylated DNA. Thermal cycling was identical
to the above conditions, except the annealing temperature for the
primers was 64°C.
 | Table I.Primer sets for nested
methylation-specific polymerase chain reaction. |
Table I.
Primer sets for nested
methylation-specific polymerase chain reaction.
| Primer | Sequence
(5′-3′) | Size (bp) |
|---|
| MLH1UF |
GTAGATGTTTTATTAGGGTTGT | 113 |
| MLH1UR |
CACCTCATCATAACTACCCACA |
|
| MLH1MF |
GTAGACGTTTTATTAGGGTCGC | 113 |
| MLH1MR |
CCTCATCGTAACTACCCGCG |
|
| MLH1ExF |
GAGTAGTTTTTTTTTTAGGAGTGA | 192 |
| MLH1ExR |
ATAAAACCCTATACCTAATCTATC |
|
NGS analysis
For NGS analysis, we examined 5 blood samples from
suspected LS patients. NGS analysis was performed on the MiSeq
sequencer (Illumina, Inc., San Diego, CA, USA), according to the
manufacturer's protocol, using the HNPCC MASTR PlusKit (Agilent
Technologies, Inc., Santa Clara, CA, USA). The kit was used with
the complementary MASTR Plus product, the molecular identifier
(MID) for Illumina Miseq kit [1-48] (Agilent Technologies, Inc.).
HNPCC MASTR Plus identifies single nucleotide variants and copy
number variation in MLH1, MSH2, MSH6, PMS2 and the 3′
untranslated region (UTR) of EPCAM, genes associated with
HNPCC. In the first step of library preparation, we amplified all
targeted regions in separate multiplex PCR amplifications for each
sample. We used 50 ng of genomic DNA per reaction. The multiplex
PCR products for each sample were then combined together into one
tube and this amplicon library was purified from small residual DNA
fragments with Agencourt AM Pure XP beads (Beckman Coulter, Inc.,
Brea, CA, USA). PCR products were diluted 1:1,000. In the second
step, universal PCR amplification tagged amplicons with MID
adaptors. For each sample, we prepared a universal PCR with a
unique combination of MID p5-p7 primer mix sequences. The average
amplicon size after universal PCR was 469 base pairs (bp). The
number of cycle reads was 2×251 bp. After this PCR, we performed
electrophoresis on a chip with an Agilent 2100 Bioanalyzer (Agilent
Technologies, Inc.) for quality control. We then purified the
tagged amplicon library again with Agencourt AM Pure XP beads,
determined the library concentrations and pooled equal volumes of
the libraries into the final library. Concentrations of the
obtained tagged amplicons were measured with the Qubit™ dsDNA HS
Assay kit (Thermo Fisher Scientific, Inc.). The library pool was
processed using kits with all necessary consumables and a reagent
cartridge for sequencing. For sequencing, we used Miseq Reagent
Nano kit v.2 (Illumina, Inc.) with a 500 megabase (Mb) capacity.
The obtained data were evaluated by the Variant Effect Predictor
software (https://www.ensembl.org). Only mutations
reported in Polyphen, SIFT and Cosmic were taken into account, and
silent or intronic mutations were not reported.
Results
Specimens and MSI PCR analysis
From a total of 300 patients identified with CRC,
103 (34%) were women, 163 (54%) were men and the gender was unknown
for 34 (11.3%) cases (Table II).
Patient age ranged from 28 to 94 years (average 60.5).
Histopathological parameters of the cases, including histological
grade, morphological subtype and localisation, were known. Most
carcinomas were classified as adenocarcinomas except: 1 (0.3%)
synchronous carcinoma, 1 (0.3%) undifferentiated carcinoma, 2
(0.6%) medullar carcinomas, 4 (1.3%) signet ring cell carcinomas
and 36 (12%) mucinous carcinomas (Table
II). Fifty-three (18%) cases were grade 3, 136 (45%) were grade
2 and 39 (13%) were grade 1 (Table
II).
| Table II.Clinicopathological features of
cohort. |
Table II.
Clinicopathological features of
cohort.
| Clinicopathological
features |
|---|
| Gender | N |
|
Female | 103 |
|
Male | 163 |
| Not
determined | 34 |
| Grade |
|
| Grade
1 | 39 |
| Grade
2 | 136 |
| Grade
3 | 53 |
| Not
determined | 77 |
| Localization |
|
| Colon
ascendens/descendens | 155 |
|
Rectum | 59 |
|
Caecum | 39 |
|
Rectosigmoideum | 13 |
| Not
determined | 34 |
| Morphological
subtype |
|
|
Synchronous carcinoma | 1 |
|
Medullar carcinoma | 2 |
| Signet
ring cell | 4 |
|
Mucinous carcinoma | 36 |
|
Undifferentiated
carcinoma | 1 |
|
Adenocarcinoma NOS | 222 |
| Not
determined | 34 |
From our cohort (n=300), we captured 33 (11%) MSI-H
and 4 (1.3%) MSI-L cases. In 263 (88%) samples, we did not detect
MSI. These samples were evaluated as MSS. The results of MMR IHC
and MSI testing were shown to be largely concordant. Genetic
examination of MSI was confirmed in 33 cases based on IHC
examination. This examination included 4 MSI-L cases grouped as MSS
carcinomas, but according to genetic analysis they belonged to the
MSI-positive tumour group. Although we stratified more patients
suspected for LS, we were only able to obtain 5 blood samples. The
blood samples, which were from suspected LS patients based on
immunohistochemical and molecular analyses, were submitted for
genetic counselling and NGS analysis.
Sanger sequencing for BRAF and nested
methylation PCR
In 37 (12.3%) samples with a positive MSI status, we
subsequently detected the BRAF V600E mutation. There was no
significant association between BRAF-and MSI-positive
statuses. We focused on detection of the BRAF
c.1799T>Amutation in the samples. Twenty-four (64.9%) samples
did not have the c.1799T>A mutation in BRAF exon 15. To
further stratify patients, we continued the analysis of the 24
samples negative for the BRAF mutation (Fig. 1). Since the presence of the above
mutation generally indicates sporadic CRC, only negative samples
were subjected to the more detailed analysis. Due to the low DNA
concentration (<50 ng/µl) required for bisulfite treatment, it
was not possible to analyse the presence of MLH1 methylation
for 1 sample. Eleven (45.8%) samples were positive for MLH1
methylation while 12 (50%) samples were without methylation. We
also determined MLH1 methylation status in 13
BRAF-mutation-positive samples, which were excluded from
additional testing for LS. From these 13 samples, 12 cases (92%)
were also positive for MLH1 methylation (Table III).
| Table III.Table of MSI cases with MSI
polymerase chain analysis and BRAF mutation analysis. |
Table III.
Table of MSI cases with MSI
polymerase chain analysis and BRAF mutation analysis.
| Case | MSI | BRAF | Methylation |
|---|
| 1 | MSI-H | − | + |
| 2 | MSI-H | + | + |
| 3 | MSI-H | − | + |
| 4 | MSI-H | + | + |
| 5 | MSI-H | − | − |
| 6 | MSI-H | − | + |
| 7 | MSI-H | − | − |
| 8 | MSI-H | + | + |
| 9 | MSI-L | − | − |
| 10 | MSI-H | − | − |
| 11 | MSI-H | + | Not determined |
| 12 | MSI-H | − | Not determined |
| 13 | MSI-H | − | − |
| 14 | MSI-H | + | + |
| 15 | MSI-H | − | + |
| 16 | MSI-H | + | + |
| 17 | MSI-H | + | + |
| 18 | MSI-H | − | + |
| 19 | MSI-H | + | + |
| 20 | MSI-H | − | − |
| 21 | MSI-H | − | − |
| 22 | MSI-H | − | − |
| 23 | MSI-H | + | + |
| 24 | MSI-H | − | − |
| 25 | MSI-H | − | − |
| 26 | MSI-H | − | − |
| 27 | MSI-H | − | + |
| 28 | MSI-H | − | + |
| 29 | MSI-H | − | + |
| 30 | MSI-H | + | + |
| 31 | MSI-H | + | + |
| 32 | MSI-L | − | + |
| 33 | MSI-L | − | + |
| 34 | MSI-H | + | + |
| 35 | MSI-H | − | + |
| 36 | MSI-H | + | + |
| 37 | MSI-L | − | − |
NGS analysis to detect germline
mutations
To confirm LS, we utilised NGS analysis with a Miseq
system. From the available blood samples (n=5), we detected 2
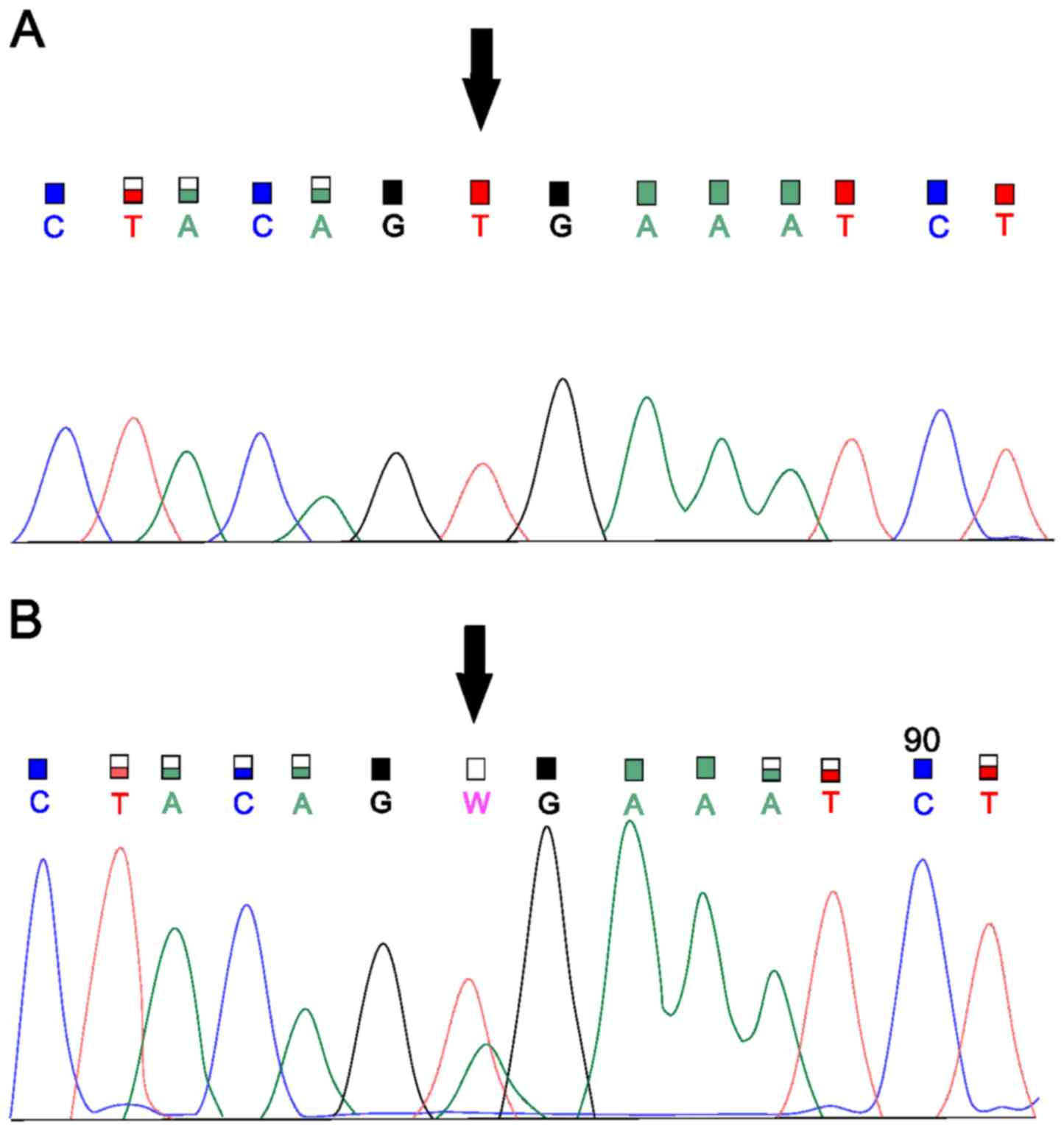
samples with suspected pathogenic germline mutations. In the first
sample, we captured the previously unpublished deletion of the four
nucleotides 1627_1630del AAAG (Glu544Lysfs*26) in
MSH6, a mutation with a high impact on the gene (based on
Assembly GRCh37). According to IHC, we assumed there was some
variant in MSH6, because the protein was not detected. This
patient was positive for MSI and negative for the BRAF
mutation and MLH1 methylation. This frameshift variant was
found in MSH6 exon 4. We verified the deletion by Sanger
sequencing (Fig. 2). In the sample
with the newly detected MSH6 deletion, we detected 42
additional nonpathogenic variants, most of which were missense
(66%), synonymous (8%) or frameshift (6%) variants with benign or
uncertain significance. Our results were verified by a commercial
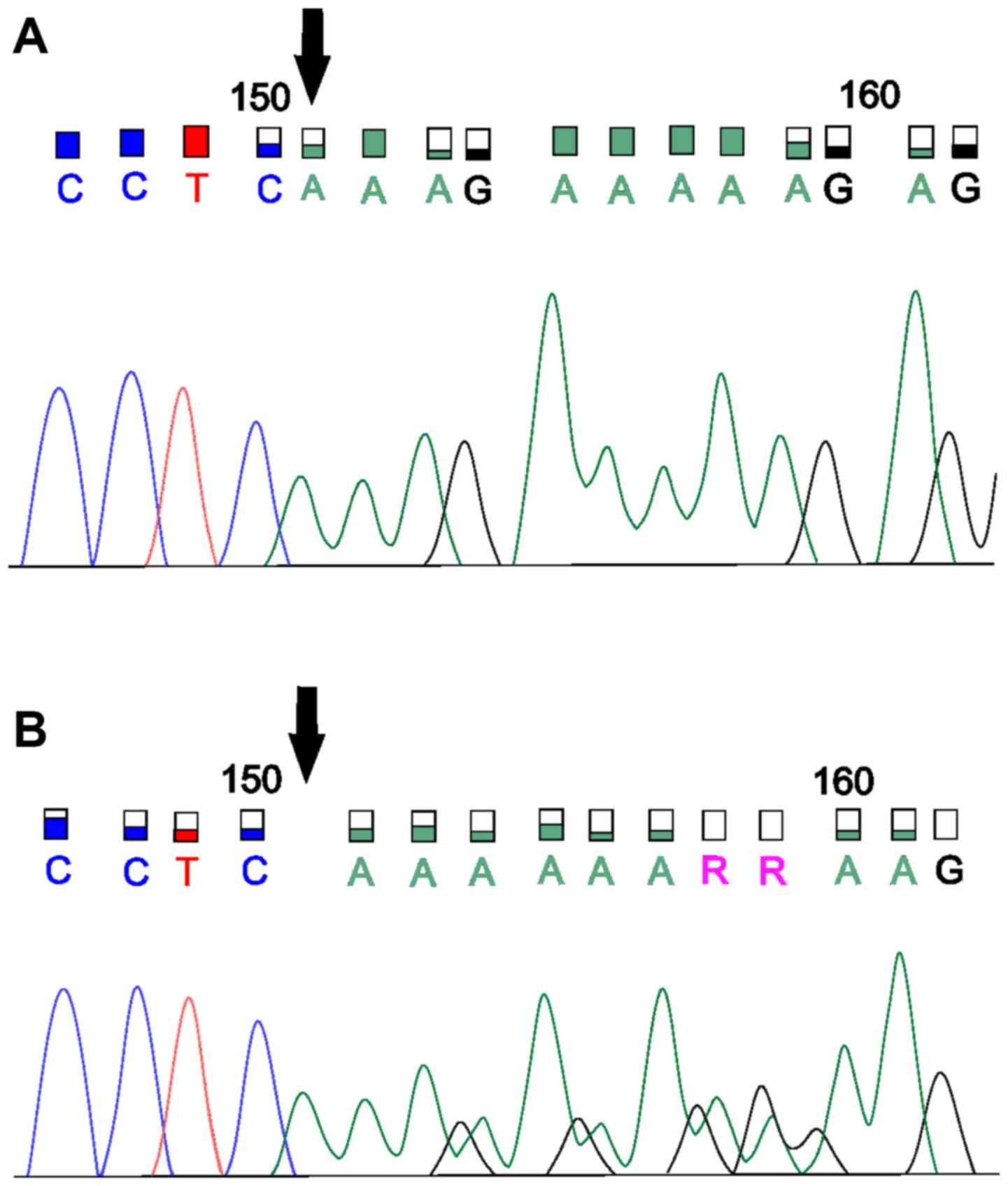
laboratory. In another sample, we confirmed the pathogenic
stop-gain variant MMR_c.1030C>T (Gln344Ter) for
MSH2 according to a five-tiered classification from the
International Agency for Research on Cancer (based on Assembly
GRCh37). This variant leads to the stop codon p.Q344X. We were not
able to verify this mutation by Sanger sequencing. In this case,
there were also mainly missense variants (81%), followed by
synonymous (11%) and stop-gain variants (8%). These variants were
classified as benign or of uncertain significance. We also
recommended verification by a commercial laboratory, but that
result is unknown to us. In the 3 other blood samples, we
identified several variants with uncertain significance.
Discussion
The MSI pathway affects CRC, particularly in terms
of genetic instability such as oncogene activation or tumour
suppressor gene inactivation. The aim of our study was to improve a
stratification procedure for Slovak CRC patients and to determine
the significance of the utilised procedures. We focused on the
possibilities of stratification, which is important for rapid
selection of appropriate prevention and treatment especially for
patients with hereditary forms of the disease. Research shows that
patients with LS have earlier disease onset compared to sporadic
CRC patients (17). A useful
screening tool for capturing LS has been MSI detection (7) since suspected LS cases usually exhibit
MSI. MSI testing with MMR deficiency has 93% sensitivity, but it
does not predict which MMR gene(s) is/are altered (18). Nevertheless, MSI testing is effective
for screening but there is significant uncertainty surrounding what
balance of sensitivity and specificity will be achieved in clinical
practice (17).
According to some authors, Bethesda criteria are
sensitive but not very specific for MSI status. Hereditary forms of
the disease occur in patients with a family history of the same
affinity or adenoma (19). Screening
MMR genes is a gold standard for LS diagnosis, and MSI has a role
in CRC patient stratification. It is a part of targeted screening
for LS patients, and MSI status is recommended as the first step
(20) because screening all MMR
genes is more expensive, demanding and time consuming (21). Determining MSI status can also be
used as a predictor for treatment (22). Recent studies showed that patients
with MMR-deficient tumours are more responsive to programmed cell
death-1 (PD-1) blockade than MMR-competent tumours, so patients
with MMR deficiency may benefit from anti-PD-1 therapy (23). Differentiation between hereditary and
sporadic MSI CRC is currently the basic diagnostic step for patient
stratification (8). Carcinomas with
MSI represent approximately 15% of all CRC cases, LS accounts for
3% and sporadic MSI carcinomas represent 12% (24). From our specimens of the 300 CRC
cases, 37 (12.3%) were MSI positive. To exclude sporadic MSI
carcinomas from further testing, we chose to detect the BRAF
V600E mutation by Sanger sequencing. According to Snowsill et
al (25), cascade testing is
used in every strategy for LS patients. Additionally, strategies
that utilise MSI and BRAF testing are the most cost
effective. This cascade strategy was also used by Buchanan et
al (26) to identify LS
patients, and it is recommended in study of Cohen et al
(27) together with MLH1
methylation testing. BRAF mutations were not detected in
cases with a germline mutation in MLH1, MSH2 or MSH6
(28). The BRAF V600E
mutation and MLH1 methylation are associated with sporadic
MSI carcinomas, but they are not associated with MMR deficiency in
LS (29). Adar et al
(30) stated that the presence of
the V600E mutation in tumours had a positive predictive value for
sporadic cancer, but negative predictive value for MLH1
methylation; the presence of the BRAF V600E mutation
excludes LS diagnosis. Seppälä et al (31) declared that MSI carcinomas had better
prognosis compared to MSS, and BRAF-positive tumours were
associated with poor prognosis.
The third stratification step in the MSI-positive
group without the BRAF V600E mutation was detection of
methylation in the most commonly methylated MMR gene, namely
MLH1. Previous studies indicated the presence of alterations
in MMR genes, which led to the loss of gene product expression and
development of malignancy. The role of epigenetic changes,
especially aberrant DNA methylation, not only in polyposis sporadic
carcinomas but also in hereditary tumours, is not yet accurately
known (3). Approximately one third
of carcinomas are thought to occur due to epigenetics (32). In the process of patient
stratification, detection of MLH1 promoter region
methylation serves to exclude sporadic forms of carcinoma from
further testing to allow the selection of suspected hereditary
tumours. The primary target sequence for DNA methylation is
5′-CpG-3′ dinucleotides. Promoter CpG-islands are normally
protected from methylation, while CpG dinucleotides that are not
associated with CpG-islands are heavily methylated (10). However, in rare cases of LS,
MLH1 hypermethylation can serve as a second hit (33). Based on these findings, we introduced
an NGS analysis that clearly confirmed the presence of pathological
changes in DNA and provided a comprehensive view of genetic
information.
Heterozygous germline variants in MMR genes are
responsible for LS. Patients with LS have one functional allele of
the MMR gene and the second allele carries the germline mutation.
Malignant tumours result from the second (defective) allelic copy,
which becomes nonfunctional because of other somatic mutations or
methylation (34). Only a few LS
cases with homozygous MMR mutations were described previously;
these patients exhibited early cancer onset (33). The majority of mutations are located
in MSH2 and MLH1, followed by MSH6 (35,36).
Indeed, almost 90% of LS cases have a germline mutation in
MLH1 or MSH2 (37).
The hot spot areas for the appearance of MMR gene variants are
MSH6 exon 4 and MSH2 exon 3 or 12. MSH6 and
PMS2 mutations also prevail in the Icelandic population
(38), but genetic drift can
influence the appearance of some mutations. In a study from 2007,
missense MMR gene mutations were common in LS (39). Usually, we identify chromosomal
mutations at position 3p21 of MLH1, 2p16 of MSH2,
7p22 of PMS2 and 2p16 of MSH6 (34). In a 2017 study, the authors declared
that MSH6 mutations accounted for approximately 18% of LS
cases (40). In our study of Slovak
CRC patients, we stratified the subjects, and in suspected LS cases
we tried to find germline mutations. In the first suspected case,
we identified a novel missense mutation, a deletion in MSH6
exon 6. This mutation, c.1627_1630AAAG (p.Glu544Lysfs), should be
included in future studies. In the second suspected LS case, we
detected a previously reported nonsense mutation (c.1030C>T
(p.Q344X)) in MSH2 (41,42). While we identified this pathogenic
variant by NGS, we were unable to verify it by the Sanger
sequencing. Nevertheless, all tested patients were recommended for
genetic counselling and diagnosis from a certified commercial
diagnostic laboratory.
Undetermined clinical information, limited sample
size for NGS analysis and NGS method were limitations of this
study. We were unable to get more information about patients and
about their family history. However, our findings provide
stratification for routine diagnostics. NGS analysis in our study
had a few limitations. For this method only 5 blood samples from 12
suspected LS cases were available. For next studies is necessary to
get more blood samples. Although, we identified a novel deletion
probably associated with LS and detected known mutation in
MSH2 gene, our findings are insufficient to prove the
association to LS. These limitations demonstrated that other
information about patients are important, especially family history
and results of IHC. However, based on our findings we recommend
testing for all CRC patients, also without unrecorded family
history.
Known mutations in hereditary CRC occur with a low
frequency and thus it is difficult to predict penetrance. There is
also heterogeneity in phenotypic expression. We found some new
variants of uncertain clinical significance, and so it was
difficult to predict the variant impacts on the genes (38). Our findings provide a few useful and
fast stratification steps for routine diagnostics. Indeed, this
study was conducted to address the need for clearer patient
stratification for suspected LS cases and for clearer information
about diagnostic results based on MSI testing. A targeted NGS panel
for LS evaluated the ability to detect germline mutation. Our
results directly demonstrated the greatly contribute to rapid
patient diagnosis and optimal treatment settings. We recommend a
cascade strategy (MSI for MMR genes, BRAF and MLH1
testing) for determination of sporadic MSI-positive cancer and
LS-like cancer. We recommend this strategy despite of study
limitation-NGS analysis. We did not have MSI-H, MSI-L, MSS
categories of patients in NGS analysis necessary for LS validation.
On the other hand, detection of a new deletion was confirmed by the
commercial laboratory.
A cascade screening strategy is suitable for
patients who meet the Amsterdam criteria or revised Bethesda
guidelines, and so we recommend use of this screening for
stratification of all patients with CRC in addition to cases with a
negative or unrecorded family history. In the future, we would like
to continue with our study. Experiment is a suggestion for future
studies in area of LS. Also new detected deletions should be as a
suggestion for further studies. We need more knowledges about this
variant because it was previously not described in ClinVar, Human
Gene Mutation Database, Ensembl and InSight databases. Based on our
findings, new screening strategies are necessary and LS needs to be
further studied. In future we would like to study new insights into
patient testing.
Acknowledgements
The authors would like to thank Dr Jana Kršiaková
(M-Genetik, Martin, Slovakia) for assisting with patient
recruitment.
Funding
The present study was supported by the Biomedical
Center Martin Grant (grant no. ITMS 26220220187), which was
co-financed by EU sources. The study was also supported by the
project ‘Genomic Profile And Transcriptional Signature Of
Colorectal Cancer’ (grant no. APVV-16-0066) and the project ‘Pilot
study of utility of DNA mutational analysis of tissue and so-called
liquid biopsy in the diagnosis and prediction of therapeutic
response in two malignancies’ (grant no. APVV-14-0273).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
IK designed the study and wrote the manuscript. IK,
MK, KJ, TB, BM, AV and SM conducted the experiments and analyzed
the data. ZL and LP designed the study, and compiled and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All patients were informed about the present study
and provided written informed consent. This research was a part of
projects that were approved by the Ethical Committee at Jessenius
Faculty of Medicine in Martin (Martin, Slovakia).
Patient consent for publication
The patients provided written informed consent for
the publication of the present study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bettington M, Walker N, Clouston A, Brown
I, Leggett B and Whitehall V: The serrated pathway to colorectal
carcinoma: Current concepts and challenges. Histopathology.
62:367–386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Worthley DL and Leggett BA: Colorectal
cancer: Molecular features and clinical opportunities. Clin Biochem
Rev. 31:31–38. 2010.PubMed/NCBI
|
|
3
|
Stadler ZK, Battaglin F, Middha S,
Hechtman JF, Tran C, Cercek A, Yaeger R, Segal NH, Varghese AM,
Reidy-Lagunes DL, et al: Reliable detection of mismatch repair
deficiency in colorectal cancers using mutational load in
next-generation sequencing panels. J Clin Oncol. 34:2141–2147.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llinàs-Arias P and Esteller M: Epigenetic
inactivation of tumour suppressor coding and non-coding genes in
human cancer: An update. Open Biol. 7(170152)2017.
|
|
5
|
Al-Sohaily S, Biankin A, Leong R,
Kohonen-Corish M and Warusavitarne J: Molecular pathways in
colorectal cancer. J Gastroenterol Hepatol. 27:1423–1431. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sharma SG and Gulley ML: BRAF mutation
testing in colorectal cancer. Arch Pathol Lab Med. 134:1225–1228.
2010.PubMed/NCBI
|
|
7
|
Meldrum C, Doyle MA and Tothill RW:
Next-generation sequencing for cancer diagnostics: A Practical
perspective. Clin Biochem Rev. 32:177–195. 2011.PubMed/NCBI
|
|
8
|
Serrati S, De Summas S, Pilato B,
Petriella D, Lacalamita R, Tommasi S and Pinto R: Next-generation
sequencing: Advances and applications in cancer diagnosis. Onco
Targets Ther. 9:7355–7365. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Buhard O, Cattaneo F, Wong YF, Yim SF,
Friedman E, Flejou JF, Duval A and Hamelin R: Multipopulation
analysis of polymorphisms in five mononucleotide repeats used to
determine the microsatellite instability status of human tumors. J
Clin Oncol. 24:241–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Søreide K: High-fidelity of five
quasimonomorphic mononucleotide repeats to high-frequency
microsatellite instability distribution in early-stage
adenocarcinoma of the colon. Anticancer Res. 31:967–971.
2011.PubMed/NCBI
|
|
11
|
Benlloch S, Payá A, Alenda C, Bessa X,
Andreu M, Jover R, Castells A, Llor X, Aranda FI and Massutí B:
Detection of BRAF V600E mutation in colorectal cancer: comparison
of automatic sequencing and real-time chemistry methodology. J Mol
Diagn. 8:540–543. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jasek K, Buzalkova V, Minarik G, Stanclova
A, Szepe P, Plank L and Lasabova Z: Detection of mutations in the
BRAF gene in patients with KIT and PDGFRA wild-type
gastrointestinal stromal tumors. Virchows Arch. 470:29–36. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Minárik G, Plank L, Lasabová Z, Szemes T,
Burjanivová T, Szépe P, Buzalková V, Porubský D and Sufliarsky J:
Spectrum of mutations in gastrointestinal stromal tumor patients-a
population based study from Slovakia. APMIS. 121:539–548. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
House MG, Guo M, Iacobuzio-Donahue C and
Herman JG: Molecular progression of promoter methylation in
intraductal mucinous neoplasm (IPMN) of the pancreas.
Carcinogenesis. 24:193–198. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lasabova Z, Tilandyova P, Kajo K, Zubor P,
Burjanivova T, Danko J and Plank L: Hypermethylation of the GSTP1
promoter region in breast cancer is associated with prognostic
clinicopathological parameters. Neoplasma. 57:35–40. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Herman JG, Umar A, Polyak K, Graff JR,
Ahuja N, Issa JPJ, Markowitz S, Willson JK, Hamilton SR, Kinzler
KW, et al: Incidence and functional consequences of hMLH1 promoter
hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA.
95:6870–6875. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coelho H, Jones-Hughes T, Snowsill T,
Briscoe S, Huxley N, Frayling IM and Hyde C: A systematic review of
test accuracy studies evaluating molecular micro-satellite
instability testing for the detection of individuals with lynch
syndrome. BMC Cancer. 17:8362017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hendriks YM, de Jong AE, Morreau H, Tops
CM, Vasen HF, Wijnen JT, Breuning MH and Bröcker-Vriends AH:
Diagnostic approach and management of lynch syndrome (hereditary
nonpolyposis colorectal carcinoma): A guide for clinicians. CA
Cancer J Clin. 56:213–225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kheirelseid EAH, Miller N and Kerin MJ:
Molecular biology of colorectal cancer: Review of the literature.
Am J Mol Biol. 3:72–80. 2013. View Article : Google Scholar
|
|
20
|
Schofield L, Watson N, Grieu F, Li WQ,
Zeps N, Harvey J, Stewart C, Abdo M, Goldblatt J and Iacopetta B:
Population-based detection of Lynch syndrome in young colorectal
cancer patients using microsatellite instability as the initial
test. Int J Cancer. 124:1097–1102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hampel H, Frankel WL, Martin E, Arnold M,
Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J,
et al: Screening for the Lynch syndrome (hereditary nonpolyposis
colorectal cancer). N Engl J Med. 352:1851–1860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ribic CM, Sargent DJ, Moore MJ, Thibodeau
SN, French AJ, Goldberg RM, Hamilton SR, Laurent-Puig P, Gryfe R,
Shepherd LE, et al: Tumor microsatellite-instability status as a
predictor of benefit from fluorouracil-based adjuvant chemotherapy
for colon cancer. N Engl J Med. 349:247–257. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gatalica Z, Vranic S, Xiu J, Swensen J and
Reddy S: High microsatellite instability (MSI-H) colorectal
carcinoma: A brief review of predictive biomarkers in the era of
personalized medicine. Fam Cancer. 15:405–412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Snowsill T, Huxley N, Hoyle M,
Jones-Hughes T, Coelho H, Cooper C, Frayling I and Hyde C: A
systematic review and economic evaluation of diagnostic strategies
for Lynch syndrome. Health Technol Assess. 18:1–406. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Buchanan DD, Clendenning M, Rosty C,
Eriksen SV, Walsh MD, Walters RJ, Thibodeau SN, Stewart J, Preston
S, Win AK, et al: Tumor testing to identify lynch syndrome in two
Australian colorectal cancer cohorts. J Gastroenterol Hepatol.
32:427–438. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cohen R, Buhard O, Cervera P, Hain E,
Dumont S, Bardier A, Bachet JB, Gornet JM, Lopez-Trabada D, Dumont
S, et al: Clinical and molecular characterisation of hereditary and
sporadic metastatic colorectal cancers harbouring microsatellite
instability/DNA mismatch repair deficiency. Eur J Cancer.
86:266–274. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng G, Bell I, Crawley S, Gum J, Terdiman
JP, Allen BA, Truta B, Sleisenger MH and Kim YS: BRAF mutation is
frequently present in sporadic colorectal cancer with methylated
hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin
Cancer Res. 10:191–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suraweera N, Duval A, Reperant M, Vaury C,
Furlan D, Leroy K, Seruca R, Iacopetta B and Hamelin R: Evaluation
of tumor microsatellite instability using five quasimonomorphic
mononucleotide repeats and pentaplex PCR. Gastroenterology.
123:1804–1811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Adar T, Rodgers LH, Shannon KM, Yoshida M,
Ma T, Mattia A, Lauwers GY, Iafrate AJ and Chung DC: A tailored
approach to BRAF and MLH1 methylation testing in a universal
screening program for Lynch syndrome. Mod Pathol. 30:440–447. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Seppälä TT, Böhm JP, Friman M, Lahtinen L,
Väyrynen VM, Liipo TK, Ristimäki AP, Kairaluoma MV, Kellokumpu IH,
Kuopio TH and Mecklin JP: Combination of microsatellite instability
and BRAF mutation status for subtyping colorectal cancer. Br J
Cancer. 112:1966–1975. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Snover DC: Update on the serrated pathway
to colorectal carcinoma. Hum Pathol. 42:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wolf AI, Buchanan AH and Farkas LM:
Historical review of Lynch syndrome. J Coloproctol (Rio J).
33:2013.
|
|
34
|
Le S, Ansari U, Mumtaz A, Malik K, Patel
P, Doyle A and Khachemoune A: Lynch syndrome and muir-torre
syndrome: An update and review on the genetics, epidemiology, and
management of two related disorders. Derma Online J. 23:22017.
|
|
35
|
Peltomäki P and Vasen H: Mutations
associated with HNPCC predisposition-Update of ICG-HNPCC/INSiGHT
mutation database. Dis Markers. 20:269–276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goldberg Y, Kedar I, Kariiv R, Halpern N,
Plesser M, Hubert A, Kaduri L, Sagi M, Lerer I, Abeliovich D, et
al: Lynch syndrome in high risk Ashkenazi Jews in Israel. Fam
Cancer. 13:65–73. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cravo M, Afonso AJ, Lage P, Albuquerque C,
Maia L, Lacerda C, Fidalgo P, Chaves P, Cruz C and Nobre-Leitão C:
Pathogenicity of missense and splice site mutations in hMSH2 and
hMLH1 mismatch repair genes: Implications for genetic testing. Gut.
50:405–412. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Haraldsdottir S, Rafnar T, Frankel WL,
Einarsdottir S, Sigurdsson A, Hampel H, Snaebjornsson P, Masson G,
Weng D, Arngrimsson R, et al: Comprehensive population-wide
analysis of Lynch syndrome in Iceland reveals founder mutations in
MSH6 and PMS2. Nat Commun. 8:147552017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lagerstedt Robinson K, Liu T, Vandrovcova
J, Halvarsson B, Clendenning M, Frebourg T, Papadopoulos N, Kinzler
KW, Vogelstein B, Peltomäki P, et al: Lynch syndrome (hereditary
nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst.
99:291–299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Houlleberghs H, Goverde A, Lusseveld J,
Dekker M, Bruno MJ, Menko FH, Mensenkamp AR, Spaander MCW, Wagner
A, Hofstra RMW and Te Riele H: Suspected Lynch syndrome associated
MSH6 variants: A functional assay to determine their pathogenicity.
PLoS Genet. 13:e10067652017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bartosova Z, Fridrichova I, Bujalkova M,
Wolf B, Ilencikova D, Krizan P, Hlavcak P, Palaj J, Lukac L,
Lukacova M, et al: Novel MLH1 and MSH2 germline mutations in the
first HNPCC families identified in Slovakia. Hum Mutat. 21:4492003.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rey JM, Noruzinia M, Brouillet JP, Sarda
P, Maudelonde T and Pujol P: Six novel heterozygous MLH1, MSH2, and
MSH6 and one homozygous MLH1 germline mutations in hereditary
nonpolyposis colorectal cancer. Cancer Genet Cytogenet.
155:149–151. 2004. View Article : Google Scholar : PubMed/NCBI
|