Introduction
Clear cell sarcoma-like tumor of the
gastrointestinal tract (CCSLTGT), also known as an ‘osteoclast-rich
tumor of the gastrointestinal tract with features resembling clear
cell sarcoma (CCS) of soft parts’, is a rare and malignant tumor
entity that occurs exclusively within the wall of the
gastrointestinal tract (1). In
contrast to CCS of the soft tissue (previously known as melanoma of
the soft tissue), CCSLTGT was initially described as a distinct
entity by Zambrano et al (2)
in 2003 from a series of 6 cases that were characterized
histologically by the presence of osteoclast-like giant cells
(OLGCs) and immunohistochemically by the absence of
melanocyte-specific markers. An increasing number of cases support
that CCSLTGT is a distinctive tumor entity, and not a variant of
CCS of the soft tissue (3–10). However, the pathological nature of
CCSLTGT is distinguishable from CCS of the soft tissue in that it
always arises in tendons and aponeuroses, and shows melanocytic
differentiation at the light microscopic, ultrastructural and
protein levels (1,10). In 2012, Stockman et al
(6) proposed to re-designate this
tumor entity as a ‘malignant gastrointestinal neuroectodermal
tumor’ (GNET) instead of a CCSLTGT, and this term has been
increasingly accepted by pathologists (7–9,11–13).
Based on recent studies, cases that were previously reported as
soft tissue-type CCS of the gastrointestinal tract (CCS-GI) lacking
melanocytic differentiation may be appropriately categorized as
CCSLTGT or GNET, although a GNET remains a controversial tumor
entity (1–4,6–11,13). To
the best of our knowledge, only 47 cases that may represent a GNET
have been reported in the English or Chinese languages, including
31 of which appear to be reported as a CCSLTGT or GNET and 16 that
correspond to CCS-GI lacking melanocytic differentiation. Table I summarizes the clinicopathological
and cytogenetic features of all 47 previous cases (2–9,11–24). Due
to its rarity and histological similarity, a GNET may be easily
misdiagnosed as a variety of neoplasms, including adenocarcinoma, a
gastrointestinal stromal tumor (GIST), a neuroendocrine tumor, CCS
and a malignant peripheral nerve sheath tumor (MPNST) (1). The present study reports a case of a
GNET of the ileum with intra-abdominal granulomatous nodules in a
30-year-old woman who was initially misdiagnosed with a poorly
differentiated carcinoma by intra-operative frozen section
diagnosis.
 | Table I.Summary of clinicopathological
features of 47 cases that were previously reported as CCSLTGT, GNET
or CCS-GI lacking melanocytic differentiation. |
Table I.
Summary of clinicopathological
features of 47 cases that were previously reported as CCSLTGT, GNET
or CCS-GI lacking melanocytic differentiation.
| Authors
(publication) | CCSLTGT or GNET
case | Age, years | Sex | Location | S100 | SOX-10 | HMB-45/Melan-A | EM findings | Genetic
findings | Follow-up | Additional
findings | (Refs.) |
|---|
| Zambrano et
al (2003) | Yes | 15 | F | Jejunum | + | ND | − | No melanosomes | t(12;22)
(q13;q12) | DOD at 16
months | OLGC | (2) |
|
| Yes | 21 | F | Jejunum | + | ND | − | ND | ND | DOD at 12
months | OLGC/LN met. |
|
|
| Yes | 35 | F | Ileum | + | ND | − | No melanosomes | ND | Liver met. at 12
months | OLGC |
|
|
| Yes | 37 | F | Ileum | + | ND | − | ND | ND | Lost | OLGC |
|
|
| Yes | 13 | M | Stomach | + | ND | − | No melanosomes | ND | Recurrence at 8
months | OLGC |
|
|
| Yes | 32 | M | Ileum | + | ND | − | ND | ND | NA | OLGC/LN met. |
|
| Huang et al
(2006) | Yes | 40 | M | Stomach | + | ND | − | ND | ND | NA | OLGC | (4) |
| Antonescu et
al (2006) | No | 81 | F | Colon | + | ND | − | No melanosomes | EWSR1-CREB1 | Intra-abdominal and
Liver met. at 60 months | LN met. | (14) |
|
| No | 42 | F | Ileum | + | ND | − | ND | EWSR1-CREB1 | NA | OLGC |
|
|
| No | 42 | F | Ileum | + | ND | − | No melanosomes | EWSR1-CREB1 | NA | Mesenteric
met. |
|
| Friedrichs et
al (2005) | Yes | 41 | M | Jejunum | + | ND | − | ND | EWSR1-ATF1 | Liver met. at 6
months | OLGC | (3) |
| Venkataraman et
al (2005) | No | 21 | F | Ileum | + | ND | − | No melanosomes | EWSR1
rearrangement | NA |
| (15) |
| Granville et
al (2006) | No | 16 | M | Ileum | + | ND | − | Rare
pre-melanosomes | EWSR1-ATF1 | DOD at 11
months | OLGC | (16) |
| Comin et al
(2007) | No | 31 | F | Ileum | + | ND | − | ND | EWSR1
rearrangement | NA | LN met. | (17) |
| Joo et al
(2009) | No | 60 | M | Ileum | + | ND | − | ND | EWSR1
rearrangement | NA | OLGC/LN and Liver
met. | (18) |
|
| No | 46 | M | Jejunum | + | ND | − | ND | No EWSR1
rearrangement | NA | LN
met./Ig-G4-scle.dis |
|
| Lagmay et al
(2009) | No | 10 | F | Stomach | + | ND | − | ND | EWSR1-ATF1 | NED at 4
months | LN and liver
met. | (19) |
| Terazawa et
al (2009) | No | 20+ | F | Ileum | + | ND | ND | ND | EWSR1-ATF1 | AWD at 24
months | OLGC | (20) |
| Shenjere et
al (2012) | No | 53 | F | Ileum | + | ND | − | No melanosomes | EWSR1-ATF1 | NA | OLGC/LN met. | (21) |
|
| No | 26 | F | Small and large
bowel | + | ND | − | ND | EWSR1-CREB1 | NA | Mesenteric
met. |
|
|
| No | 66 | M | Small bowel | + | ND | − | ND | EWSR1-CREB1 | NA | LN met. |
|
| Stockman et
al (2012) | Yes | 30 | F | Jejunum | + | + | − | Dense-core
granules | EWSR1-ATF | AWD at 21
months |
| (6) |
|
| Yes | 35 | M | Jejunum | + | + | − | Dense-core
granules | EWSR1-ATF1 | DOD at 18
months | OLGC |
|
|
| Yes | 33 | M | Ileum | + | + | − | Dense-core
granules | EWSR1-CREB1 | AWD at 1.5
months | OLGC |
|
|
| Yes | 50 | F | Stomach | + | + | − | Dense-core
granules | EWSR1-ATF1 | AWD at 20
months |
|
|
|
| Yes | 20 | F | Small bowel | + | + | − | Dense-core
granules | EWSR1
rearrangement | NED at 20
months |
|
|
|
| Yes | 52 | M | Ileum | + | + | − | ND | EWSR1 and
FUS-negative | DOD at 22
months | OLGC |
|
|
| Yes | 46 | M | Stomach | + | + | − | ND | EWSR1
rearrangement | NA | OLGC |
|
|
| Yes | 34 | F | Stomach | + | + | − | ND | EWSR1-ATF1 | DOD at 19
months | OLGC |
|
|
| Yes | 37 | F | Ileum | + | + | ND | ND | ND | NA | OLGC |
|
|
| Yes | 77 | F | Colon | + | + | − | ND | EWSR1-ATF1 | DOD at 106
months | OLGC |
|
|
| Yes | 31 | M | Colon | + | + | − | ND | ND | DOD at 3
months | OLGC |
|
|
| Yes | 17 | M | Small bowel | + | + | − | ND | EWSR1
rearrangement | NA |
|
|
|
| Yes | 60 | M | Ileum | + | + | − | ND | EWSR1-ATF1 | AWD at 36
months |
|
|
|
| Yes | 60 | F | Jejunum | + | + | − | ND | EWSR1-CREB1 | NED at 41
months |
|
|
|
| Yes | 56 | M | Stomach | + | + | − | ND | EWSR1-CREB1 | NA |
|
|
|
| Yes | 28 | F | Small bowel | + | + | − | ND | EWSR1
rearrangement | DOD at 23
months |
|
|
| Lasithiotakis et
al (2013) | No | 49 | F | Jejunum | + | ND | − | ND | EWSR1
rearrangement | NED at 20
months |
| (22) |
| Kong et al
(2014) | Yes | 17 | M | Stomach | + | ND | − | ND | EWSR1
rearrangement | NED at 10
months | OLGC | (7) |
| Thway et al
(2014) | Yes | 33 | M | Small bowel | + | ND | − | ND | EWSR1-CREB1 | DOD at 7
months | LN
met./hepatoblastoma history | (5) |
| Zhao et al
(2014) | Yes | 33 | F | Ileum | + | ND | − | ND | EWSR1
rearrangement | NED at 12
months |
| (8) |
| Insabato et
al (2015) | Yes | 29 | M | Stomach | + | ND | − | No melanosomes | EWSR1
rearrangement | AWD at 74
months | Ewing's Sarcoma
history | (11) |
| Boland et al
(2016) | Yes | 46 | F | Stomach | + | + | − | ND | EWSR1-ATF1 | NA | Oncocytic
variant | (12) |
| Alyousef et
al (2017) | Yes | 18 | M | Jejunum | + | ND | − | ND | EWSR1
rearrangement | DOD at 48
months | OLGC | (13) |
| Li et al
(2005) | No | 40 | M | Stomach | + | ND | − | No melanosomes | ND | NED at 9
months | OLGC/LN met. | (23) |
| Huang et al
(2014) | No | 45 | F | Colon | + | ND | − | ND | EWSR1
rearrangement | Liver met. at 16
months | OLGC/LN met. | (24) |
| Kansal et al
(2017) | Yes | 55 | F | Jejunum | + | ND | − | ND | ND | NA |
Neurofilament(+) | (9) |
Case report
A 30-year-old woman presenting with acute abdominal
pain and occasional vomiting was admitted to the Department of
General Surgery, 924th (181st) Hospital of the Chinese People's
Liberation Army (Guilin, Guangxi, China) in November 2017. The
patient had a history of 10 kg of weight loss over the past 6
months and an appendectomy 6 months previously.
Physical examination revealed a mid-abdominal bulge
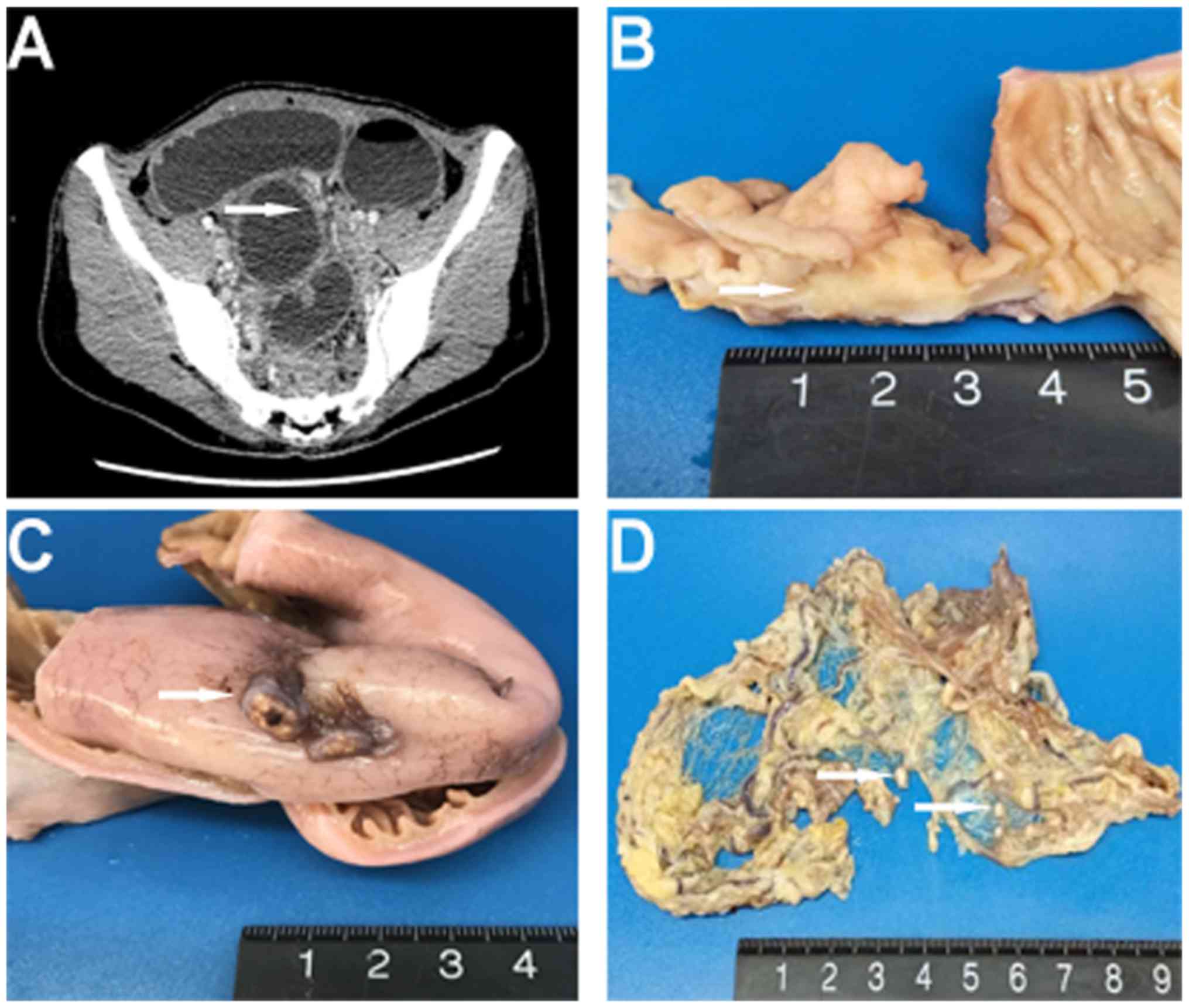
with visible intestinal peristalsis. A computed tomography (CT)
scan of the abdomen and pelvis showed a segmental wall thickening
and dilatation of the distal ileum, evidence of intestinal
obstruction (Fig. 1A). The patient
underwent an exploratory laparotomy, and a mass located in the
distal ileum, as well as multiple gray-white nodules adhering to
the omentum majus and ileal serosa, were identified. An excision of
the segmental ileum and partial omentum majus was performed.
As the patient was in an unstable condition, further
imaging examinations besides the CT scan, including magnetic
resonance imaging and position emission tomography-computed
tomography, were not performed. The patient presented with symptoms
of acute intestinal obstruction and was subjected to an immediate
exploratory laparotomy. The mass in the ileum was initially
misdiagnosed as a poorly differentiated carcinoma based on an
intra-operative frozen section diagnosis; the growth pattern of the
tumor was nested and sheet-like with the presence of
intra-abdominal small nodules.
Macroscopic examination revealed a 3.5×2×1.8-cm
annular mass within the ileum wall; the cut surface was gray-white
and well-circumscribed (Fig. 1B).
Multiple gray-white nodules were adhered to the ileal serosa and
omentum that appear similar in nature to metastatic carcinoma
nodules (Fig. 1C and D). For the
microscopic examination, surgical specimens were fixed in neutral
buffered 10% formalin overnight at room temperature and
paraffin-embedded. Subsequently, 3-µm sections were stained with
hematoxylin for 5 min and eosin for 40 sec at room temperature.
Under light microscopy (DM3000; Leica Microsystems GmbH, Wetzlar,
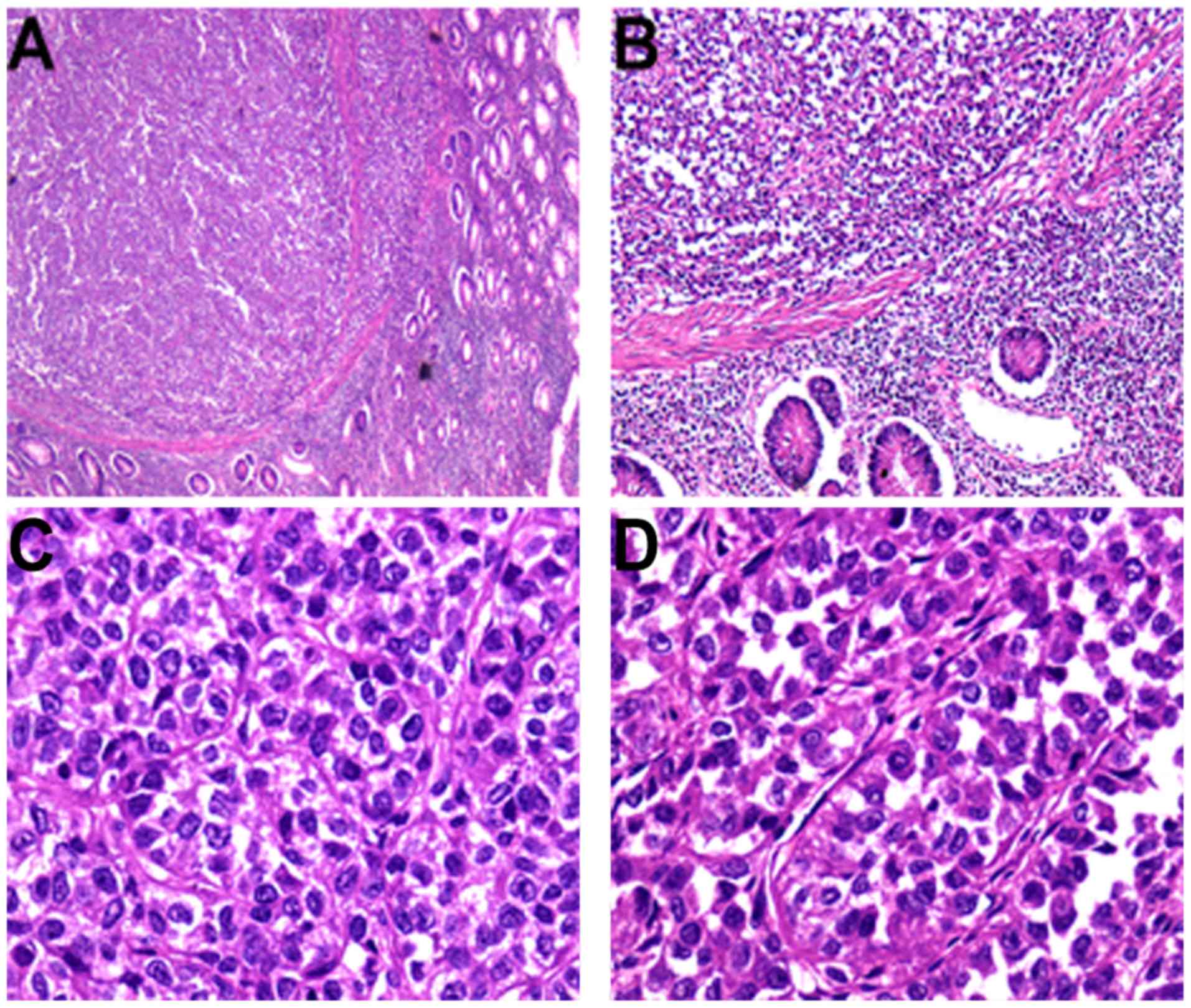
Germany), the tumor was situated in the muscularis propria and
extended into the mucosa and serosa (Fig. 2A and B). Tumor cells were
predominantly arranged in nested and pseudopapillary patterns with
eosinophilic cytoplasm, oval or polygonal vesicular nuclei and
prominent nucleoli (Fig. 2C and D).
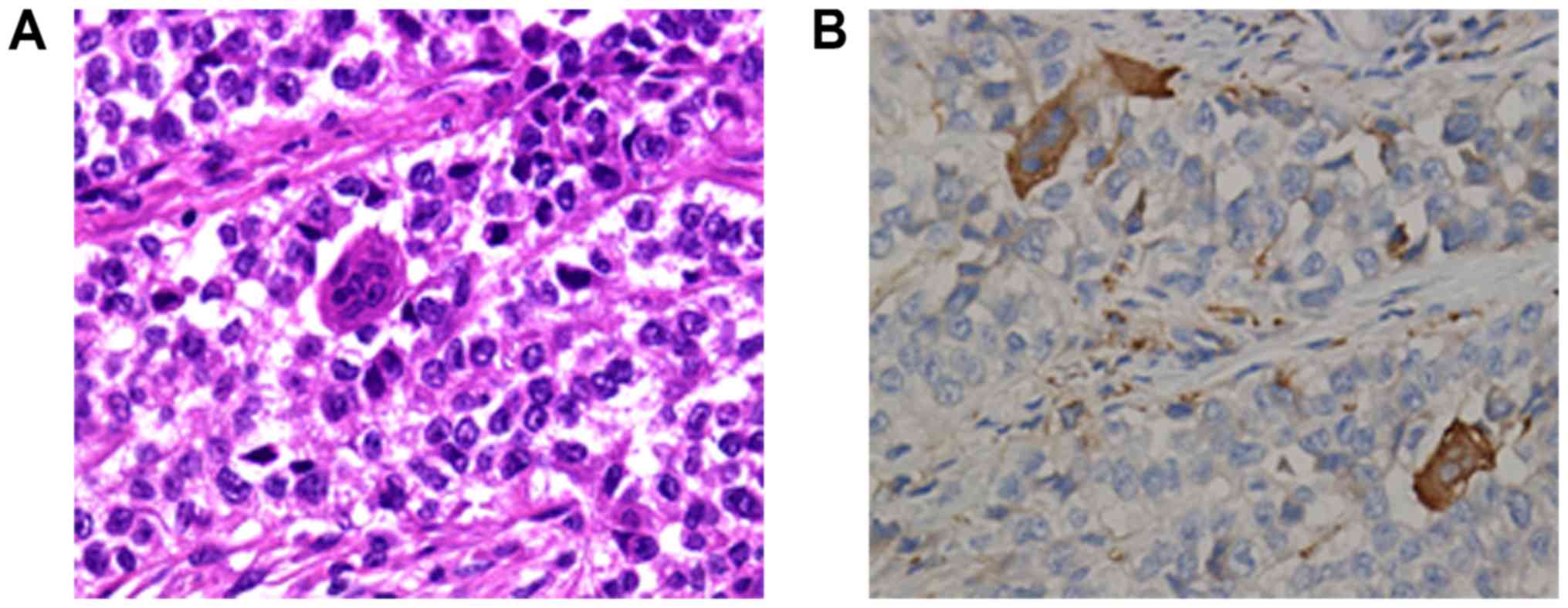
Cluster of differentiation (CD)68-positive, scattered OLGCs were
identified (Fig. 3A and B). Necrosis
and mitotic figures (in 8/10 high-powered fields) were also noted.
There was no tumor involvement in the surgical margin, regional
lymph nodes or liver. The gray-white nodules that adhered to the
omentum and ileal serosa were identified as a chronic granulomatous
inflammation.
The differential diagnosis included a variety of
epithelial and mesenchymal tumors, and tests using a panel of
immunohistochemical markers were performed. The immunohistochemical
assay was performed as previously described (8). Images were acquired using a light
microscope DM3000 (Leica Microsystems GmbH) and the antibodies used
in this case study were listed in Table
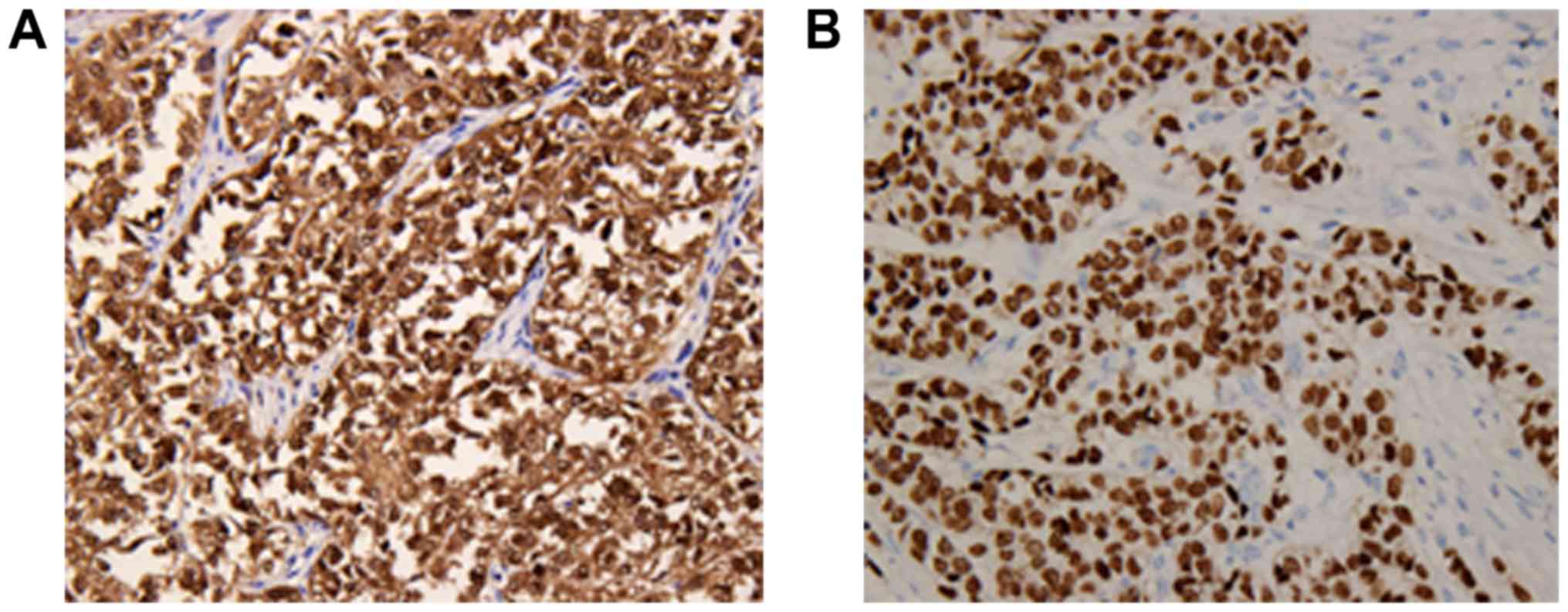
II. The neoplastic cells showed strong diffuse expression of
S-100 and SOX-10 protein (Fig. 4A and
B), and positive staining for Vimentin and CD56, but no
staining for pan-cytokeratin AE1/AE3, cytokeratin (CK)7, CK20,
homeobox protein CDX-2 (CDX-2), synaptophysin, chromogranin-A,
CD117, anoctamin-1 (DOG-1), CD34, human melanoma black-45 (HMB-45),
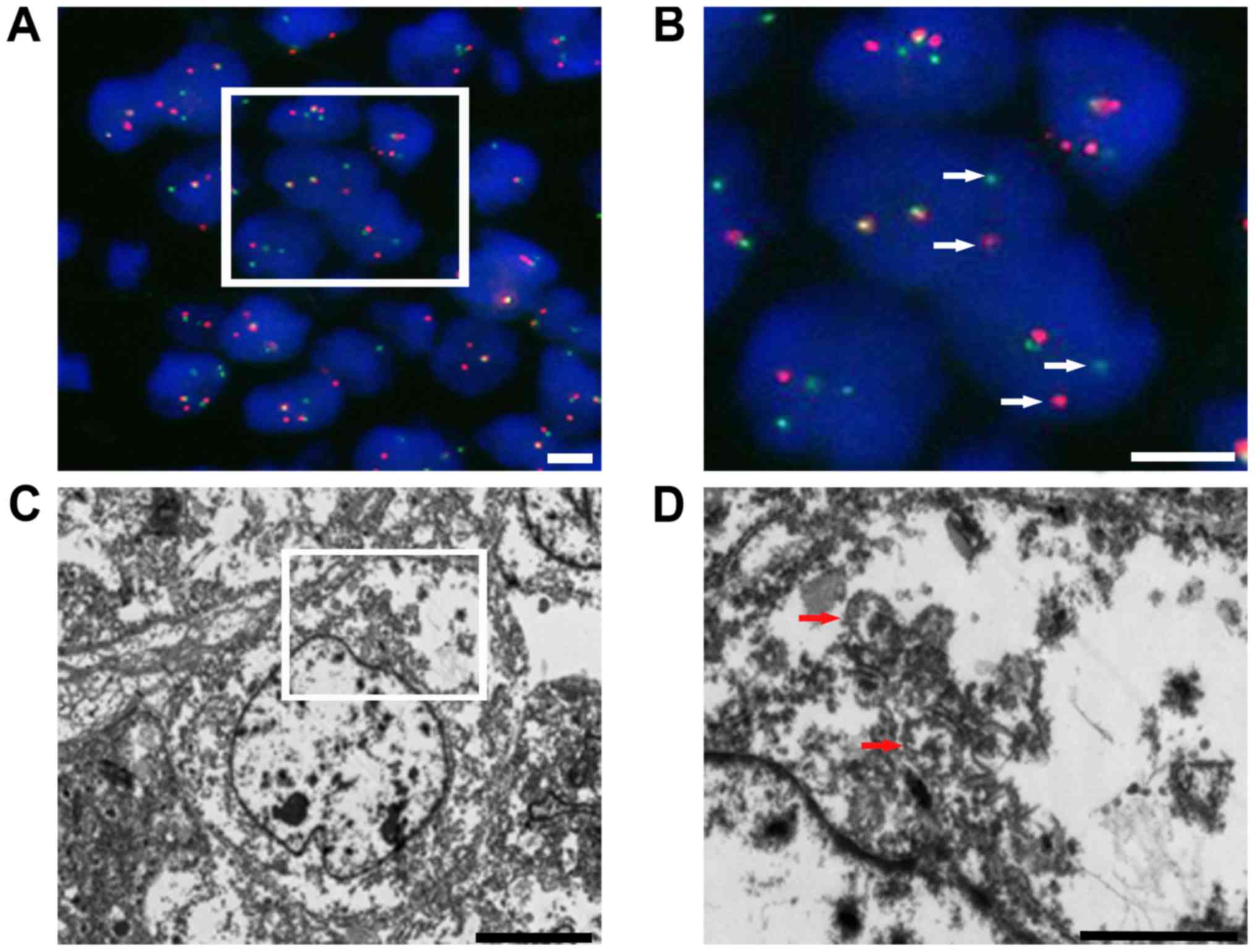
Melan-A, smooth muscle actin, CD3 and CD20. Fluorescence in
situ hybridization (FISH) analysis was performed as previously
described (5) with an Ewing sarcoma
breakpoint region 1 (EWSR1) break-apart probe that revealed the
splitting signal in 164 out of 200 nuclei, i.e., a significant
separation of the red and green signals, indicating the presence of
EWSR1 gene rearrangement (Fig. 5A and
B). Ultrastructurally, no typical melanosomes were identified,
but there were isolated dense-core secretory granules in the tumor
cell cytoplasm. The nuclei were irregularly shaped, and the
nucleoli were dense and occasionally prominent (Fig. 5C and D).
| Table II.Antibodies used in the
immunohistochemical analysis. |
Table II.
Antibodies used in the
immunohistochemical analysis.
| Antibodies | Catalogue
number | Supplier | Dilution |
|---|
|
Pan-cytokeratin | M351529-2 | Dako | 1:500 |
| Cytokeratin 7 | M701829-2 | Dako | 1:400 |
| Cytokeratin 20 | M701929-2 | Dako | 1:400 |
| S-100 | ab4066 | Abcam | 1:600 |
| SOX-10 | ab212843 | Abcam | 1:600 |
| Vimentin | M072501-2 | Dako | 1:800 |
| CD56 | M730401-2 | Dako | 1:500 |
| Synaptophysin | M731529-2 | Dako | 1:300 |
| Chromogranin-A | M086929-2 | Dako | 1:300 |
| CD117 | M3260 | Spring Bioscience;
Roche | 1:200 |
| DOG-1 | ab53212 | Abcam | 1:300 |
| CDX-2 | 06970940001 | Roche | 1:300 |
| CD34 | M716529-2 | Dako | 1:200 |
| CD3 | 05493315001 | Roche | 1:200 |
| CD20 | 05493234001 | Roche | 1:200 |
| HMB-45 | M063429-2 | Dako | 1:500 |
| Melan-A | M719629-2 | Dako | 1:200 |
| Smooth muscle
actin | 06770886001 | Roche | 1:200 |
| Secondary
antibodies | P044701-2 &
P044801-2 | Dako | 1:1,000 |
The patient received 4 cycles of adjuvant
chemotherapy with ifosfamide (400 mg/day for 5 days) plus
epirubicin (40 mg/day for 1 day) following the intestinal segment
resection, as described previously (8,11). The
patient remained alive without tumor recurrence or metastasis
during the 6-month follow-up period from December 2017 to May 2018.
The patient received imaging examinations every 6 months and to
determine her prognosis an intensive follow-up schedule was
required.
Discussion
Since CCSLTGT was first described by Zambrano et
al (2) in 2003, it has remained
a controversial entity. In 1985, Alpers and Beckstead (25) reported a case of a malignant
neuroendocrine tumor of the jejunum with OLGCs, which particularly
resembled CCSLTGT histologically. However, an increasing number of
cases support CCSLTGT as an independent tumor entity that lacks
melanocytic differentiation at the light microscopic,
ultrastructural and protein levels (3–10). In
2012, Stockman et al (6)
proposed that this tumor entity should be re-designated as a GNET,
a term which has achieved increasing acceptance (7–9,11–13).
GNETs are rare and aggressive malignant neoplasms that
predominantly occur in younger and middle-aged adults (median age,
35 years) without sex predominance (Table I). In a review of the literature, all
reported cases of GNET arose within the abdominal cavity,
frequently involving the small intestine, stomach or colon
(2–9,11–24).
GNETs appear to progress aggressively with preferential metastases
to the liver and/or mesenteric lymph nodes (2,5,14,17–19,21,23,24).
Patients often present with abdominal pain, intestinal obstruction
or incidental image findings of an abdominal mass. Occasionally
non-specific symptoms, including weight loss and anemia, are
associated (1).
The etiology of GNETs is unknown. A total of 2 cases
of GNETs document a patient history of hepatoblastoma or Ewing's
sarcoma in early childhood, suggesting that genetic aberration in
the embryonic stage could be regarded as a risk for the oncogenesis
of this tumor (5,11). A single case occurred as a secondary
malignancy following irradiation for neuroblastoma in infancy,
suggesting that radiotherapy may also be regarded as a risk factor
for the development of this tumor later in life (26). Intra-abdominal granulomatous
inflammation in the present case, and immunoglobulin-4-related
sclerosing inflammation in another case have been reported with
this tumor, suggesting that immune factors may participate in its
progression (18). Furthermore, it
was found that the granulomatous nodules in the current case were
formed of abundant CD68-positive macrophages, indicating that
immune cells, particularly macrophages, may be associated with the
development of this tumor. Due to the absence of
melanocyte-specific markers and the significant expression of
SOX-10 protein, an important transcriptional factor responsible for
the development of the neural crest, GNETs are hypothesized to
originate from gastrointestinal neuroectodermal precursor cells
that are unable to differentiate into the melanocytic lineage
through uncertain etiology (6).
Macroscopically, GNETs typically arise within the
muscularis propria of the gastrointestinal tract, and often extend
into the submucosa and subserosa with a well-circumscribed and
pushing border (1,6). Certain tumors manifest as a polypoid
mass (2). These growth patterns
frequently lead to an ulcerated mucosa, or a thickening or
stenosing gastrointestinal wall (1,2,6). Histologically, tumor cells frequently
present with a solid, nested, pseudoalveolar or fascicular growth
pattern, occasionally forming an uncommon pseudopapillary,
microcystic or rosette-like architecture (1,2,6–8). The
majority of tumor cells are composed of medium and large-sized
ovoid, epithelioid or spindle cells with eosinophilic or clear
cytoplasm, occasionally with an oncocytic cytoplasm (1,6,12). The nuclei are often located centrally
and are polygonal. The nucleoli are not usually prominent, while
the nuclei occasionally display numerous macronucleoli (1,2,6). The presence of OLGCs and the absence of
melanin pigment detected by Fontana stain are characteristic
features distinguishing GNETs from other mesenchymal tumors of the
gastrointestinal tract, particularly soft tissue-type CCS
involvement of the gastrointestinal tract (1,4,6,14). CCS
may also feature multinucleated tumor-derived giant cells, but the
OLGCs in GNETs are not tumor-derived, and invariably appear to be
CD68-positive (2). As presented in
Table I, the OLGC feature was not
observed in a number of the cases of GNETs (5,6,8,9,11,12,14,17–19,21,22).
Immunohistochemically, S-100 protein positivity has
been found in all reported cases. Melanocyte-specific markers
(HMB-45, Melan-A and tyrosinase) are negative in the majority of
cases, indicating a deficiency in melanocytic differentiation. At
least one of the neuroendocrine markers (including chromogranin-A,
synaptophysin, neuron-specific enolase and CD56) is positive in the
majority of cases. A 100% positivity rate for SOX-10 protein
expression was found in all cases where SOX-10 immunostaining was
performed, further supporting the hypothesis that GNET arises from
a primitive neural crest cell lineage (6,12). GIST
markers, including CD117, DOG-1 and CD34, were negative in all
cases. The GNETs were also negative for desmin, smooth muscle actin
and pan-cytokeratin AE1/AE3, and just 1 case was reported to be
focally positive CAM5.2 (6).
Ultrastructurally, no melanosomes or melanosome-like structures
were generally identified, although rare pre-melanosomes (stage I)
were observed in 1 case (16). The
neoplastic cells commonly showed clear secretory vesicles,
dense-core secretory granules or multiple interdigitating cell
processes (6,14).
Genetically, GNETs are characterized by EWSR1
(22q12.2) gene rearrangement in the majority of investigated cases;
common fusion partners are the cAMP responsive element binding
protein 1 or activating transcription factor 1 CREB1 or ATF1 genes
(5,6,12,19–21).
However, Stockman et al (6)
and Joo et al (18) reported
that EWSR1 gene rearrangement was not detected by FISH in their
cases, indicating that other genetic events may also be associated
with GNET tumorigenesis. Fused in sarcoma (FUS; 16p11.2), a gene
that shares extensive nucleotide sequence homology with EWSR1, was
proposed by Stockman et al (6) as an alternative gene, but FUS had no
signal for gene rearrangement detection in all cases analyzed.
EWSR1 gene rearrangement has been identified in other distinctive
tumors, including Ewing sarcoma, hyalinizing clear cell carcinoma
of the salivary gland, myoepithelial carcinoma, extraskeletal
myxoid chondrosarcoma, myxoid liposarcoma, angiomatoid fibrous
histiocytoma and desmoplastic small round cell tumors (27–30).
Therefore, EWSR1 gene rearrangement is not a specific criterion for
GNETs, but can aid in confirming the diagnosis of a GNET.
In the patients with follow-up data, regional lymph
nodes and liver metastases were common (2,3,6,23). As
GNET cases are rare, no 5-year survival rate is available. The
reported cases include survival periods ranging from 3 to 106
months (6). The consensus treatment
for a GNET is a surgical resection of the involved bowel segment
followed by regular image monitoring for recurrence and metastasis
(1). Although 2 patients received
chemotherapy in previously reported cases, chemotherapy is not
regarded as an evidence-based practice for this tumor entity
(8,11).
The differential diagnosis of a CCSLTGT or GNET
includes adenocarcinoma, a GIST, a neuroendocrine tumor, CCS
involving the gastrointestinal tract, metastatic melanoma, an
MPNST, a malignant granular cell tumor and synovial sarcoma.
Microscopically, a poorly differentiated carcinoma usually has a
nested and sheet pattern composed of epithelial tumor cells,
potentially leading to a misdiagnosis. A panel of epithelial
markers of the gastrointestinal tract, including pan-cytokeratin
AE1/AE3, CK20 and CDX-2, will be useful for distinguishing poorly
differentiated carcinomas from GNETs. Although GISTs can display a
variety of morphologies, occasionally mimicking the structure and
morphology of GNETs, routine immunohistochemistry can exclude the
diagnosis of a GIST. GISTs usually express at least one marker from
CD117, DOG-1 and CD34, whereas GNETs are negative for all these
markers (31). Although the
expression of neuroendocrine markers is often found in GNETs, the
OLGCs and genetic translocation features are useful for excluding
neuroendocrine tumors (6).
Metastatic melanoma usually presents with a history of previous
skin melanoma and generally expresses HMB-45, Melan-A or
tyrosinase, and lacks EWSR1 rearrangement (1,6). Soft
tissue-type CCS typically occurs at tendons and aponeuroses, and
CCS involving the gastrointestinal tract is extremely rare
(13). CCS commonly displays
tumor-derived giant cells rather than non-tumor OLGCs, is positive
for HMB-45 and/or Melan-A, and contains melanin pigment, as
detected by Fontana stain (1,10).
SOX-10 is also positive in certain cases of CCS, so SOX-10
immunoreactivity is less useful (32). Genetic analysis is also not
beneficial for distinguishing CCS from GNETs due to the presence of
similar genetic hallmarks, whereas electron microscopy analysis
presenting numerous melanosomes in varying developmental stages is
of value in diagnosing CCS (33).
The mass of an MPNST may be connected to a nerve, and typically
occurs in a patient with a history of neurofibroma or schwannoma.
The epithelioid MPNST may show a similar histology to a GNET, while
it often shows a strong and diffuse immunostaining for S-100
protein when compared with GNETs. Malignant granular cell tumors
are exceedingly rare and may display similar immunophenotypes to
GNETs. It is occasionally difficult to make a definitive diagnosis
based on morphology, but genetic analysis is valuable in
distinguishing GNETs from malignant granular cell tumors (12). Although synovial sarcomas focally
express S-100 protein in up to 40% of cases, potentially confusing
the diagnosis, epithelial markers, including EMA and cytokeratins,
are commonly focally detectable in various types of synovial
sarcoma (34). This tumor is
characterized by the t(X;18)(p11;q11) translocation that results in
the SS18 nBAF chromatin remodeling complex subunit-SSX family
member 2 gene fusion, which is found exclusively in synovial
sarcoma (35). Transducin-like
enhancer of split 1 was also recently identified as a robust
diagnostic biomarker for synovial sarcoma and correlates with
t(X;18) (36).
In conclusion, the present study reports a case of a
GNET of the ileum with intra-abdominal granulomatous nodules, an
uncommon accompanying finding. A GNET is a rare tumor that shares
some features with certain other neoplasms. Unfamiliarity with the
features of GNETs by surgical pathologists can easily lead to a
misdiagnosis. Therefore, it is necessary to comprehensively assess
the clinical, gross, morphological, immunohistochemical, genetic
and even ultrastructural characteristics for a definitive diagnosis
of a GNET.
Acknowledgements
Not applicable.
Funding
The collection of data from FISH analysis and
electron microscopy in this study was supported by the National
Natural Science Foundation of China (grant no. 81360320) and the
Scientific Research and Technology Development Program of Guilin
(grant nos. 20180107-12 and 2016012702-3).
Availability of data and material
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
GXH made substantial contributions to acquisition of
the patient data and drafting the manuscript. QYC contributed to
the interpretation of the patient data. LLZ performed the FISH and
electron microscopy, and contributed to the data analysis. HC
contributed to the histological assessment and case diagnosis. HPZ
performed the histological examination. XFL performed the
immunochemical analysis. FT designed the study and reviewed the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Written informed consent for publication of clinical
details was obtained from the patient.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CCSLTGT
|
clear cell sarcoma-like tumor of the
gastrointestinal tract
|
|
CCS
|
clear cell sarcoma
|
|
OLGC
|
osteoclast-like giant cells
|
|
GNET
|
malignant gastrointestinal
neuroectodermal tumor
|
|
CCS-GI
|
soft tissue-type CCS of the
gastrointestinal tract
|
|
GIST
|
gastrointestinal stromal tumor
|
|
MPNST
|
malignant peripheral nerve sheath
tumor
|
|
FISH
|
fluorescence in situ
hybridization
|
|
EWSR1
|
Ewing sarcoma breakpoint region 1
|
|
FUS
|
fused in sarcoma
|
References
|
1
|
Wang J and Thway K: Clear cell
sarcoma-like tumor of the gastrointestinal tract: An evolving
entity. Arch Pathol Lab Med. 139:407–412. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zambrano E, Reyes-Mugica M, Franchi A and
Rosai J: An osteoclast-rich tumor of the gastrointestinal tract
with features resembling clear cell sarcoma of soft parts: Reports
of 6 cases of a GIST simulator. Int J Surg Pathol. 11:75–81. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Friedrichs N, Testi MA, Moiraghi L, Modena
P, Paggen E, Plötner A, Wiechmann V, Mantovani-Löffler L,
Merkelbach-Bruse S, Buettner R and Wardelmann E: Clear cell
sarcoma-like tumor with osteoclast-like giant cells in the small
bowel: Further evidence for a new tumor entity. Int J Surg Pathol.
13:313–318. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang W, Zhang X, Li D, Chen J, Meng K,
Wang Y, Lu Z and Zhou X: Osteoclast-rich tumor of the
gastrointestinal tract with features resembling those of clear cell
sarcoma of soft parts. Virchows Arch. 448:200–203. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thway K, Judson I and Fisher C: Clear cell
sarcoma-like tumor of the gastrointestinal tract, presenting as a
second malignancy after childhood hepatoblastoma. Case Rep Med.
2014:9843692014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Stockman DL, Miettinen M, Suster S,
Spagnolo D, Dominguez-Malagon H, Hornick JL, Adsay V, Chou PM,
Amanuel B, Vantuinen P and Zambrano EV: Malignant gastrointestinal
neuroectodermal tumor: Clinicopathologic, immunohistochemical,
ultrastructural, and molecular analysis of 16 cases with a
reappraisal of clear cell sarcoma-like tumors of the
gastrointestinal tract. Am J Surg Pathol. 36:857–868. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kong J, Li N, Wu S, Guo X, Gu C and Feng
Z: Malignant gastrointestinal neuroectodermal tumor: A case report
and review of the literature. Oncol Lett. 8:2687–2690. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao Z, Zhang D, Li W, Zhang L, Li Z and
Zhou J: Primary malignant neuroectodermal tumor of the ileum with
predominantly uncommon pseudopapillary architecture. Int J Clin Exp
Pathol. 7:8967–8971. 2014.PubMed/NCBI
|
|
9
|
Kansal S and Rao S: Malignant
gastrointestinal neuroectodermal tumor: A unique rare neoplasm.
Indian J Surg Oncol. 8:630–633. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kosemehmetoglu K and Folpe AL: Clear cell
sarcoma of tendons and aponeuroses, and osteoclast-rich tumour of
the gastrointestinal tract with features resembling clear cell
sarcoma of soft parts: A review and update. J Clin Pathol.
63:416–423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Insabato L, Guadagno E, Natella V, Somma
A, Bihl M, Pizzolorusso A, Mainenti PP, Apice G and Tornillo L: An
unusual association of malignant gastrointestinal neuroectodermal
tumor (clear cell sarcoma-like) and Ewing sarcoma. Pathol Res
Pract. 211:688–692. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boland JM and Folpe AL: Oncocytic variant
of malignant gastrointestinal neuroectodermal tumor: A potential
diagnostic pitfall. Hum Pathol. 57:13–16. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alyousef MJ, Alratroot JA, ElSharkawy T,
Shawarby MA, Al Hamad MA, Hashem TM and Alsayyah A: Malignant
gastrointestinal neuroectodermal tumor: A case report and review of
the literature. Diagn Pathol. 12:292017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Antonescu CR, Nafa K, Segal NH, Dal Cin P
and Ladanyi M: EWS-CREB1: A recurrent variant fusion in clear cell
sarcoma-association with gastrointestinal location and absence of
melanocytic differentiation. Clin Cancer Res. 12:5356–5362. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Venkataraman G, Quinn AM, Williams J and
Hammadeh R: Clear cell sarcoma of the small bowel: A potential
pitfall. Case report. APMIS. 113:716–719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Granville L, Hicks J, Popek E, Dishop M,
Tatevian N and Lopez-Terrada D: Visceral clear cell sarcoma of soft
tissue with confirmation by EWS-ATF1 fusion detection. Ultrastruct
Pathol. 30:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Comin CE, Novelli L, Tornaboni D and
Messerini L: Clear cell sarcoma of the ileum: Report of a case and
review of literature. Virchows Arch. 451:839–845. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Joo M, Chang SH, Kim H, Gardner JM and Ro
JY: Primary gastrointestinal clear cell sarcoma: Report of 2 cases,
one case associated with IgG4-related sclerosing disease, and
review of literature. Ann Diagn Pathol. 13:30–35. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lagmay JP, Ranalli M, Arcila M and Baker
P: Clear cell sarcoma of the stomach. Pediatr Blood Cancer.
53:214–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Terazawa K, Otsuka H, Morita N, Yamashita
K and Nishitani H: Clear-cell sarcoma of the small intestine
detected by FDG-PET/CT during comprehensive examination of an
inflammatory reaction. J Med Invest. 56:70–75. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shenjere P, Salman WD, Singh M, Mangham
DC, Williams A, Eyden BP, Howard N, Knight B and Banerjee SS:
Intra-abdominal clear-cell sarcoma: a report of 3 cases, including
1 case with unusual morphological features, and review of the
literature. Int J Surg Pathol. 20:378–385. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lasithiotakis K, Protonotarios A, Lazarou
V, Tzardi M and Chalkiadakis G: Clear cell sarcoma of the jejunum:
A case report. World J Surg Oncol. 11:172013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li DJ, Zhang XH, Huang WB, Meng K and Zhou
XJ: An osteoclast-rich tumor of the gastrointestinal tract with
features resembling clear cell sarcoma of soft parts: A case report
and review of the literature. Zhonghua Bing Li Xue Za Zhi.
34:757–758. 2005.(In Chinese). PubMed/NCBI
|
|
24
|
Huang HF, L Q, Bu H, Chen M, Chen HJ, Lin
YY and Zhang HY: Clear cell sarcoma of gastrointestinal tract:
Clinicopathologic analyses and review of literatures. J Clin Exp
Pathol. 30:383–388. 2014.
|
|
25
|
Alpers CE and Beckstead JH: Malignant
neuroendocrine tumor of the jejunum with osteoclast-like giant
cells. Enzyme histochemistry distinguishes tumor cells from giant
cells. Am J Surg Pathol. 9:57–64. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang JC, Chou AJ, Oeffinger KC, La Quaglia
MP and Wolden SL: Clear cell sarcoma of the gastrointestinal tract
after very low-dose therapeutic radiation therapy: A case report. J
Pediatr Surg. 47:1943–1945. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Thway K and Fisher C: Tumors with
EWSR1-CREB1 and EWSR1-ATF1 fusions: The current status. Am J Surg
Pathol. 36:e1–e11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Antonescu CR, Katabi N, Zhang L, Sung YS,
Seethala RR, Jordan RC, Perez-Ordoñez B, Have C, Asa SL, Leong IT,
et al: EWSR1-ATF1 fusion is a novel and consistent finding in
hyalinizing clear-cell carcinoma of salivary gland. Genes
Chromosomes Cancer. 50:559–570. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Antonescu CR, Dal Cin P, Nafa K, Teot LA,
Surti U, Fletcher CD and Ladanyi M: EWSR1-CREB1 is the predominant
gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes
Cancer. 46:1051–1060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanas MR, Rubin BP, Tubbs RR, Billings SD,
Downs-Kelly E and Goldblum JR: Utilization of fluorescence in situ
hybridization in the diagnosis of 230 mesenchymal neoplasms: An
institutional experience. Arch Pathol Lab Med. 134:1797–1803.
2010.PubMed/NCBI
|
|
31
|
West RB, Corless CL, Chen X, Rubin BP,
Subramanian S, Montgomery K, Zhu S, Ball CA, Nielsen TO, Patel R,
et al: The novel marker, DOG1, is expressed ubiquitously in
gastrointestinal stromal tumors irrespective of KIT or PDGFRA
mutation status. Am J Pathol. 165:107–113. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Karamchandani JR, Nielsen TO, van de Rijn
M and West RB: Sox10 and S100 in the diagnosis of soft-tissue
neoplasms. Appl Immunohistochem Mol Morphol. 20:445–450. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mukai M, Torikata C, Iri H, Mikata A,
Kawai T, Hanaoka H, Yakumaru K and Kageyama K: Histogenesis of
clear cell sarcoma of tendons and aponeuroses. An
electron-microscopic, biochemical, enzyme histochemical, and
immunohistochemical study. Am J Pathol. 114:264–272.
1984.PubMed/NCBI
|
|
34
|
Pelmus M, Guillou L, Hostein I,
Sierankowski G, Lussan C and Coindre JM: Monophasic fibrous and
poorly differentiated synovial sarcoma: immunohistochemical
reassessment of 60 t(X;18)(SYT-SSX)-positive cases. Am J Surg
Pathol. 26:1434–1440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ladanyi M, Antonescu CR, Leung DH,
Woodruff JM, Kawai A, Healey JH, Brennan MF, Bridge JA, Neff JR,
Barr FG, et al: Impact of SYT-SSX fusion type on the clinical
behavior of synovial sarcoma: A multi-institutional retrospective
study of 243 patients. Cancer Res. 62:135–140. 2002.PubMed/NCBI
|
|
36
|
Knosel T, Heretsch S, Altendorf-Hofmann A,
Richter P, Katenkamp K, Katenkamp D, Berndt A and Petersen I: TLE1
is a robust diagnostic biomarker for synovial sarcomas and
correlates with t(X;18): Analysis of 319 cases. Eur J Cancer.
46:1170–1176. 2010. View Article : Google Scholar : PubMed/NCBI
|