Introduction
Breast cancer is the most common type of cancer in
women, accounting for 25% of all cases of cancer (1). According to gene expression profiles,
the molecular features of breast cancer are divided as follows:
Human epidermal growth factor receptor 2-positive, a basal subtype
and two luminal subtypes (2). In
addition, infiltration of various types of immune cells is commonly
observed in breast cancer. For instance, emerging evidence has
demonstrated that lymphocytic infiltration in breast cancer is
generally associated with a favorable prognosis (3–6).
Interleukin 15 receptor α (IL15RA) is a component of
the IL15R, which shares a β and γ subunit with the IL2 receptor
(7). IL15-dependent signaling
regulates the activation and proliferation of T cells and natural
killer cells, and modulates the adaptive immune response (8). Marra et al (9) demonstrated that IL15 and IL15RA are
frequently upregulated in breast cancer and breast cancer cell
lines. The authors suggested that upregulation of IL15 and IL15RA
induces an antitumor immune response by activating peripheral blood
mononuclear cell synthesis, and subsequently improves the prognosis
of patients with lymphocyte-enriched breast cancer. However, the
underlying mechanisms of the effects of IL15RA expression on breast
cancer remain unknown.
The present study aimed to investigate the molecular
mechanisms underlying the antitumor effects induced by IL15RA
upregulation and to identify a gene signature for breast cancer
prognosis. Since IL15RA/cg09290866 hypermethylation leads to the
suppression of IL15RA expression, and hypomethylation elevates the
expression of IL15RA, the present study aimed to highlight the
differentially expressed genes (DEGs) detected in hypermethylated
and hypomethylated IL15RA breast cancer samples compared to normal
samples. Univariate Cox regression analysis identified the
prognosis-associated DEGs. In silico analyses of the
expression profiles of these prognosis-associated DEGs enabled the
generation of a four-gene signature, capable of predicting the odds
of patient outcome, in the training and test sets. The four-gene
signature may allow the classification of patients with IL15RA
hypermethylation in the training set into a high-risk group and a
low-risk group with significantly different survival times. In
addition, the DEGs between the two risk groups were identified, and
their possible biological roles were studied using Gene Ontology
(GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analyses.
Materials and methods
The Cancer Genome Atlas (TCGA) and
Gene Expression Omnibus (GEO) datasets
The present study acquired the gene expression and
methylation data of 316 breast cancer samples and 21 normal tissue
samples from The Cancer Genome Atlas (TCGA) data portal (https://portal.gdc.cancer.gov/projects)
based on Illumina Methylation 450 platform and Illumina HiSeq 2000
RNA Sequencing (Illumina, Inc., San Diego, CA, USA). These samples
were defined as the training set (TCGA set). The corresponding
clinical characteristics are shown in Table I.
 | Table I.Clinical summary of patients in TCGA
and GEO datasets. |
Table I.
Clinical summary of patients in TCGA
and GEO datasets.
| Clinical
features | TCGA (n=316) | GSE39004
(n=57) |
|---|
| Pathologic_M
(M0/M1/MX) | 299/9/8 | – |
| Pathologic_N
(N0/N1/N2/N3/NX) | 167/95/25/21/8 | – |
| Pathologic_T
(M1/M2/M3/M4/MX) | 84/187/28/16 | – |
| Pathologic_stage
(I/II/III/IV/–) | 57/185/52/9/13 | 3/40/14/0/0 |
| Age (>60/≤60/–,
years) | 154/162 | 17/38/2 |
| ER_Status
(positive/negative/–) | 238/69/9 | 29/28 |
| HER2_Status
(positive/negative/–) | 56/243/17 | – |
| PR_Status
(positive/negative/–) | 201/106/9 | – |
| Radiation_therapy
(Yes/No/–) | 6/18/292 | – |
|
Pharmaceutical_therapy (Yes/No) | 11/14/291 | – |
| Hormone_therapy
(Yes/No) | – | 23/33/1 |
| Mortality
(Dead/Alive) | 36/280 | 24/33 |
| Overall survival
days (months, mean ± SD) | 29.07±34.73 | 57.61±38.03 |
The GSE37751 dataset (10), including the gene expression data of
human breast tumor samples, and the GSE39004 dataset, (10) consisting of DNA methylation profiles
of the same human breast tumor samples, were downloaded from the
National Center of Biotechnology Information (NCBI) GEO database
(http://www.ncbi.nlm.nih.gov/geo/). A
total of 57 samples with paired gene expression and DNA methylation
data were selected as the test set (GEO set). Clinical
characteristics of the test set are displayed in Table I.
DEGs screening
According to the methylation value of
IL15RA/cg09290866 (β-value), samples from the training set were
categorized into a hypermethylation group (β-value≥0.3) and a
hypomethylation group (β-value<0.3). A false discovery rate
(FDR)<0.05 and |log fold change (FC)|>0.585 were considered
as the strict cutoff thresholds, and the DEGs were subsequently
screened by comparing the DEGs in the hypermethylation or
hypomethylation samples with those in the normal samples using
Limma package (11) (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
in R3.1.0 language. The overlapping DEGs in both hypermethylation
and hypomethylation samples were selected for further analysis.
Association of DEGs with patient
survival
Univariate Cox regression analysis (12) was performed to evaluate the
association of the overlapping DEGs with patient survival using
survival package (https://cran.r-project.org/web/packages/survival/index.html)
in R3.1.0 language. The DEGs that were significantly associated
with patient survival were identified (log-rank P<0.05) and
ordered.
Risk score
The risk score was calculated for each patient based
on the linear combination of expression levels of the DEGs with the
regression coefficient as follows:
Risk score = βgene 1 × exprgene 1 + βgene 2 ×
exprgene 2 +···+ βgene n × exprgene n
where ‘β gene n’ describes the estimated regression
coefficient of the gene n derived from the univariate Cox
regression analysis, and ‘exprgene n’ describes the expression
level of the gene n.
By considering the median risk score of the
hypermethylation group as the cutoff value, the hypermethylation
samples in the training set were divided into a high-risk and a
low-risk subgroup. By applying the β-value derived from the
training set, the samples in the test set were also classified by
risk score into a high-risk and a low-risk subgroup.
Statistical analysis
The overall survival (OS) time of the risk groups
was analyzed using Kaplan-Meier (KM) survival analysis followed by
log-rank test. Multivariate Cox regression analysis and data
stratification analysis were conducted to determine the association
of risk score with other clinical characteristics in TCGA and GEO
sets. P<0.05 was considered to indicate a statistically
significant difference. Hazard ratios (HR) and 95% confidence
intervals (CI) were calculated.
Functional enrichment analysis
In the training set, the DEGs between the high-risk
and the low-risk groups were also identified using the Limma
package, with the threshold set at FDR<0.05. Correlations of the
DEGs selected with risk score were characterized using cor function
(https://www.rdocumentation.org/packages/stats/versions/3.4.1/topics/cor)
in R language. The DEGs with correlation coefficient 0–1 were
defined as positive DEGs, whereas the DEGs with correlation
coefficient −1–0 were defined as negative DEGs. The positive or
negative DEGs were ranked according to their r. The top 20 positive
DEGs and the top 20 negative DEGs underwent GO (13) function and KEGG (14) pathway enrichment analyses using the
Database for Annotation, Visualization and Integrated Discovery
(DAVID) (15). There are three types
of GO terms, including biological processes (BP), cellular
compartment (CC), and molecular function (MF). GO terms and KEGG
pathways with FDR<0.05 were considered as significant functional
annotations.
Results
Identification of DEGs
There were 226 hypermethylation samples
(β-value≥0.3), 90 hypomethylation samples (β-value<0.3) and 21
normal samples in the training set. A total of 326 overlapping DEGs
(FDR<0.05, |logFC|>0.585) were identified in the
hypomethylation and hypermethylation samples relative to the normal
samples.
Development of a four-gene
signature-based risk-scoring model
The genes associated with prognosis were selected
from the overlapping DEGs previously identified in the training set
using univariate Cox regression analysis. These genes were then
used to build a risk-scoring model for prognosis. Eventually, the
SRC homology 3 (SH3) domain, SH3 and cysteine rich domain 2
(STAC2), proline rich 11 (PRR11), homeobox C11 (HOXC11) and
nucleolar and spindle associated protein 1 (NUSAP1) genes were
selected to calculate the risk score based on their Cox regression
coefficients as follows:
Risk score = −1.744862×Exp STAC2 +
−0.969505×Exp KCND3 + 1.404081×Exp HOXC11 +
−4.4925×Exp NUSAP1
A risk score was assigned to each patient in the
hypermethylation group. By using the 50th percentile cutoff of risk
score as the threshold, all hypermethylation samples were
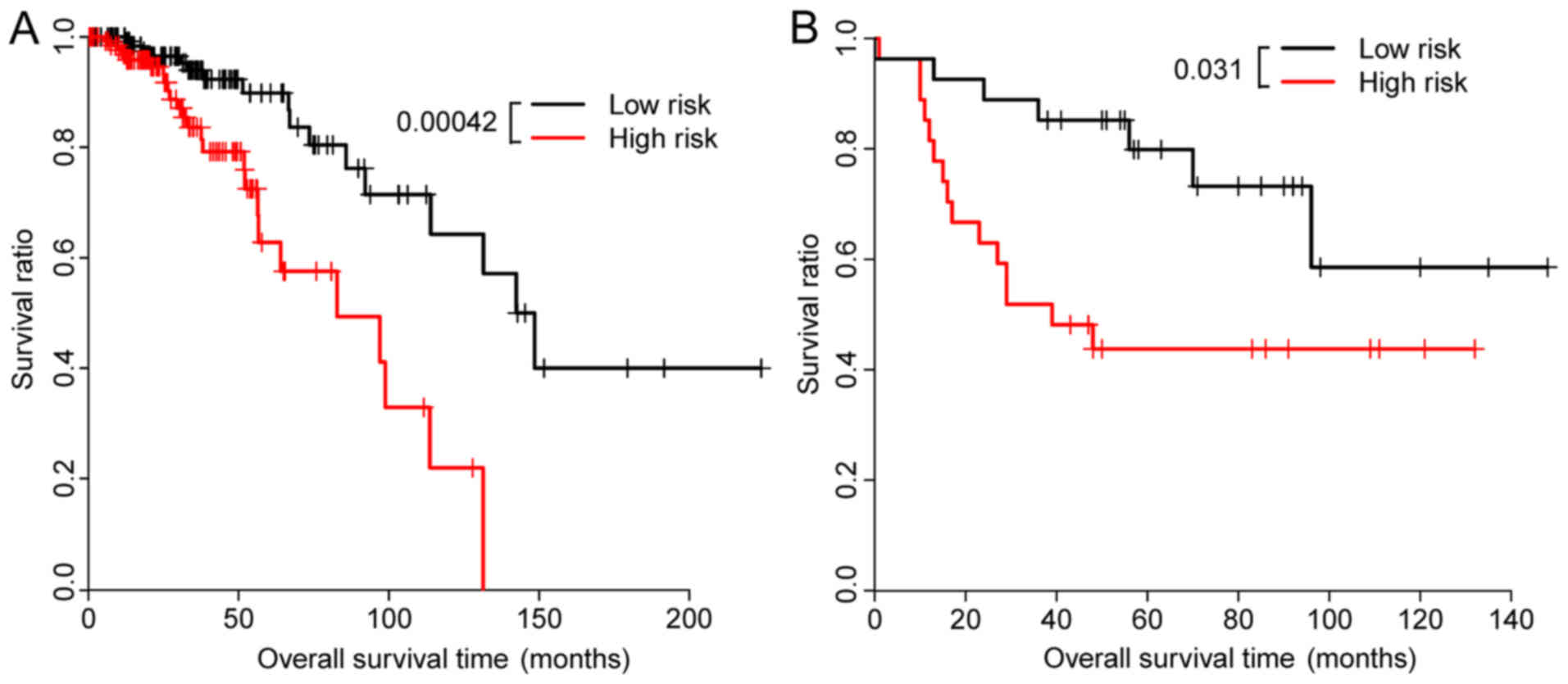
classified into a high-risk and a low-risk subgroup. The low-risk
subgroup exhibited significantly elongated survival time compared
with the high-risk subgroup (median OS: 647 days vs. 92 days;
P=0.00042; Fig. 1A).
To validate the prognostic performance of the
four-gene signature in the test set, the patients with breast
cancer in the test set were also dichotomized into low-risk and
high-risk subgroups using the 50th percentile cutoff of risk score
as the threshold. Similarly, better survival was observed in the
low-risk group compared with the high-risk group (median OS: 1,792
days vs. 1,209 days; P=0.031; Fig.
1B).
Expression of the four prognostic
genes in different risk groups
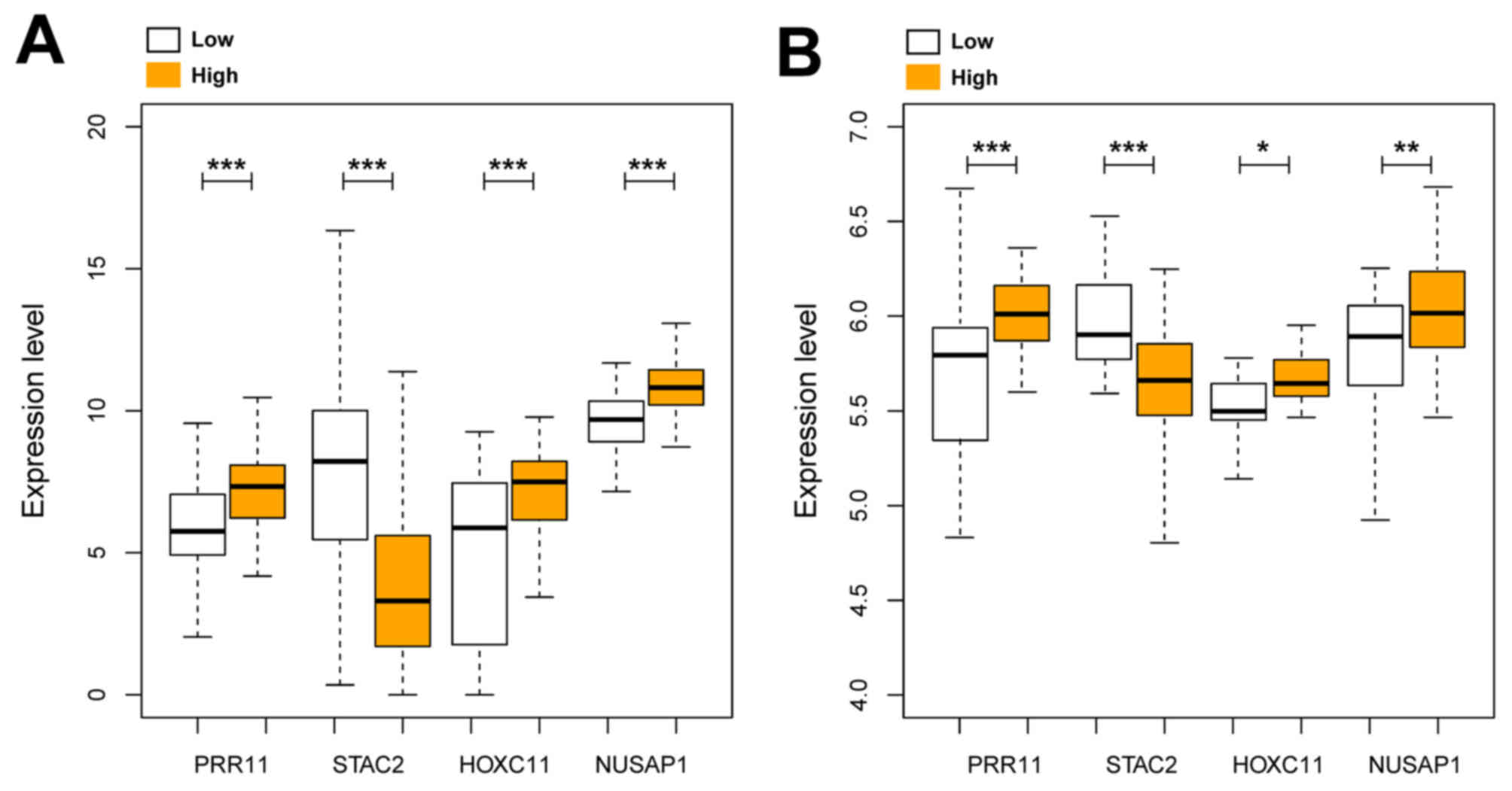
As shown in Fig. 2,
the high-risk group presented significantly increased expression
levels of PRR11, HOXC11 and NUSAP1, and significantly decreased
expression of STAC2 compared to the low-risk subgroup in the
training set (Fig. 2A; PRR11,
P<0.005; STAC2, P<0.005; HOXC11, P<0.005; NUSAP1,
P<0.005) and the test set (Fig.
2B; PRR11, P<0.005; STAC2, P<0.005; HOXC11, P<0.05;
NUSAP1, P<0.001).
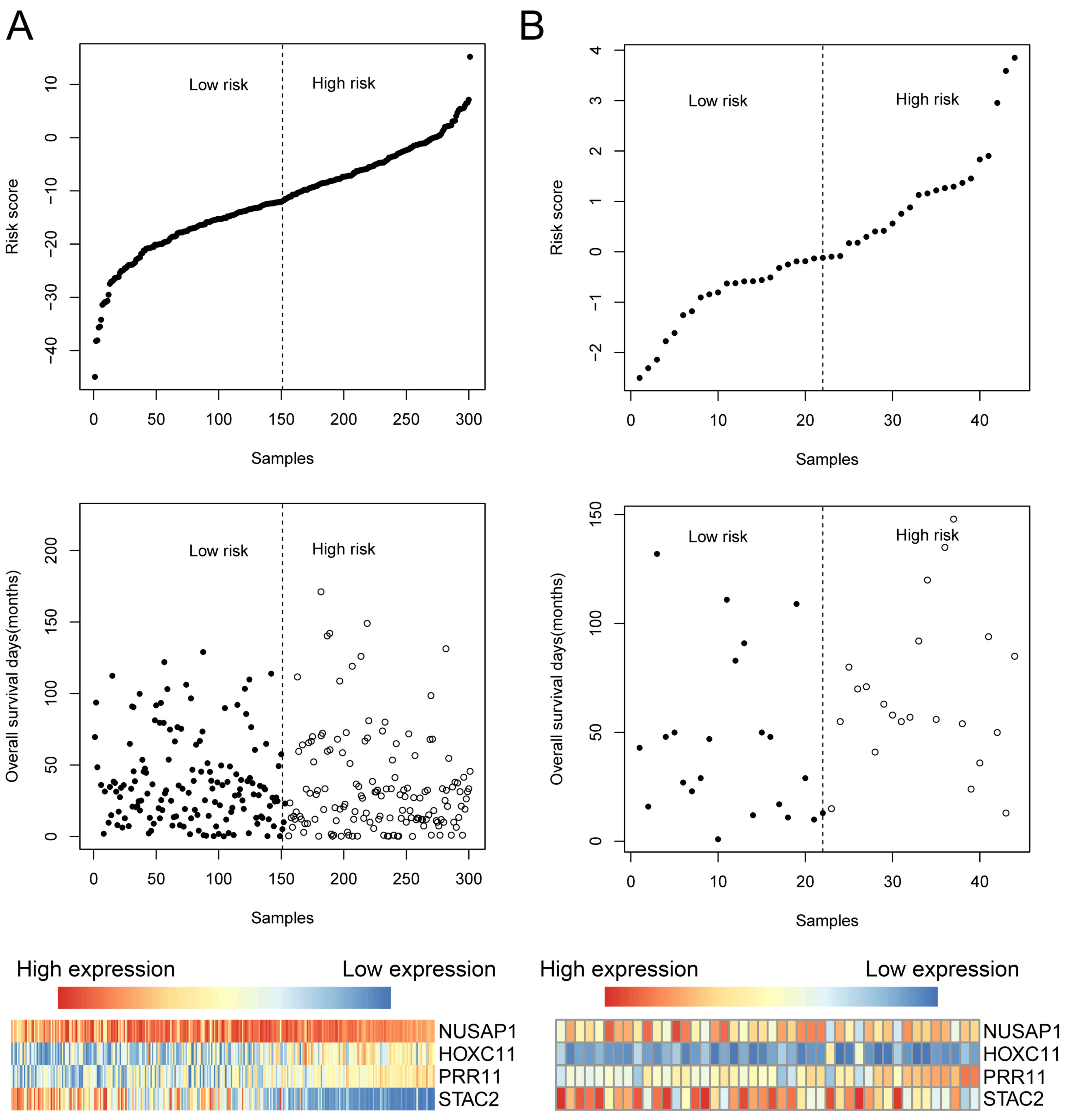
Fig. 3 displayed the
four-gene risk score distribution, OS time of patients and a
heatmap showing the expression of STAC2, PRR11, HOXC11 and
NUSAP1 in the training set (Fig.
3A) and the test set (Fig. 3B).
In the group of patients with high-risk score, PRR11, HOXC11
and NUSAP1 were upregulated, whereas STAC2 was
downregulated.
Four-gene signature is an independent predictor for
OS. The results of univariate Cox regression analysis for the
training set and the test set demonstrated that the four-gene risk
score (training set: P=3.01×10−05, HR=1.086,
CI=1.044–1.128; test set: P=0.0288, HR=0.827, CI=0.582–1.175) and
the pathological stage (training set: P=0.00472, HR=1.813,
CI=1.200–2.738; test set: P=0.0448, HR=2.646, CI=1.023–3.845) were
significantly associated with the survival of patients with breast
cancer (Table II). Furthermore, the
four-gene risk score (training set: P=0.0032, HR=1.069,
CI=1.023–1.117; test set: P=0.0205, HR=0.780, CI=0.534–1.141) and
the pathological stage (training set: P=0.0047, HR=1.198,
CI=0.487–2.947; test set: P=0.0663, HR=2.458, CI=0.941–3.42) were
independent predictors of survival according to the multivariate
Cox regression analysis, which included clinical variables for both
sets (Table II).
| Table II.Regression coefficients from
univariate and multivariate Cox regression analysis. |
Table II.
Regression coefficients from
univariate and multivariate Cox regression analysis.
| A, TCGA |
|---|
|
|---|
|
| Univariate Cox | Multivariable
Cox |
|---|
|
|
|
|
|---|
| Variable | P-value | HR (CI 95%) | P-value | HR (CI 95%) |
|---|
| pathologic_T | 0.156 | 1.398
(0.881–2.220) | – | – |
| ER_Status | 0.549 | 0.771
(0.328–1.808) | – | – |
| HER2_Status | 0.731 | 0.831
(0.288–2.396) | – | – |
| pathologic_M | 0.014 | 3.480
(1.284–9.430) | 0.094 | 2.941
(1.676–2.947) |
| pathologic_N | 0.017 | 1.517
(1.077–2.136) | 0.072 | 1.009
(0.656–1.846) |
| Age | 0.048 | 1.030
(1.000–1.060) | 0.126 | 1.026
(0.993–1.061) |
| PR_Status | 0.025 | 0.426
(0.202–0.897) | 0.053 | 0.304
(0.132–0.698) |
|
pathologic_stage | <0.005 | 1.813
(1.200–2.738) | <0.005 | 1.198
(0.487–2.947) |
| Risk score | <0.005 | 1.086
(1.044–1.128) | <0.005 | 1.069
(1.023–1.117) |
|
| B,
GSE39004 |
|
|
| Univariate
Cox | Multivariable
Cox |
|
|
|
|
|
Variable | P-value | HR (CI) | P-value | HR (CI) |
|
| Ethnicity | 0.185 | 0.515
(0.193–1.376) | – | – |
| ER_Status | 0.171 | 0.521
(0.205–1.326) | – | – |
| Triple | 0.818 | 1.128
(0.402–3.167) | – | – |
| Grade | 0.302 | 1.424
(0.728–2.787) | – | – |
|
Neoadjuvant-therapy | 0.621 | 0.601
(0.0797–1.527) | – | – |
|
Hormone-therapy | 0.262 | 0.581
(0.225–1.501) | – | – |
| Chemotherapy | 0.204 | 0.547
(0.216–1.387) | – | – |
| Stage | 0.045 | 2.646
(1.023–3.845) | 0.0663 | 2.458
(0.941–3.42) |
| Age | 0.016 | 1.039
(1.007–1.071) | 0.0125 | 1.036
(1.008–1.128) |
| Risk score | 0.029 | 0.827
(0.582–1.175) | 0.0205 | 0.780
(0.534–1.141) |
Correlation analysis between risk
score and clinical characteristics of patients
To explore whether the prognostic power of the
four-gene signature was independent of other clinical features,
including age, pathologic_M, N, stage and progesterone receptor
(PR) status, the data stratification analysis was carried out for
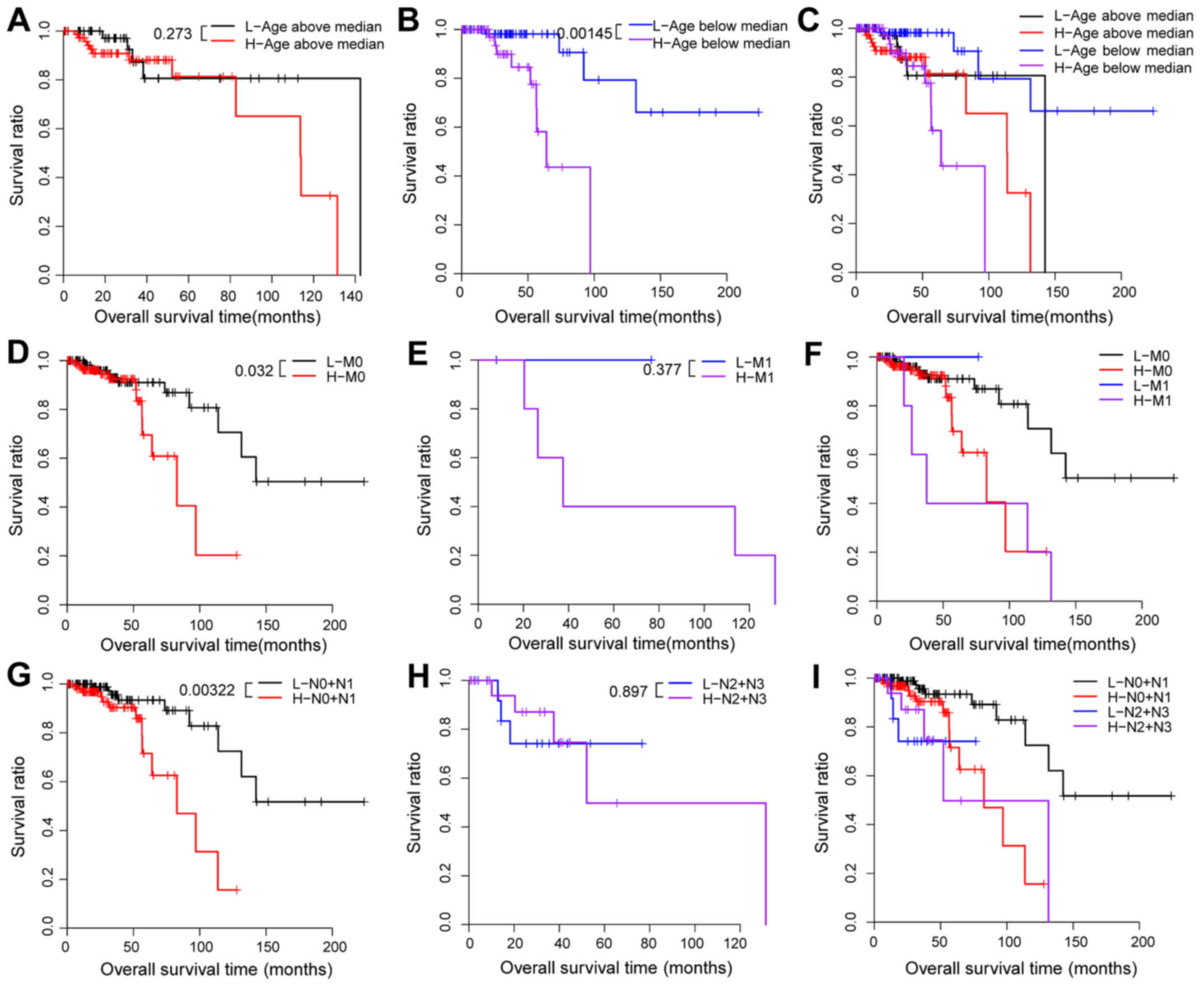
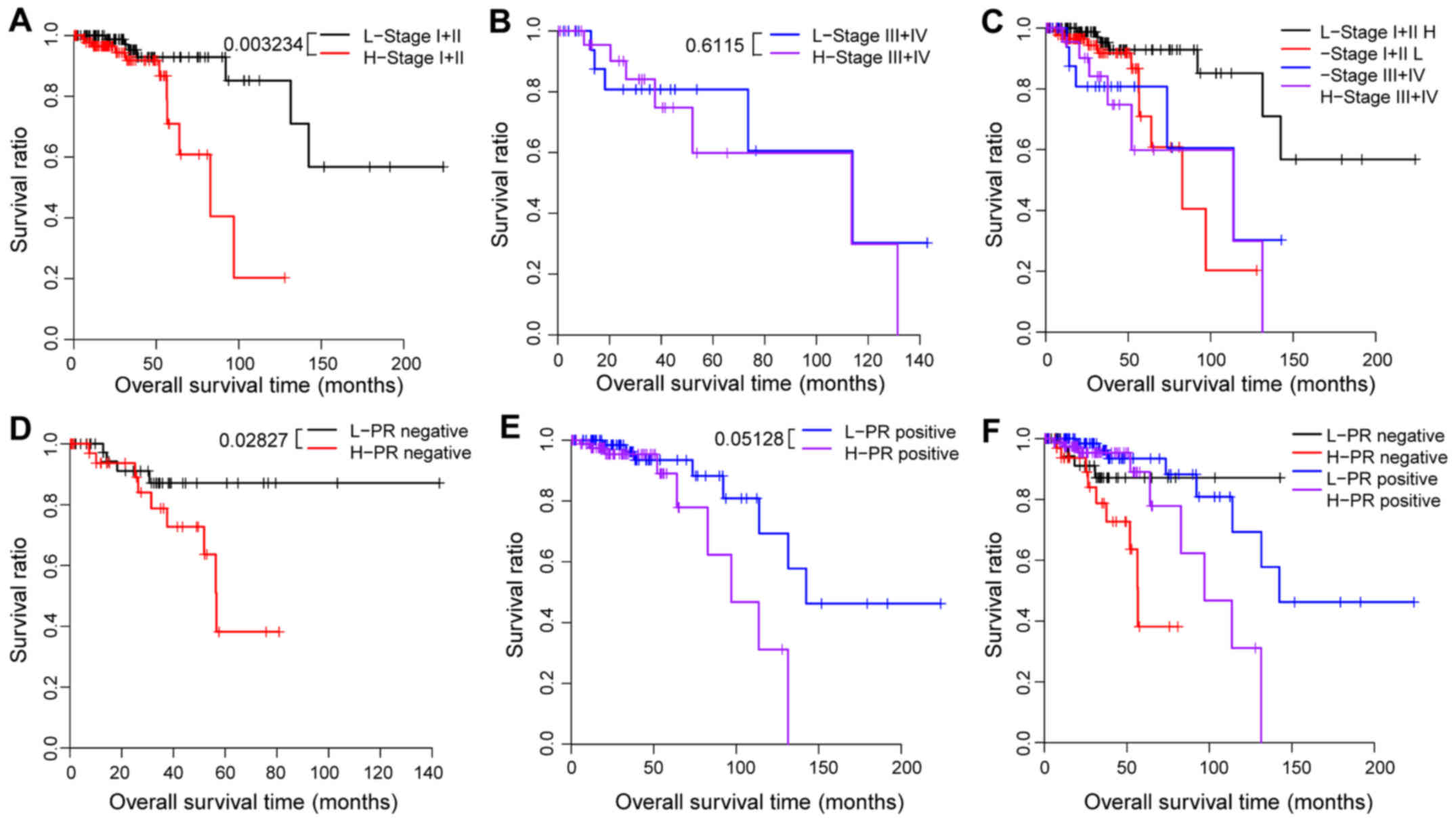
each clinical feature in the training set (Table III and Figs. 4 and 5). Firstly, all hypermethylated samples of
the training set were stratified by age into an older dataset
(>60 years, n=132) and a younger dataset (≤60 years, n=132). In
addition, the older dataset was classified into a high-risk group
and a low-risk group: however, no significant difference in OS time
was observed between the two risk groups (P=0.273; Table III; Fig.
4A and C). The younger dataset was classified according to the
risk score and divided into a high-risk and a low-risk group with
significantly different OS time (P=0.0015; Table III, Fig.
4B and C). Subsequently, all hypermethylated samples were
stratified by pathologic_M into an M0 dataset (n=284) and an M1
dataset (n=9). The M0 dataset was divided according to the risk
score into a high-risk and a low-risk group. Difference in OS time
between the two risk groups was significant (P=0.032; Table III, Fig.
4D and F). Nevertheless, the risk score that classified the M1
dataset into a high-risk and a low-risk group did not present
significant difference in the OS time (P=0.3766; Table III, Fig.
4E and F). In addition, all hypermethylated samples were
stratified by pathologic_N into an N0 + N1 dataset (n=227) and an
N2 + N3 dataset (n=37). A significant difference in OS time between
the high-risk and the low-risk group was observed in the N0 + N1
dataset (P=0.0032; Table III,
Fig. 4G and I) but not in the N2 +
N3 dataset (P=0.897; Table III,
Fig. 4H and I). Based on the
pathologic_stage, these samples were categorized into a stage I +
II dataset (n=215) and a stage III + IV dataset (n=49). In the
stage I + II dataset, the difference in OS time was significantly
different between the two risk groups (P=0.0032; Table III, Fig.
5A). However, in the stage III + IV dataset, the survival time
was not significantly different between the high-risk and the
low-risk group (P=0.6115; Table
III, Fig. 5B and C). These
samples were eventually grouped into a PR_positive (n=173) and a
PR_negative dataset (n=91) according to their PR_status. Difference
in OS time between the high-risk and the low-risk group was
significant in the PR_positive dataset (P=0.0283; Table III, Fig.
5D) but not in the PR_negative dataset (P=0.0513; Table III, Fig.
5E and F). These results indicated that the predictive capacity
of the four-gene risk score for prognosis may be associated with
the aforementioned clinical features.
| Table III.Results of data stratification
analysis for the training set. |
Table III.
Results of data stratification
analysis for the training set.
| Variable | P-value |
|---|
| Age (>60 years,
n=132) | 0.273 |
| Age (<60 years,
n=132) | <0.005 |
| pathologic_M (M0,
n=284) | 0.032 |
| pathologic_M (M1,
n=9) | 0.377 |
| pathologic_N (N0 +
N1, n=227) | <0.005 |
| pathologic_N (N2 +
N3, n=37) | 0.897 |
| pathologic_stage (I
+ II, n=215) | <0.005 |
| pathologic_stage
(III + IV, n=49) | 0.612 |
| PR_Status
(Positive, n=173) | <0.005 |
| PR_Status
(Negative, n=91) | 0.051 |
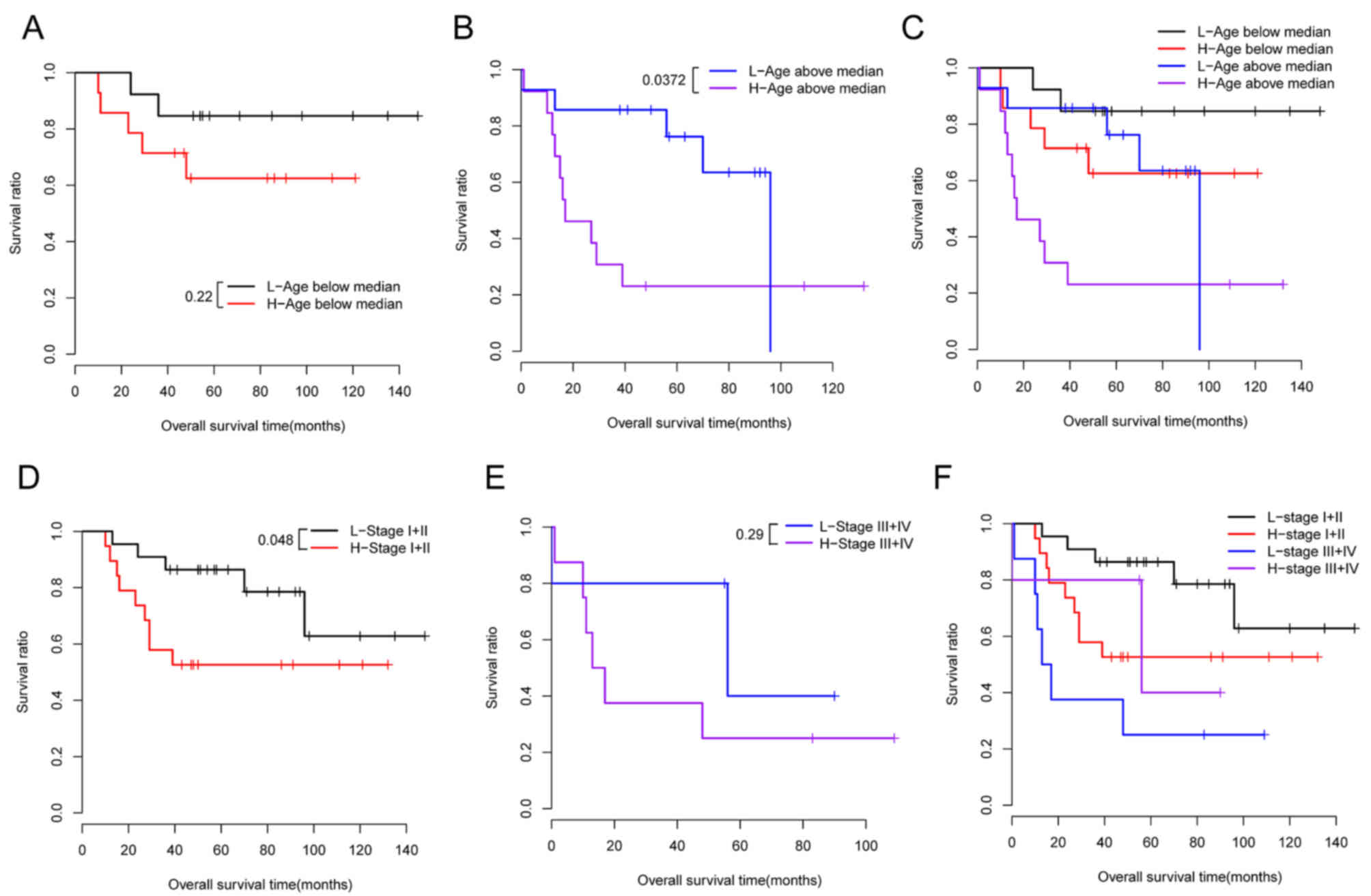
Data stratification analysis was also conducted for
the GEO dataset. All breast cancer samples in the GEO dataset were
stratified by age and pathological stage (Fig. 6). The difference in the OS time
between the high-risk and the low-risk group was not significant in
the younger dataset (P=0.22; Fig.
6A), however, was significant in the older dataset (P=0.0372;
Fig. 6B and C). In addition, the
difference in the OS time between the high-risk and the low-risk
group was significant in the stage I + II dataset (P=0.048;
Fig. 6D), however, was not
significant in the stage III + IV dataset (P=0.29; Fig. 6E and F).
Functional analysis
In order to identify the molecular mechanisms
underlying the prognostic performance of this four-gene signature,
the present study investigated the DEGs between the high-risk and
the low-risk group of the IL15RA hypermethylated samples in the
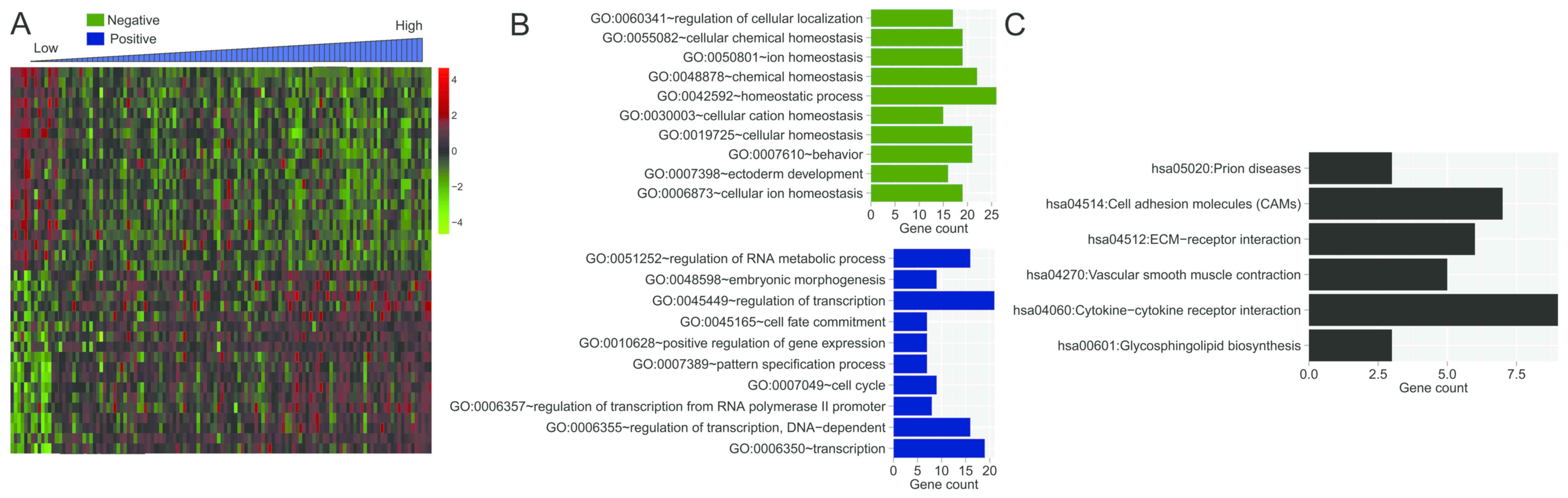
training set. Subsequently, among the 308 DEGs retrieved, 224 were
negatively associated with the risk score (negative DEGs) and 84
were positively associated with the risk score (positive DEGs).
These positive or negative DEGs were ranked according to the
correlation coefficient. A list of the top 20 positive DEGs and top
20 negative DEGs is shown in Table
IV, and includes STAC2, PRR11 and HOXC11. Hierarchical cluster
analysis revealed that the gene expression pattern of high-risk
samples was different from that of low-risk samples (Fig. 7A). In order to determine the possible
biological roles of the top 20 positive and top 20 negative DEGs,
GO function and KEGG pathway enrichment analysis were carried out.
As shown in Fig. 7B, positive DEGs
were mainly associated with several transcription-related GO BP
terms, whereas negative DEGs were mostly associated with many
cellular homeostasis-related GO BP terms. These genes were
significantly associated with several KEGG pathways, such as
‘ECM-receptor interaction’ pathway and ‘cell adhesion molecules
(CAMs)’ pathway (Fig. 7C).
| Table IV.Top 20 negative DEGs and top 20
positive DEGs. |
Table IV.
Top 20 negative DEGs and top 20
positive DEGs.
| A, Negative
DEGs |
|---|
|
|---|
| Gene | Correlation
coefficients |
|---|
| STAC2 | −0.626 |
| SFRP1 | −0.470 |
| MSRA | −0.435 |
| KCNMB1 | −0.425 |
| SAA1 | −0.421 |
| KRT17 | −0.419 |
| PDLIM4 | −0.409 |
| TRIM29 | −0.409 |
| KRT14 | −0.409 |
| TMEM220 | −0.408 |
| SPRY2 | −0.406 |
| KRT5 | −0.400 |
| TP63 | −0.398 |
| SAA2 | −0.394 |
| COL17A1 | −0.391 |
| KLK5 | −0.389 |
| KRT15 | −0.389 |
| TINAGL1 | −0.384 |
| NTRK2 | −0.382 |
| ID4 | −0.381 |
|
| B, Positive
DEGs |
|
| Gene | Correlation
coefficients |
|
| PRR11 | 0.562 |
| INTS2 | 0.523 |
| SERINC5 | 0.417 |
| HOXC11 | 0.406 |
| TAOK1 | 0.401 |
| UHMK1 | 0.383 |
| BRIP1 | 0.374 |
| GAS2L3 | 0.370 |
| CASC5 | 0.358 |
| CCNT1 | 0.358 |
| DDI2 | 0.353 |
| CLSPN | 0.345 |
| HOXC13 | 0.342 |
| CENPI | 0.338 |
| HOXC10 | 0.333 |
| SHCBP1 | 0.331 |
| EPN3 | 0.329 |
| CCNE2 | 0.323 |
| ZBTB37 | 0.322 |
| E2F8 | 0.319 |
Discussion
Breast cancer is a common type of cancer in woman
(16). It has been reported that
high expression of IL15RA contributes to better survival outcomes
for patients with breast cancer (9).
The present study explored the molecular mechanisms underlying the
positive effect of IL15RA upregulation on patient prognosis. The
results detected 326 DEGs in the hypomethylation and
hypermethylation samples compared to normal samples. A four-gene
signature was then identified and risk score for prognosis was
calculated. The gene signature consisted of STAC2, PRR11,
HOXC11 and NUSAP1. The four-gene signature-based risk
score enabled classification of patients with IL15RA
hypermethylated breast cancer in the training set into a high-risk
and a low-risk group that had significantly different survival
times. The prognostic performance of this four-gene signature-based
risk score was verified in the GEO set. Furthermore, the four-gene
risk score was a significant prognostic factor in the multivariate
Cox regression analysis. These results suggested that the use of
this four-gene signature may allow for prognostic prediction in
patients with breast cancer. This also suggested that IL15RA may
affect the development and progression of breast cancer partly via
modulating the expression of these DEGs, thus influencing the
prognosis of patients.
In the present study, the prognostic gene signature
for breast cancer included STAC2, PRR11, HOXC11 and
NUSAP1. STAC2 belongs to a small family of SH3 and
cysteine-rich containing adaptor proteins (STAC1, STAC2 and STAC3),
and is expressed in various types of tissue (17). Few reports concern STAC2 in breast
cancer. PRR11 belongs to the family of proline-rich proteins. Zhou
et a (18) reported that
PRR11 promotes the invasion of breast cancer cells by regulating
epithelial-to-mesenchymal transition (EMT). The findings from the
present study suggested that IL15RA may regulate EMT by controlling
PRR11 expression. HOXC11 is a member of the homeobox
family of genes, which encode transcription factors involved in
morphogenesis (19). It has been
demonstrated that HOX genes serve important roles in cancer
development by regulating several biological processes, including
apoptosis, differentiation and angiogenesis (20). Makiyama et al (21) reported that HOXC11 expression
is decreased in breast cancer tissues compared to non-cancerous
tissues. Soon et al (22)
revealed that the interaction of HOXC11 with the steroid receptor
coactivator SRC-1 is responsible for the development of endocrine
therapy resistance in breast cancer and suggested that nuclear
HOXC11 is associated with survival. Nucleolar-spindle associated
protein (NuSAP1), encoded by the NUSAP1 gene, is a
microtubule- and chromatin-binding protein that is critical for the
spindle assembly in the cytokinesis stages of mitosis (23). Subsequently, the dysregulated
expression of NuSAP1 leads to cancer development (24). A growing body of evidence also
reported that NuSAP1 is associated with various types of cancer,
including cervical cancer, lung adenocarcinoma and prostate cancer
(25–27). Furthermore, it has been demonstrated
that elevated expression of NuSAP1 is predictive of poor prognosis
in triple-negative breast cancer (28). The present study also demonstrated
that NuSAP1 was associated with survival of patients with breast
cancer.
In the present study, the DEGs between the high-risk
and the low-risk group of the training set were also investigated
in order to determine the underlying mechanisms of the four-gene
signature to predict prognosis. Among these DEGs, the top 20 DEGs
positively or negatively associated with risk score were further
selected. Results of GO analysis revealed that the top 20 positive
DEGs were mainly involved in transcription-related biological
processes, whereas the top 20 negative DEGs were mostly associated
with cellular homeostasis-related biological processes. These DEGs
were significantly associated with a number of signaling pathways,
including ‘ECM-receptor interaction’ and ‘cell adhesion molecule
(CAMs)’ pathways. It has been demonstrated that extracellular
matrix (ECM) is involved in breast cancer development and
progression (29). In addition,
Emery and Tripathi reported that dysregulation of the adhesion and
ECM pathways serves a role in breast cancer progression (30). A recent study by He et al
(31) identified the role of
ECM-receptor interaction in the crosstalk between tumor stroma and
the peripheral blood mononuclear cells in breast cancer by using an
integrated bioinformatics approach. The findings from the present
study indicated that IL15RA may exert an effect on breast cancer
development and progression partly by modulating ‘ECM-receptor
interaction’ and ‘cell adhesion molecules (CAMs)’ pathways.
In conclusion, the present study suggested that
IL15RA may affect breast cancer development and progression by
regulating expression of STAC2, PRR11, HOXC11 and
NUSAP1, and ‘ECM-receptor interaction’ and ‘cell adhesion
molecules (CAMs)’ pathways. A four-gene signature was proposed for
the prediction of prognosis in patients with breast cancer. The
present study improved the understanding of the potent biological
roles of IL15RA in the development of breast cancer. Further
validation of this prognostic four-gene signature in a large cohort
of patients with breast cancer, and experimental validation of
these microarray-based results are required.
Acknowledgements
Not applicable.
Funding
The present study was supported by Suzhou Commission
of Health and Family Planning (grant no. LCZX201713) and Foundation
of Nanjing Medical University (no. 2017NJMU164).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HY and LZ performed data analyses and wrote the
manuscript. JHC, JS, WS, BL and JDZ contributed significantly to
data analyses and manuscript revision. SYY and JQ conceived and
designed the study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mcguire S: World Cancer Report 2014.
Geneva, Switzerland: World Health Organization, International
Agency for Research on Cancer, WHO Press, 2015. Adv Nutr.
7:418–419. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Denkert C, Loibl S, Noske A, Roller M,
Müller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R,
Hanusch C, et al: Tumor-associated lymphocytes as an independent
predictor of response to neoadjuvant chemotherapy in breast cancer.
J Clin Oncol. 28:105–113. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Adams S, Gray RJ, Demaria S, Goldstein L,
Perez EA, Shulman LN, Martino S, Wang M, Jones VE, Saphner TJ, et
al: Prognostic value of tumor-infiltrating lymphocytes in
triple-negative breast cancers from two phase III randomized
adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Onc.
32:2959–2966. 2014. View Article : Google Scholar
|
|
5
|
Dieci MV, Mathieu MC, Guarneri V, Conte P,
Delaloge S, Andre F and Goubar A: Prognostic and predictive value
of tumor-infiltrating lymphocytes in two phase III randomized
adjuvant breast cancer trials. Ann Oncol. 26:1698–1704. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mahmoud SM, Paish EC, Powe DG, Macmillan
RD, Grainge MJ, Lee AH, Ellis IO and Green AR: Tumor-infiltrating
CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin
Oncol. 29:1949–1955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waldmann T: The contrasting roles of IL-2
and IL-15 in the life and death of lymphocytes: Implications for
the immunotherapy of rheumatological diseases. Arthritis Res.
4:S161–S167. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lodolce JP, Burkett PR, Koka RM, Boone DL
and Ma A: Regulation of lymphoid homeostasis by interleukin-15.
Cytokine Growth Factor Rev. 13:429–439. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Marra P, Mathew S, Grigoriadis A, Wu Y,
Kyle-Cezar F, Watkins J, Rashid M, De Rinaldis E, Hessey S,
Gazinska P, et al: IL15RA drives antagonistic mechanisms of cancer
development and immune control in lymphocyte-enriched
triple-negative breast cancers. Cancer Res. 74:4908–4921. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Terunuma A, Putluri N, Mishra P, Mathé EA,
Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, et al:
MYC-driven accumulation of 2-hydroxyglutarate is associated with
breast cancer prognosis. J Clin Invest. 124:398–412. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang P, Wang Y, Hang B, Zou X and Mao JH:
A novel gene expression-based prognostic scoring system to predict
survival in gastric cancer. Oncotarget. 7:55343–55351.
2016.PubMed/NCBI
|
|
13
|
Thomas PD: The gene ontology and the
meaning of biological function. Methods Mol Biol. 1446:15–24. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kanehisa M, Sato Y, Kawashima M, Furumichi
M and Tanabe M: KEGG as a reference resource for gene and protein
annotation. Nucleic Acids Res. 44:D457–D462. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for Annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Desantis C, Ma J, Bryan L and Jemal A:
Breast cancer statistics, 2013. CA Cancer J Clin. 64:52–62. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nelson BR, Wu F, Liu Y, Anderson DM,
McAnally J, Lin W, Cannon SC, Bassel-Duby R and Olson EN: Skeletal
muscle-specific T-tubule protein STAC3 mediates voltage-induced
Ca2+ release and contractility. Proc Natl Acad Sci USA.
110:11881–11886. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou F, Liu H, Zhang X, Shen Y, Zheng D,
Zhang A, Lai Y and Li H: Proline-rich protein 11 regulates
epithelial-to-mesenchymal transition to promote breast cancer cell
invasion. Int J Clin Exp Pathol. 7:8692–8699. 2014.PubMed/NCBI
|
|
19
|
Holland PWH: Evolution of homeobox genes.
Wiley Interdisciplinary Rev Dev Biol. 2:31–45. 2013. View Article : Google Scholar
|
|
20
|
Bhatlekar S, Fields JZ and Boman BM: HOX
genes and their role in the development of human cancers. J Mol Med
(Berl). 92:811–823. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Makiyama K, Hamada J, Takada M, Murakawa
K, Takahashi Y, Tada M, Tamoto E, Shindo G, Matsunaga A, Teramoto
K, et al: Aberrant expression of HOX genes in human invasive breast
carcinoma. Oncol Rep. 13:673–679. 2005.PubMed/NCBI
|
|
22
|
Soon YH, Bane F, Hughes E and Young LS:
Protein bomarkers Ki67, HOXC10 and HOXC11 for the prediction of
response to endocrine treatment in breast cancer. BMC Proc.
9:A522015. View Article : Google Scholar
|
|
23
|
Ribbeck K, Groen AC, Santarella R,
Bohnsack MT, Raemaekers T, Köcher T, Gentzel M, Görlich D, Wilm M,
Carmeliet G, et al: NuSAP, a Mitotic RanGTP target that stabilizes
and cross-links microtubules. Mol Biol Cell. 17:2646–2660. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iyer J, Moghe S, Furukawa M and Tsai MY:
What's Nu(SAP) in mitosis and cancer? Cell Signal. 23:991–998.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bidkhori G, Narimani Z, Ashtiani SH,
Moeini A, Nowzari-Dalini A and Masoudi-Nejad A: Reconstruction of
an integrated genome-scale co-expression network reveals key
modules involved in lung adenocarcinoma. PLoS One. 8:e675522013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Espinosa AM, Alfaro A, Roman-Basaure E,
Guardado-Estrada M, Palma Í, Serralde C, Medina I, Juárez E,
Bermúdez M, Márquez E, et al: Mitosis is a source of potential
markers for screening and survival and therapeutic targets in
cervical cancer. PLoS One. 8:e559752013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gulzar ZG, Mckenney JK and Brooks JD:
Increased expression of NuSAP in recurrent prostate cancer is
mediated by E2F1. Oncogene. 32:70–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen L, Yang L, Qiao F, Hu X, Li S, Yao L,
Yang XL and Shao ZM: High levels of nucleolar spindle-associated
protein and reduced levels of BRCA1 expression predict poor
prognosis in triple-negative breast cancer. PLoS One.
10:e01405722015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Giussani M, Merlino G, Cappelletti V,
Tagliabue E and Daidone MG: Tumor-extracellular matrix
interactions: Identification of tools associated with breast cancer
progression. Semin Cancer Biol. 35:3–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Emery LA, Tripathi AC, King C, Kavanah M,
Mendez J, Stone MD, de las Morenas A, Sebastiani P and Rosenberg
CL: Early dysregulation of cell adhesion and extracellular matrix
pathways in breast cancer progression. Am J Pathol. 175:1292–1302.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He L, Wang D, Wei N and Guo Z: Integrated
bioinformatics approach reveals crosstalk between tumor stroma and
peripheral blood mononuclear cells in breast cancer. Asian Pac J
Cancer Prev. 17:1003–1008. 2016. View Article : Google Scholar : PubMed/NCBI
|