Distinctive clinicopathological features of Von Hippel‑Lindau‑associated hereditary renal cell carcinoma: A single‑institution study
- Authors:
- Published online on: March 1, 2019 https://doi.org/10.3892/ol.2019.10091
- Pages: 4600-4606
-
Copyright: © Hong et al. This is an open access article distributed under the terms of Creative Commons Attribution License.
Abstract
Introduction
Von Hippel-Lindau (VHL) disease (Online Mendelian Inheritance in Man no. 193300; http://www.omim.org/entry/193300) is an autosomal dominant inherited multisystemic tumor syndrome, which causes mutations of the tumor suppressor gene VHL located on chromosome 3p25-26 (1). The incidence of VHL syndrome is ~1/36,000–52,000 births worldwide, with a penetrance of ~90% at 65 years old in families with a history of VHL (2–4). VHL clinical manifestations include central nervous system hemangioblastoma, retinal hemangioblastoma, renal cell carcinoma (RCC), pheochromocytoma (PHEO), pancreatic cysts or tumors, endolymphatic sac tumors and epididymal or broad ligament cystadenoma (5,6). Based on these clinical symptoms, VHL syndrome is classified as clinical type I (PHEO-free) or clinical type II (PHEO). In addition, type II is subclassified as type IIA (absence of RCC), type IIB (presence of RCC) and type IIC (PHEO only) (7–9). The kidney is one of the numerous organs affected by VHL syndrome, and the incidence of RCC or renal cysts in VHL syndrome is 30–60% (10). The majority of patients with VHL have multiple bilateral kidney lesions and require numerous surgeries across their lifetime. However, repeated surgeries increase the risk of renal dysfunction or uremia, which eventually lead to dialysis treatment or renal transplantation. It is therefore crucial to establish the optimal time point for surgery, in order to minimize the number of procedures, to preserve kidney integrity as long as possible and to improve patient quality of life. Medical advances and a deeper comprehension of VHL disease have allowed median survival time to increase from 49 to 62 years (3,11). A better understanding of the clinicopathological characteristics and prognosis of VHL-associated hereditary RCC may therefore aid clinicians in making more rational treatment decisions for patients with VHL.
In the present study, 27 patients diagnosed with VHL according to established clinical criteria and genetic tests were included. Clinical characteristics were collected, and pathological features were reevaluated according to the latest diagnostic criteria in order to improve our understanding of VHL syndrome.
Materials and methods
Patients and tumor samples
A total of 27 patients with VHL who underwent radical or partial nephrectomy at Peking University First Hospital (Beijing, China) between January 2010 and April 2018 were included in the present study. Patients were definitively diagnosed with VHL according to clinical criteria and genetic tests performed on peripheral blood prior to surgery. The present study was approved by the Institutional Ethics Committee of Peking University First Hospital (Beijing, China). All patients provided written informed consent.
Patients with VHL with one of the following clinical diagnostic criteria were included: i) >1 hemangioblastoma in the central nervous system (CNS) or retina; ii) a single hemangioblastoma in the CNS or retina combined with a visceral complication, including multiple renal, pancreatic or hepatic cysts, PHEO or renal cancer, with the exception of epididymal and renal cysts; and iii) any of the aforementioned clinical symptom plus a family history of VHL (7,12). A genetic test was performed by direct sequencing of polymerase chain reaction products using multiplex ligation-dependent probe amplification (13). The results were compared with the normal sequence (13).
The clinicopathological characteristics of patients included in the present study comprised sex, age, affected organs, VHL mutation site, nucleotide and protein alterations, surgical procedure, tumor size, pathological type [according to the 2016 World Health Organization (WHO) Kidney Tumor Classification] (14), tumor stage [2010 American Joint Committee on Cancer Kidney Cancer Tumor-Node-Metastasis (TNM) Classification] (15), tumor grade [2016 WHO/International Society of Urological Pathology (ISUP)] (14) and prognosis. The tumor pathological type and grade were reevaluated according to the latest diagnostic pathology criteria.
Hematoxylin and eosin (H&E) staining and immunohistochemistry (IHC)
Tissue sections (thickeness, 4 µm) were fixed in 10% formalin-fixed at room temperature for 24–48 h. The paraffin-embedded RCC specimens were subjected to hematoxylin & eosin (H&E) staining at 4°C for 20 h, and examined by light microscopy (BX43; Olympus Corporation, Tokyo, Japan). The expression of RCC-associated molecular markers, including cluster of differentiation 10 (CD10), carbonic anhydrase 9 (CAIX), cytokeratin 7 (CK7) and Ki-67, was evaluated by IHC, as previously described (16). The primary antibodies against CD10 (cat. no. ZM-0283; 1:150), CAIX (cat. no. ZM-0161; 1:150), CK7 (cat. no. ZM-0071; 1:150) and Ki-67 (cat. no. ZM-0166; 1:150) were purchased from OriGene Technologies, Inc. (Beijing, China). Tissues were subsequently incubated with poly-peroxidase-conjugated anti-rabbit/mouse immunoglobulin G secondary antibody (cat. no., PV-9000; 200 µl/per slice; OriGene Technologies, Inc.) at room temperature for 1 h. The H&E and IHC stained sections were independently analyzed by two qualified pathologists.
The expression of RCC-associated molecular markers was semi-quantitatively assessed. A total of 10 representative fields of vision were randomly selected under high magnification (×400). The percentage of positive cells was determined as follows: % positive cells = (number of positive cells/number of total cells) ×100. The score was defined as negative (−), focally positive (+), moderately positive (++) and strongly positive (+++) for 0–5, 6–10, 11–50 and 51–100% positively stained cells, respectively. The expression levels of RCC-associated molecular markers were divided into two groups according to the aforementioned scores as follows: Low/negative expression (−, +) and high expression (++, +++).
Results
Clinical characteristics
In the present study, 27 patients with VHL from Peking University First Hospital were included (Table I). These patients comprised 13 (48.1%) men and 14 (51.9%) women. The mean age at the time of surgery was 42.0±14.9 years (range, 23.3–71.6 years). The majority of patients exhibited tumors in multiple organs (Fig. 1). According to the VHL clinical classification, 21 (77.8%) patients were defined as clinical type I, and six (22.2%) patients were defined as clinical type IIB. Additional genetic testing clarified that the number of patients who presented with VHL mutation on exon 1, 2 or 3 and on introns was nine (33.3%), seven (25.9%), 10 (37.0%) and two (7.4%), respectively. The majority of patients (14/27, 51.9%) had missense mutations, six (22.2%) had large deletions, three (11.1%) had nonsense mutations, two (7.4%) had frameshift mutations and two (7.4%) had splicing mutations. In addition, 19 patients underwent partial nephrectomy (open or laparoscopic-assisted in 12 and seven cases, respectively). Nephron-sparing surgery was the conventional therapy for VHL-associated RCC.
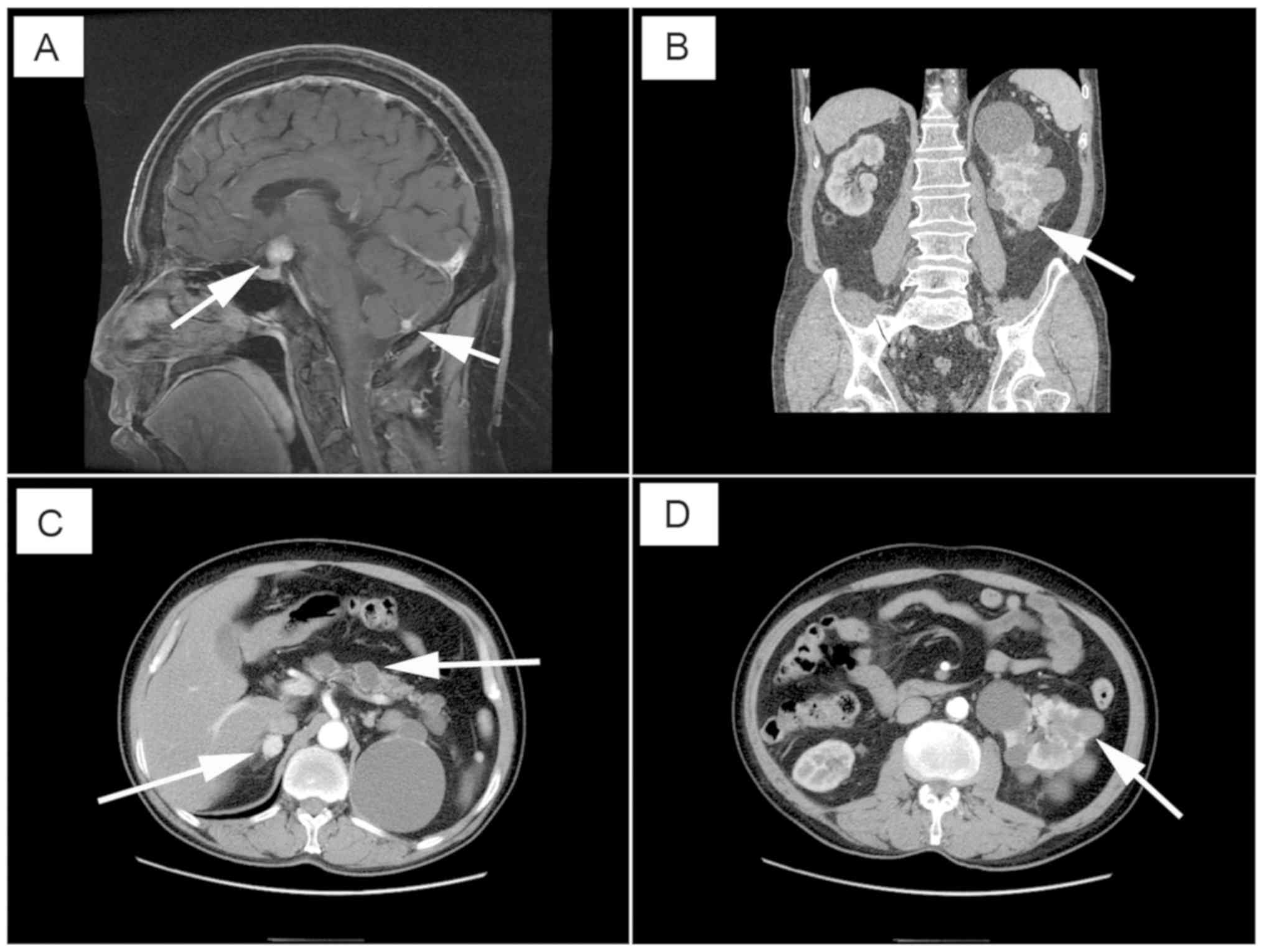
Table I.Clinical characteristics of the 27 patients with VHL- associated hereditary renal cell carcinoma. |
Pathological features
In the 27 patients with VHL, the mean size of the renal tumors was 4.3±2.0 cm (range, 1.3–9.5 cm; Table II). The pathological type of 26 (96.3%) cases was clear cell RCC (CCRCC). Open partial nephrectomy was conducted in one patient, and four renal tumors were resected. The pathological type of three tumors was CCRCC (Fig. 2), whereas the fourth tumor was diagnosed as clear cell papillary RCC (CCPRCC), according to IHC staining that indicated the following: CAIX (+++, goblet cell staining), CK7 (+++), CD10 (−) and P504S (−) (Fig. 2). The number of patients with TNM stages T1aN0M0, T1bN0M0, T2N0M0 and T3N0M0 was 14 (51.9%), seven (25.9%), two (7.4%) and four (14.8%), respectively. According to the 2016 WHO/ISUP guidelines, the number of patients with tumor grades 1, 2 and 3 was 16 (59.3%), eight (29.6%) and three (11.1%), respectively. In addition, renal cyst walls were collected during surgery. Pathological analysis of these tissues demonstrated that the renal cyst linings of seven patients were made of CCRCC cells (Fig. 3). A total of four RCC-associated molecular markers, namely CD10, CAIX, CK7 and Ki-67, were detected by IHC (Fig. 4). The expression rates of CD10, CAIX and CK7 were 63, 88.9 and 11.1%, respectively (Table II). All patients presented with low or negative Ki-67 expression levels.
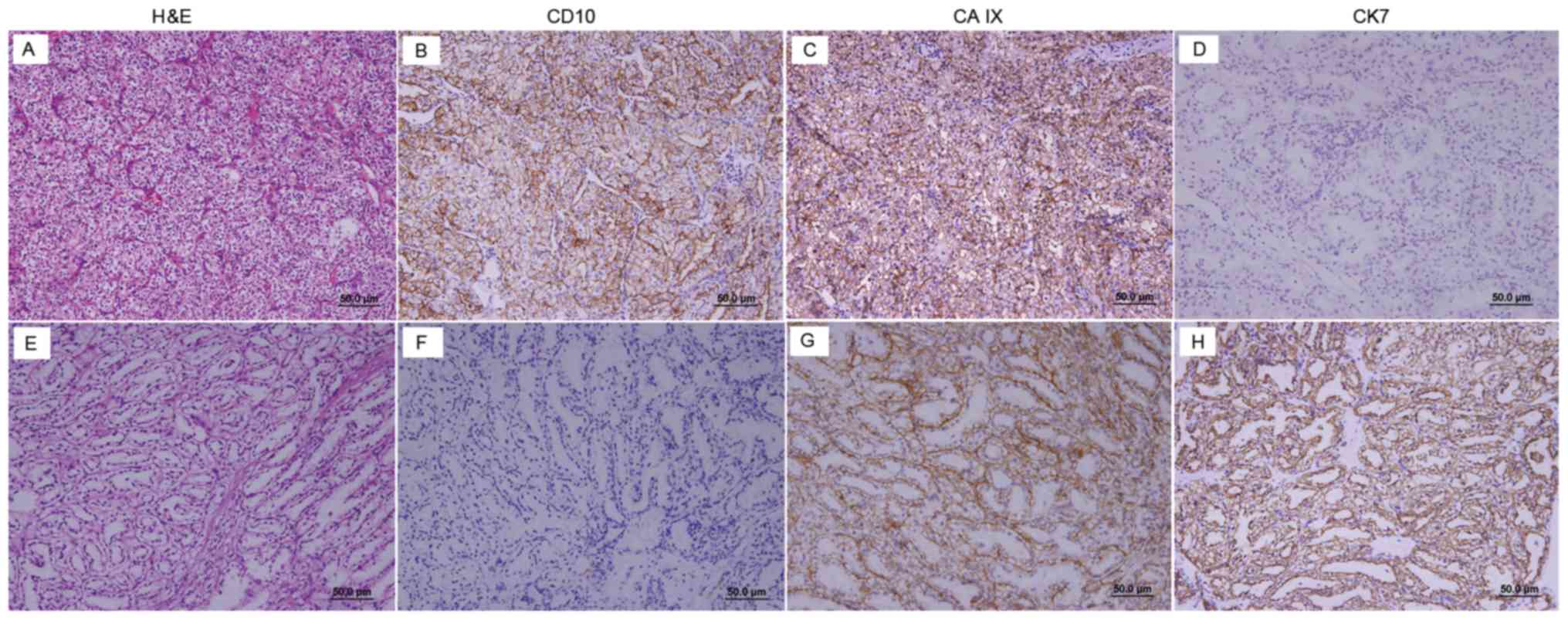
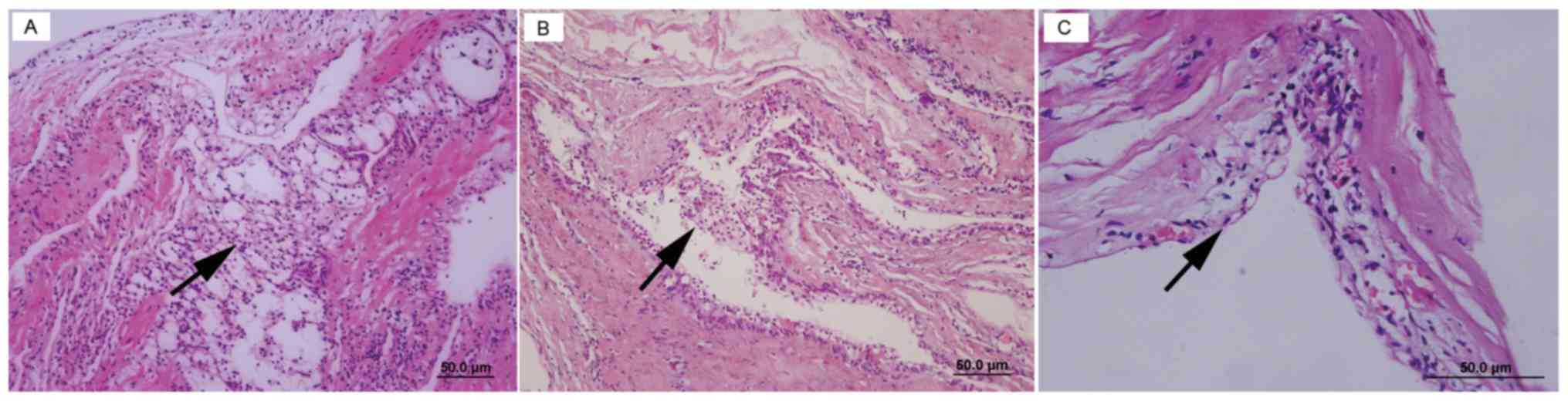
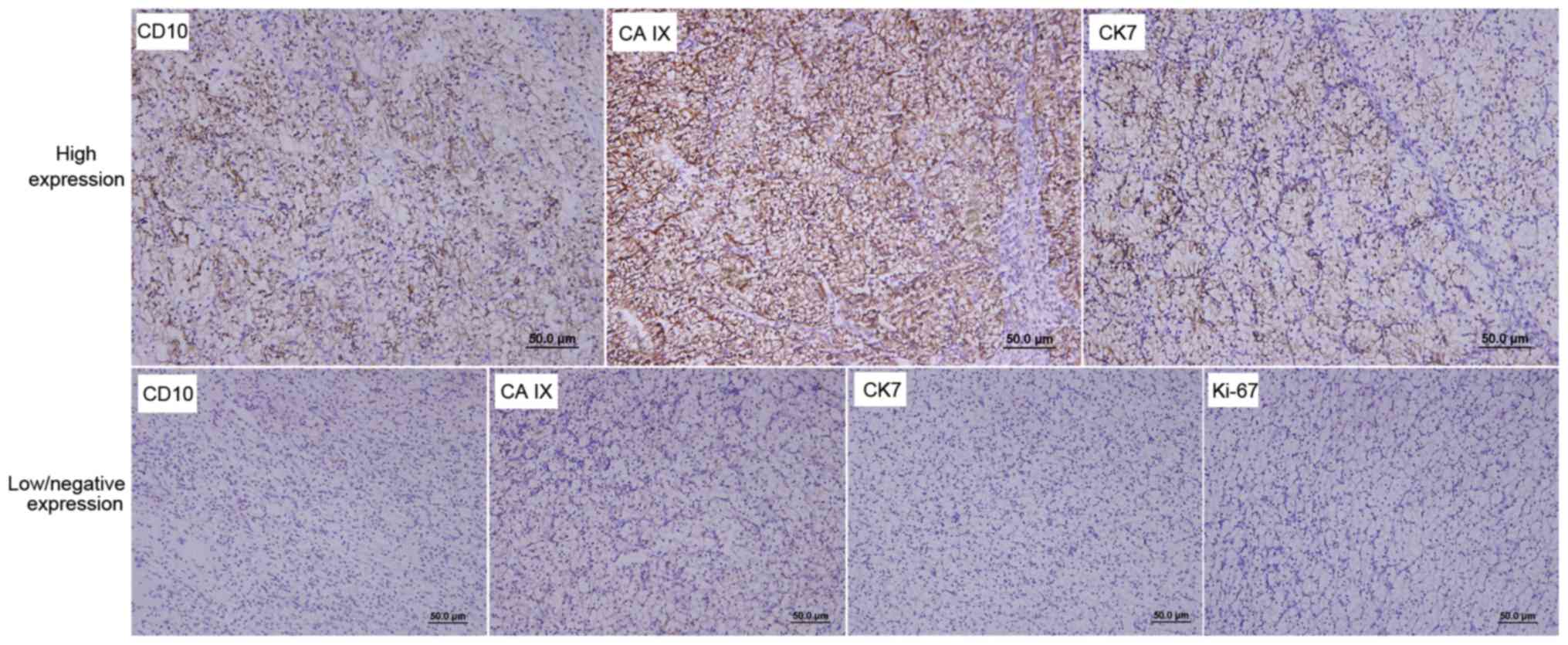
Table II.Pathological features of the 27 patients with VHL-associated hereditary renal cell carcinoma. |
Follow-up
The mean postoperative follow-up duration was 39.0±24.0 months (range, 1.7–96.5 months). No case of VHL-associated mortality was observed. The majority of patients had normal renal functions, with the exception of four patients, who presented with slightly higher creatinine or urea nitrogen levels than the upper normal limit. This mild abnormality however, had no significant effect on the patient's quality of life, as normal levels will be restored with treatment. It is therefore suggested that creatinine and urea nitrogen levels can reflect the renal function.
Discussion
VHL disease is a rare cancer syndrome that affects numerous organs. Multiple bilateral kidney tumors are the most common clinical manifestations of VHL. Previous studies have reported that the clinical characteristics and molecular mechanisms of VHL-associated RCC differ from those of sporadic RCC (17,18). In addition, the onset age of VHL-associated RCC is ~25 years old, which is earlier than that of sporadic RCC. Ning et al (19) reported that genetic anticipation is present in families with a VHL history and that telomere length shortening may be associated with this phenomenon. VHL-associated RCC is commonly multifocal, occurs in both kidneys and is combined with multiple renal cysts. In addition, simultaneous or metachronous bilateral kidney cancer can be present. Walther et al (20) reported that ~1,100 non-malignant cysts have a clear cell lining and that ~600 clear cell neoplasms are observed in the VHL-associated kidney. Furthermore, the pathological type of VHL-associated RCC is low-grade CCRCC. Due to later distant metastasis, VHL-associated RCC prognosis is moderately favorable, compared with that of sporadic RCC (17). Binderup et al (21) reported that the estimated mean life expectancy of a man with VHL is 67 years, whereas that for a woman with VHL is 60 years, which is higher than previously reported (49 years) (3). The results from our previous study demonstrated that the estimated median life expectancy is 62 years for Chinese patients with VHL disease (11). A deeper understanding of VHL syndrome, combined with the development of novel technology has therefore significantly improved the prognosis of affected patients.
In the present study, the clinicopathological characteristics of 27 patients with VHL who underwent nephrectomy at Peking University First Hospital were analyzed. The majority of patients maintained adequate renal function following surgery. Among these 27 patients, 19 underwent partial nephrectomy. Patients with VHL commonly undergo numerous renal surgeries across their lifetimes. Nephron-sparing surgery is therefore preferred for these patients. Surgeries are not completely curative, but they can prevent metastasis. However, the tumor size threshold for surgery indication is controversial. Duffey et al (22) reported that patients with VHL renal tumors of <3 cm did not present with metastasis. The 3-cm size was therefore proposed as the threshold for nephron-sparing surgery in patients with VHL. However, Gupta et al (23) reported that oncological outcomes of patients with hereditary RCC tumors of >4 cm were similar to those of patients with T1b tumors and sporadic RCC treated with partial nephrectomy. In the present study, the mean size of the largest renal tumors was 4.3 cm (range, 1.3–9.5 cm). No cases of VHL-associated mortality were observed during the mean 39 months of follow-up. Consequently, the 4-cm size may represent an appropriate threshold for nephron-sparing surgery, which may allow renal function preservation in patients with VHL and decrease the number of surgeries required across their lifetimes.
In the present study, renal cyst walls were collected from 10 patients with VHL during surgery. The pathological analyses indicated that seven renal cyst samples presented with a clear cell lining and RCC cell clusters that were scattered in the renal cyst cavities. These findings suggested that during surgery, the cyst fluid, which may contain RCC cells, should be prevented from flowing into the abdominal cavity to avoid RCC dispersion. Furthermore, the pathological characteristics of the renal tumors from patients with VHL were also evaluated according to the most recent diagnostic criteria. As previously reported, VHL-associated RCC is always of the clear cell subtype (24,25). In the present study, renal tumors with overlapping histological features of CCRCC and CCPRCC were observed. CCPRCC is a newly described renal epithelial tumor that was considered a novel RCC subtype in the 2012 ISUP Vancouver Classification of Renal Neoplasia. CCPRCC was initially reported in patients with end-stage renal disease; however, the clinicopathological features of this tumor subtype in VHL disease remain unclear. Rao et al (26) reported that CCPRCC represents a unique RCC pathological subtype and should be distinguished from CCRCC due to its different biological potentials. CCPRCC represents a rare subtype, but its prognosis is favorable (27). Conversely, the results from the present study indicated that inter and intratumor heterogeneity was present in VHL-associated hereditary RCC. VHL inactivation through genetic mutation is a common and early trigger that is followed by various additional mutations in different renal tumor lesions (28). In clinical practice, multiple site sampling is crucial to allow clinicians to better understand the biological potentials of the tumor.
Actual methods that determine risk stratification rely on tumor stage and grade. According to TNM staging, 21 patients (77.8%) in the present study were classified as T1N0M0 and 16 patients (59.3%) had a grade 1 tumor. The majority of the VHL-associated RCC cases were therefore considered as low-risk tumors due to their low TNM stage and nuclear grade. In addition, Ki-67 expression was evaluated by IHC. Ki-67 is a non-histone nuclear protein that is highly expressed during the cell cycle. The Ki-67 proliferative index indicates that cell proliferation is a predictive marker for tumor differentiation, invasion, metastasis and prognosis. In the present study, all tumors from patients with VHL exhibited low or negative Ki-67 expression. These findings suggested that when the degree of malignancy of VHL-associated RCC was relatively low, the prognosis was good.
In conclusion, VHL-associated RCC is a low-risk disease, which exhibits inter- and intratumor heterogeneity. A tumor size of ~4 cm may be considered as the threshold for nephron-sparing surgery in patients with VHL in order to reduce surgical frequency. In addition, the results from the present study suggested that, to prevent tumor cell dispersion, renal cysts should be treated with care. The present study does; however, have certain limitations, including the small patient number and its retrospective nature. Since VHL syndrome is a rare hereditary disease, it is hard to include a high number of patients with VHL in a study. In addition, kidneys can present with numerous tumors; however, only large tumors can be resected and not the tumor foci. It is therefore difficult to define recurrence in patients with VHL. Future studies will investigate the clinicopathological and survival data from a larger scale comparative study that includes patients with non-VHL-associated RCC. Understanding the clinicopathological characteristics and molecular mechanisms involved in VHL disease is crucial as it may aid clinicians in making rational treatment decisions.
Acknowledgements
The authors would like to thank Dr Dingfang Bu (Medical Experiment Center, Peking University First Hospital, Beijing, China) for providing technical assistance, Dr Qun He (Department of Urologic Pathology, Peking University First Hospital) for providing assistance with the immunohistochemistry and Dr Yuan Zhao (Department of Pathology, Beijing Chaoyang Hospital, Beijing, China) for providing assistance with immunohistochemistry.
Funding
This study was supported by the National Natural Science Foundation of China (grant no. 81572506) and the Special Health Development Research Project of Capital (grant no. 2016-2-4074).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
BH, ZZ and KG designed the study. ZZ, JZ, LC and NZ developed the methodology. JCZ, KM and JZ acquired the data. BH, JZ and KM wrote the manuscript. NZ, KG and LC were responsible for funding acquisition and project administration. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The present study was approved by the Institutional Ethics Committee of Peking University First Hospital (Beijing, China). Patients had been informed that important findings of the study would be published in a formal paper. Patients' names, ID numbers or other personally identifiable information will not be included in the paper. All patients provided written informed consent.
Patient consent for publication
All patients provided written informed consent.
Competing interests
The authors declare that they have no competing interests.
References
|
Gossage L, Eisen T and Maher ER: VHL, the story of a tumour suppressor gene. Nat Rev Cancer. 15:55–64. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Neumann HP and Wiestler OD: Clustering of features of von Hippel-Lindau syndrome: Evidence for a complex genetic locus. Lancet. 337:1052–1054. 1991. View Article : Google Scholar : PubMed/NCBI | |
|
Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT and Ferguson-Smith MA: Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 77:1151–1163. 1990. View Article : Google Scholar : PubMed/NCBI | |
|
Maher ER, Neumann HP and Richard S: von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet. 19:617–623. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Bader HL and Hsu T: Systemic VHL gene functions and the VHL disease. FEBS Lett. 586:1562–1569. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Nielsen SM, Rhodes L, Blanco I, Chung WK, Eng C, Maher ER, Richard S and Giles RH: Von Hippel-Lindau disease: Genetics and role of genetic counseling in a multiple neoplasia syndrome. J Clin Oncol. 34:2172–2181. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Nordstrom-O'Brien M, van der Luijt RB, van Rooijen E, van den Ouweland AM, Majoor-Krakauer DF, Lolkema MP, van Brussel A, Voest EE and Giles RH: Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 31:521–537. 2010.PubMed/NCBI | |
|
Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, Gnarra JR, Orcutt ML, Duh FM, Glenn G, et al: Germline mutations in the von Hippel-Lindau disease tumor suppressor gene: Correlations with phenotype. Hum Mutat. 5:66–75. 1995. View Article : Google Scholar : PubMed/NCBI | |
|
Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M and Kaelin WG Jr: von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 10:1019–1027. 2001. View Article : Google Scholar : PubMed/NCBI | |
|
Richard S, David P, Marsot-Dupuch K, Giraud S, Béroud C and Resche F: Central nervous system hemangioblastomas, endolymphatic sac tumors, and von Hippel-Lindau disease. Neurosurg Rev. 23:1–24. 2000. View Article : Google Scholar : PubMed/NCBI | |
|
Wang JY, Peng SH, Li T, Ning XH, Liu SJ, Hong BA, Liu JY, Wu PJ, Zhou BW, Zhou JC, et al: Risk factors for survival in patients with von Hippel-Lindau disease. J Med Genet. 55:322–328. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Wu P, Zhang N, Wang X, Ning X, Li T, Bu D and Gong K: Family history of von Hippel-Lindau disease was uncommon in Chinese patients: Suggesting the higher frequency of de novo mutations in VHL gene in these patients. J Hum Genet. 57:238–243. 2012. View Article : Google Scholar : PubMed/NCBI | |
|
Wang JY, Peng SH, Ning XH, Li T, Liu SJ, Liu JY, Hong BA, Qi NN, Peng X, Zhou BW, et al: Shorter telomere length increases age-related tumor risks in von Hippel-Lindau disease patients. Cancer Med. 6:2131–2141. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Moch H, Cubilla AL, Humphrey PA, Reuter VE and Ulbright TM: The 2016 WHO classification of tumours of the urinary system and male genital organs-part a: Renal, penile, and testicular tumours. Eur Urol. 70:93–105. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Kim SP, Alt AL, Weight CJ, Costello BA, Cheville JC, Lohse C, Allmer C and Leibovich BC: Independent validation of the 2010 American Joint Committee on Cancer TNM classification for renal cell carcinoma: Results from a large, single institution cohort. J Urol. 185:2035–2039. 2011. View Article : Google Scholar : PubMed/NCBI | |
|
Ning XH, Li T, Gong YQ, He Q, Shen QI, Peng SH, Wang JY, Chen JC, Guo YL and Gong K: Association between FBP1 and hypoxia-related gene expression in clear cell renal cell carcinoma. Oncol Lett. 11:4095–4098. 2016. View Article : Google Scholar : PubMed/NCBI | |
|
Neumann HP, Bender BU, Berger DP, Laubenberger J, Schultze-Seemann W, Wetterauer U, Ferstl FJ, Herbst EW, Schwarzkopf G, Hes FJ, et al: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol. 160:1248–1254. 1998. View Article : Google Scholar : PubMed/NCBI | |
|
Choyke PL, Glenn GM, Walther MM, Patronas NJ, Linehan WM and Zbar B: Von Hippel-Lindau disease: Genetic, clinical, and imaging features. Radiology. 194:629–642. 1995. View Article : Google Scholar : PubMed/NCBI | |
|
Ning XH, Zhang N, Li T, Wu PJ, Wang X, Li XY, Peng SH, Wang JY, Chen JC and Gong K: Telomere shortening is associated with genetic anticipation in Chinese Von Hippel-Lindau disease families. Cancer Res. 74:3802–3809. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Walther MM, Lubensky IA, Venzon D, Zbar B and Linehan WM: Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: Clinical implications. J Urol. 154:2010–2015. 1995. View Article : Google Scholar : PubMed/NCBI | |
|
Binderup ML, Jensen AM, Budtz-Jørgensen E and Bisgaard ML: Survival and causes of death in patients with von Hippel-Lindau disease. J Med Genet. 54:11–18. 2017. View Article : Google Scholar : PubMed/NCBI | |
|
Duffey BG, Choyke PL, Glenn G, Grubb RL, Venzon D, Linehan WM and Walther MM: The relationship between renal tumor size and metastases in patients with von Hippel-Lindau disease. J Urol. 172:63–65. 2004. View Article : Google Scholar : PubMed/NCBI | |
|
Gupta GN, Peterson J, Thakore KN, Pinto PA, Linehan WM and Bratslavsky G: Oncological outcomes of partial nephrectomy for multifocal renal cell carcinoma greater than 4 cm. J Urol. 184:59–63. 2010. View Article : Google Scholar : PubMed/NCBI | |
|
Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM and Oldfield EH: Von Hippel-Lindau disease. Lancet. 361:2059–2067. 2003. View Article : Google Scholar : PubMed/NCBI | |
|
Crespigio J, Berbel L, Dias MA, Berbel RF, Pereira SS, Pignatelli D and Mazzuco TL: Von Hippel-Lindau disease: A single gene, several hereditary tumors. J Endocrinol Invest. 41:21–31. 2018. View Article : Google Scholar : PubMed/NCBI | |
|
Rao P, Monzon F, Jonasch E, Matin SF and Tamboli P: Clear cell papillary renal cell carcinoma in patients with von Hippel-Lindau syndrome-clinicopathological features and comparative genomic analysis of 3 cases. Hum Pathol. 45:1966–1972. 2014. View Article : Google Scholar : PubMed/NCBI | |
|
Diolombi ML, Cheng L, Argani P and Epstein JI: Do clear cell papillary renal cell carcinomas have malignant potential. Am J Surg Pathol. 39:1621–1634. 2015. View Article : Google Scholar : PubMed/NCBI | |
|
Kim E and Zschiedrich S: Renal cell carcinoma in von Hippel-Lindau disease-from tumor genetics to novel therapeutic strategies. Front Pediatr. 6:162018. View Article : Google Scholar : PubMed/NCBI |