Introduction
Cancer cells have complex interrelationships with
nonmalignant cells in their tissue microenvironments. In a variety
of cancers, non-malignant cells have been shown to exhibit complex
effects on malignant cells with activities that both promote and
inhibit cancer cell growth. In addition, the tumor microenvironment
has been shown to influence the effect of anticancer drugs on
cancer cells, and in many circumstances has been shown to exert
some protective effect on cancer cells. Leukemia is a malignancy of
bone marrow origin. The bone marrow microenvironment is a complex,
highly vascular tissue. Hematopoietic cells in the marrow are
derived from hematopoietic stem cells, while nonhematopoietic
stromal cells are derived from mesenchymal stem cells (1,2).
Acute lymphoblastic leukemia (ALL) is a cancer
derived from lymphoid precursors in the bone marrow, most commonly
B cell precursors. Although it is a high grade malignancy that
grows rapidly in vivo, primary ALL cells do not grow well
in vitro, and spontaneously undergo apoptosis. However,
primary ALL cells do survive in vitro when cocultured with
nonmalignant bone marrow stromal cells (3,4). Bone
marrow stromal cells are nonhematopoietic cells in the bone marrow
that are derived from mesenchymal stem cells. Functionally they are
defined by their adherence to plastic in standard tissue culture
conditions. Phenotypically they are negative for hematopoietic cell
markers CD45, CD34, CD14, CD11b, CD79, CD19 and HLA-DR (5). The mechanisms explaining the leukemia
cells' stromal dependence are not well understood. Stromal
cell-derived chemokines are among the mechanisms studied. Work by a
number of groups has shown that the chemokine CXCL12 plays a role
in hematopoietic precursor cell homing to and retention in bone
marrow (6–9) and can affect ALL cells. There is
interest in developing leukemia therapies that target CXCL12.
In the present study we examined the impact of
interference of the CXCL12 effect on a panel of recently derived
patient-derived xenograft ALLs. While we were able to reproduce
results of others that interference with CXCL12 could affect ALL
survival, we observed considerable variation in the effect within
our panel of patient-derived xenografts. These results led us to
more broadly examine gene expression patterns in stromal cells and
leukemia cells. We discovered overexpression of pathways related to
redox reactions and metabolism. We then discovered that the stromal
cells and leukemia cells directly exchange intracellular materials
and mitochondria.
Materials and methods
ALL cells
Deidentified primary B lineage ALL cells from adult
and pediatric patients were obtained from bone marrow or peripheral
blood leukapheresis samples at time of initial diagnosis or
relapse. The samples were used under the auspices of an IRB
approved protocol. The IRB deemed that individual patient consent
was not needed since no personal identifying information was
involved and the materials were from residual lab samples that
would otherwise have been discarded. All samples were from patients
who met NCI criteria for high risk ALL. Limited clinical, genetic
and phenotypic data are available because of the deidentification
process. All specimens were human CD45 positive, human CD19
positive both before and after expansion in immunodeficient mice.
Table I contains information about
individual samples.
 | Table I.Characteristics of ALL cells used in
these studies. |
Table I.
Characteristics of ALL cells used in
these studies.
| Name | Tissue | Age | Disease phase | Cytogenetics |
|---|
| ALL A | Blood | Adult | Diagnosis | Unknown |
| ALL B | Blood | Adult | Diagnosis | Unknown |
| ALL C | Blood | Pediatric | Diagnosis | Ph + |
| ALL D | Marrow | Pediatric | Relapse | MLL-AF4 |
| ALL E | Blood | Adult | Relapse | Unknown |
| ALL F | Blood | Adult | Diagnosis | Unknown |
| ALL G | Marrow | Pediatric | Relapse | Normal |
Specimens were expanded a single time as
patient-derived xenografts in NOD-SCID mice. Only first-generation
xenograft samples were used; i.e., leukemias were not serially
passaged in mice. At 8–12 weeks of age, mice received 250 cGy total
body irradiation. Mice were injected intravenously with 5 ×
106 leukemia cells 4 h later. The spleen and bone marrow
were harvested after 8–12 weeks. To confirm engraftment of leukemic
cells, the cells were examined by flow cytometry with human CD19
and CD45 antibodies. We compared ALL cells before and after
xenograft expansion and found no differences in flow phenotype,
morphology, stromal dependence and in vitro growth
potential.
Established cell lines
SC is a human monocyte/macrophage cell line obtained
from ATCC (ATCC CRL-9855). Subsequently, STR profiling (Genetica
LabCorp, Burlington, NC, USA) and STR analysis was performed using
the ATCC STR database (https://www.atcc.org/en/STR_Database.aspx), which
demonstrated that the cell line was derived from U-937, a human
histiocytic lymphoma (ATCC CRL-1593.2). Jurkat is a human T
lymphoblastic leukemia line. K562 is a human chronic myelogenous
leukemia line. Sup T1 is a human T lymphoblastic lymphoma line.
Stromal cells
In order to have consistent and uniform marrow
stromal cell source we used a stromal cell line-derived from normal
bone marrow immortalized with the human telomerase reverse
transcriptase (TERT) gene (10).
Unless otherwise specified in the text this is the stromal line
used in an experiment. A second immortalized stromal line, HS27
(ATCC CRL-2496), derived from normal bone marrow and immortalized
with a retroviral vector containing human papilloma virus E6/E7
genes was also used (11). Short
term marrow stromal cultures were frequently used to confirm
observations made with the immortalized stromal lines. These
primary stromal cell cultures were established by placing 1–3 ml of
marrow aspirate in 10 ml RPMI supplemented with 20% FCS, MEM
non-essential amino acids 1X, sodium pyruvate 1 mM,
2-mercaptopurine 5.5 µM, penicillin/streptomycin 1X and 1 µM
hydrocortisone (R10C+H) in 25 cm2 conventional tissue culture
flasks. After 24–48 h nonadherent cells were removed. At first
passage (14–21 days) FCS was reduced to 10%. Stromal cells were
also generated in a similar manner from de-identified human skin,
fat, and placenta obtained under an IRB approved protocol from
discarded surgical material. Freshly isolated tissues were minced
to 2 mm fragments, placed in culture and handled as above. Primary
stromal cells were used within 4 passages. Stromal cells did not
express hematopoietic genes PTPRD (CD45), CD34, CD19 or CD79a. All
were adherent and expressed COL1A2 (collagen1). The stromal cells
used were not mesenchymal stem cells since they did not meet
consensus criteria (5) for
multipotent mesenchymal stem cells (i.e., ENG positive, THY-1
positive, NT5E positive).
Counting live ALL cells by flow
cytometry
A total of 5×103 stromal cells were plated in
flat-bottom 96-well plates in R10C+H. Two days later medium was
removed and viable ALL cells were added in AIM V medium (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). A total of 3×104
leukemia cells were added per well in experiments. Five days later
wells were harvested and flow cytometry was performed by 3-color
flow cytometry (huCD19-FITC+, huCD45-PE dim+, AAD-, plus 2.5×105
counting beads). To create consistency within each experiment,
sample acquisition continued until a fixed number of bead events
were collected (this number varied from experiment to experiment
but ranged between 2×104 and 3×104). For analysis gating was made
on the huCD19+huCD45+ region and the number of AAD-live cells
reported.
Vital dye transfer studies
Cells were incubated at 37°C for 30 min with calcein
AM 1 µM (BD Pharmingen; BD Biosciences, Franklin Lakes, NJ, USA) as
directed by manufacturer. Following extensive washing they were
placed in culture. One day later the cells were collected and
calcein labeled cells were identified by flow cytometry. To stain
mitochondria, cells were incubated per manufacturer's instructions
in MitoTracker Red CMXRos 25 nM (Invitrogen; Thermo Fisher
Scientific, Inc.). Labeled cells were imaged by fluorescent
microscopy. In experiments assessing the need for cell:cell
contact, stromal cells were plated on the surface of 12-well tissue
culture plates. 0.4 µm membrane cell culture Transwell inserts
(Falcon) were then placed in wells, and leukemia cells placed in
the Transwell insert.
Stromal cell treatment with siRNA
Stromal cells in 96-flat bottom well plates were
reverse transfected with 6 pmol Stealth siRNA (Invitrogen; Thermo
Fisher Scientific, Inc.) specific for the target gene or control
siRNA, using Lipofectamine RNAiMAX 0.05 µM. Wells were washed 2
days later with RPMI or AIM V and then used in ALL survival assays.
Effectiveness of target gene knockdown was measured at 48 h after
transfection by quantitative SYBR-Green RT-PCR.
Drug treatment of cultures
Drugs were added to cocultures at the time leukemia
cells were added and remained present for the full 5 days of
coculture. The concentrations used were: dexamethasone 6 ng/ml,
methotrexate 6.25 ng/ml, vincristine 3 ng/ml, 6-mercaptopurine 250
µM, plerixafor 200–400 µM, recombinant human CXCL12 240 ng/ml. Drug
interaction was analyzed using the fractional product method in
which one compares the predicted magnitude of combined drug
treatment with the actual results (12). For example, if drug X reduced the
leukemia cell population to 0.75, and drug Y to 0.75, the
fractional product prediction for combined treatment would be
0.75×0.75=0.56. If the actual effect of combined treatment was 0.4,
drug synergy (as opposed to simple additive effects) might be
present.
Gene expression analyses
RNA was prepared from 5×106 cells using Qiagen
RNeasy kits (Qiagen, Inc., Valencia, CA, USA). For experiments in
which assessment of gene expression changes after stroma
cell:leukemia cell occurred, 1.6×106 stromal cells were plated in a
25 cm2 flask and 24 h later 5×106 leukemia cells were plated. Two
days later the cells were harvested and purified by flow cytometric
sorting. Residual genomic DNA was removed by DNAse treatment and a
cDNA library was prepared. Sequencing was performed in the
University of Rochester Genomics Core Facility (Rochester, NY,
USA). Sequencing was performed using Illumina HiSeq 2500 Sequencer
(Illumina Inc., San Diego, CA, USA). Raw reads were demultiplexed
using configurebcl2fastq.pl version 1.8.4. Quality filtering and
adapter removal were performed using Trimmomatic version 0.32 with
the following parameters: ‘SLIDINGWINDOW:4:20 TRAILING:13
LEADING:13 ILLUMINACLIP:adapters.fasta:2:30:10 MINLEN:15’.
Processed/cleaned reads were then mapped to the human reference
genome (hg19) using SHRiMP version 2.2.3 with the following
parameters: ‘-qv-offset 33 -all-contigs’. Gene-level read
quantification and differential expression analysis were performed
using Cufflinks version 2.0.2 (cuffdiff) with these parameters:
‘-FDR 0.05 -u -b GENOME’ (thus employing a correction for multiple
comparisons). For further gene set enrichment analysis only those
differential expressed genes with both >0.1 fragments per
kilobase per million (FPKM) and a two-fold up or down regulation
were considered. Functional annotation clustering of differentially
expressed genes was performed using the web-based software of the
Gene Ontology Consortium (geneontology.org. PANTHER Overrepresentation test
release 20170413, GO Ontology database release 2017-06-29, Homo
sapiens reference list, and use of a Bonferroni correction for
identification of statistically significant
overrepresentation.)
Statistics
Statistical analyses were performed using R
statistical language and RStudio. Data from leukemia cell survival
assays was found to be normally distributed. In cases in which
there were more than two groups, one way analysis of variance was
performed; the Tukey post hoc test was applied to determine whether
there were significant differences between specific pairs of
experimental groups. These P-values (which adjust for multiple
comparisons) are reported in the text and figures. For studies of
CXCL12 expression and leukemia cell survival data were not normally
distributed. Percent survival leukemia on a supporting cell type
was first normalized to leukemia survival on our internal standard
stromal line, and then log transformed. CXCL12 gene expression was
expressed as FPKM, and then divided by HLA-A FKPM (to provide
internal control). This normalized expression was then log
transformed. A Spearman correlation test was then performed.
Results
Relationship between CXCL12 gene
expression and support of ALL cells
Work by a number of groups has shown that the
chemokine CXCL12 plays a role in hematopoietic precursor cell
homing to and retention in bone marrow (6–9). We
assessed CXCL12 gene transcription in 17 stromal cell lines that
supported ALL cell survival in coculture and in 7 cell lines
derived from hematopoietic cells that poorly support leukemia cell
survival in coculture (Table II has
details about these cells). We then explored correlation of percent
survival of ALL cells with CXCL12 expression (as normalized to
HLA-A expression). The Spearman correlation (rho) was 0.542
(P=0.0092) indicating a moderate positive correlation between the
two (data not shown).
| Table II.Characteristics of cells assessed for
capacity to support ALL cells in vitro. |
Table II.
Characteristics of cells assessed for
capacity to support ALL cells in vitro.
| Supports
leukemia | Cell type | Cell name |
|---|
| Yes | Marrow-derived
primary stroma | p210 |
| Yes | Marrow-derived
primary stroma | p235 |
| Yes | Marrow-derived
primary stroma | p364 |
| Yes | Marrow-derived
primary stroma | p386 |
| Yes | Marrow-derived
primary stroma | p395 |
| Yes | Marrow-derived
primary stroma | p416 |
| Yes | Marrow-derived
primary stroma | p417 |
| Yes | Subclone of hTERT
immortalized stroma line | cl_19 |
| Yes | Subclone of hTERT
immortalized stroma line | cl_23 |
| Yes | Subclone of hTERT
immortalized stroma line | cl_30 |
| Yes | Subclone of hTERT
immortalized stroma line | cl_33 |
| Yes | Adipose-derived
primary stroma | Adipose |
| Yes | Skin-derived
fibroblastic stroma line | Fibroblast |
| Yes | hTERT immortalized
stroma line | P15 |
| Yes | Placenta-derived
primary stroma | Placenta |
| Yes | Skin-derived
primary stroma | Skin |
| Yes | Skin-derived
primary stroma | Skin |
| No | B ALL | ALL C |
| No | B ALL | ALL B |
| No | B ALL | ALL A |
| No | T acute
lymphoblatsic leukemia line | Jurkat |
| No | Chronic myelogenous
leukemia line | K562 |
| No | Monocyte/macrophage
cell line | SC |
| No | Histiocytic
lymphoma | U-937 |
| No | T lymphoblast
lymphoma | SupT1 |
Interference with CXCL12 produces a
modest reduction in survival of leukemia cells
To assess a causal relationship between stromal cell
CXCL12 expression and leukemia cell survival we transfected stromal
cells with siRNA specific for CXCL12 or control siRNA. Quantitative
RT-PCR studies showed CXCL12 knockdown efficiency was typically in
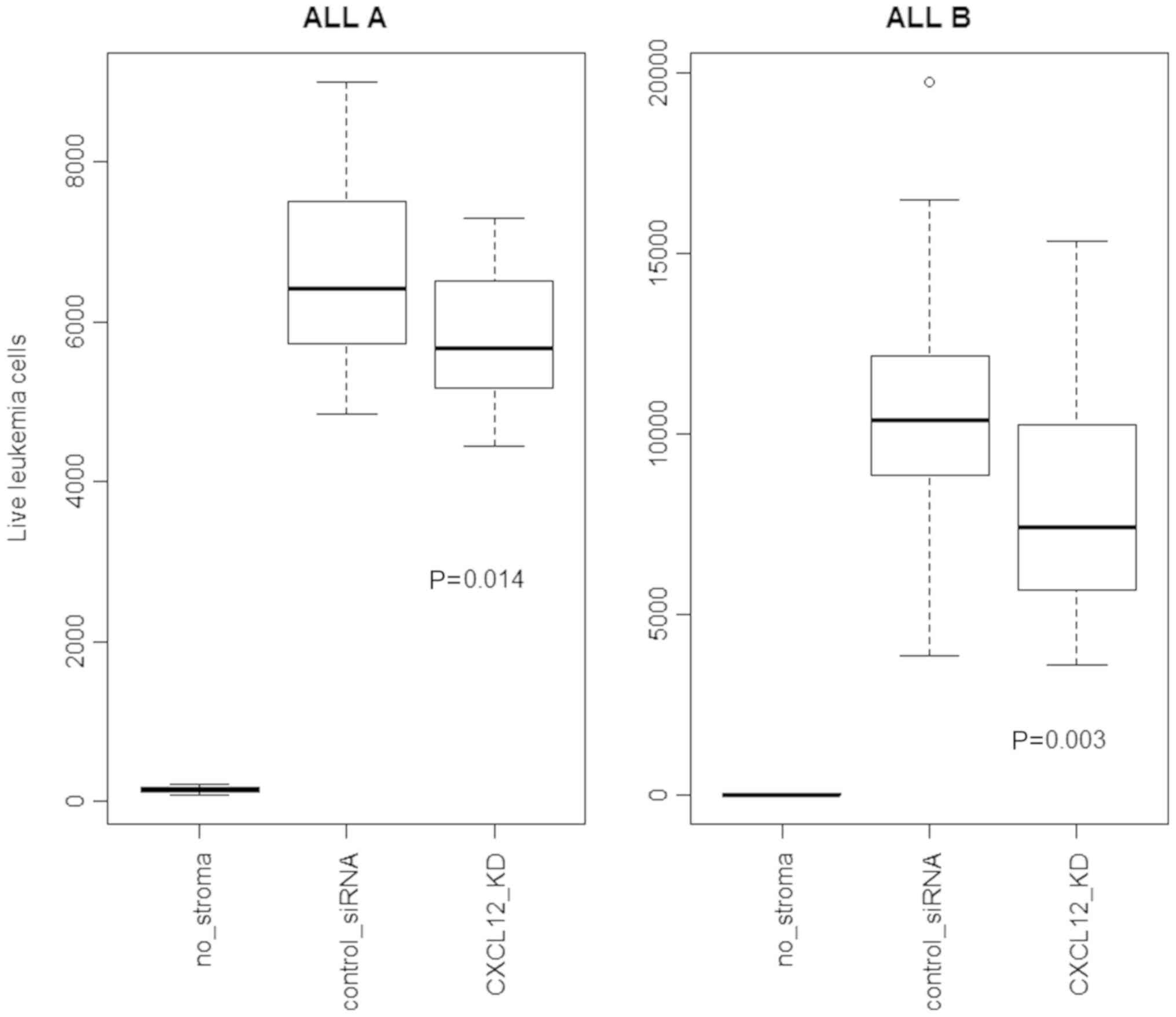
the range of 50–75% (data not shown). Fig. 1 shows a modest (10–25%) reduction of
leukemia cell survival due to CXCL12 knockdown.
It is possible that these experiments underestimated
the role of CXCL12 because siRNA knockdown was incomplete. CXCL12
interacts with the chemokine receptor CXCR4 on hematopoietic cells.
Plerixafor is a small molecule inhibitor of this interaction
(13,14). Some studies of plerixafor have shown
that it leads hematopoietic cells to mobilize away from stroma, and
can increase the antileukemia activity of some drugs. Use of
plerixafor would allow us to more fully interfere with CXCL12
effects on leukemia cells. We added plerixafor to the stromal
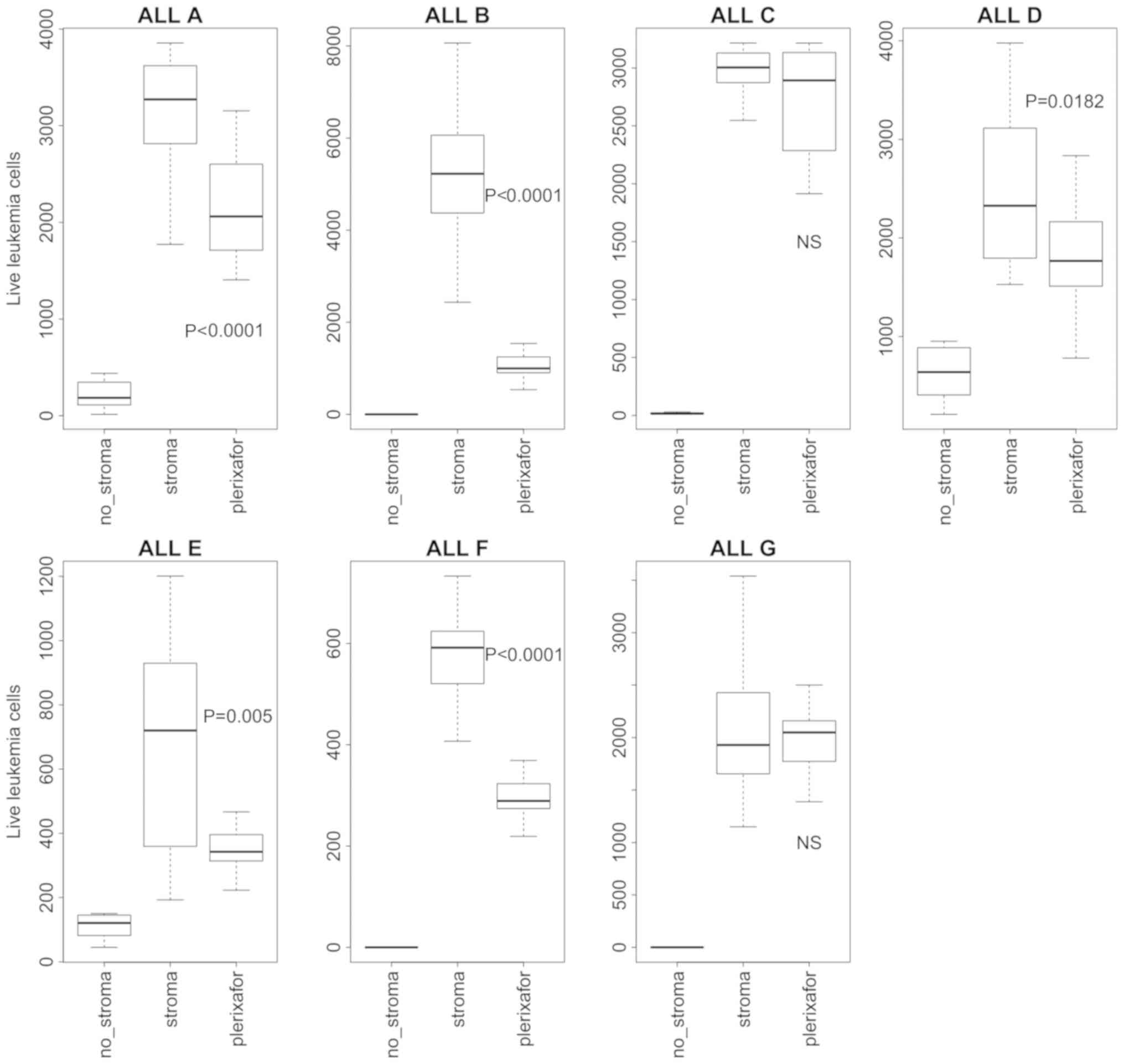
cell-leukemia cell cocultures. We found that plerixafor reduced the
survival of 5 of 7 independent ALLs (Fig. 2), but the effect ranged significantly
between leukemias (20–80%). Two of the ALLs showed no reduction in
survival. In independent experiments we found that nonmalignant
hematopoietic bone marrow cells did not exhibit the same degree of
dependence on stroma, and that plerixafor did not significantly
reduce survival of these normal marrow cells (data not shown).
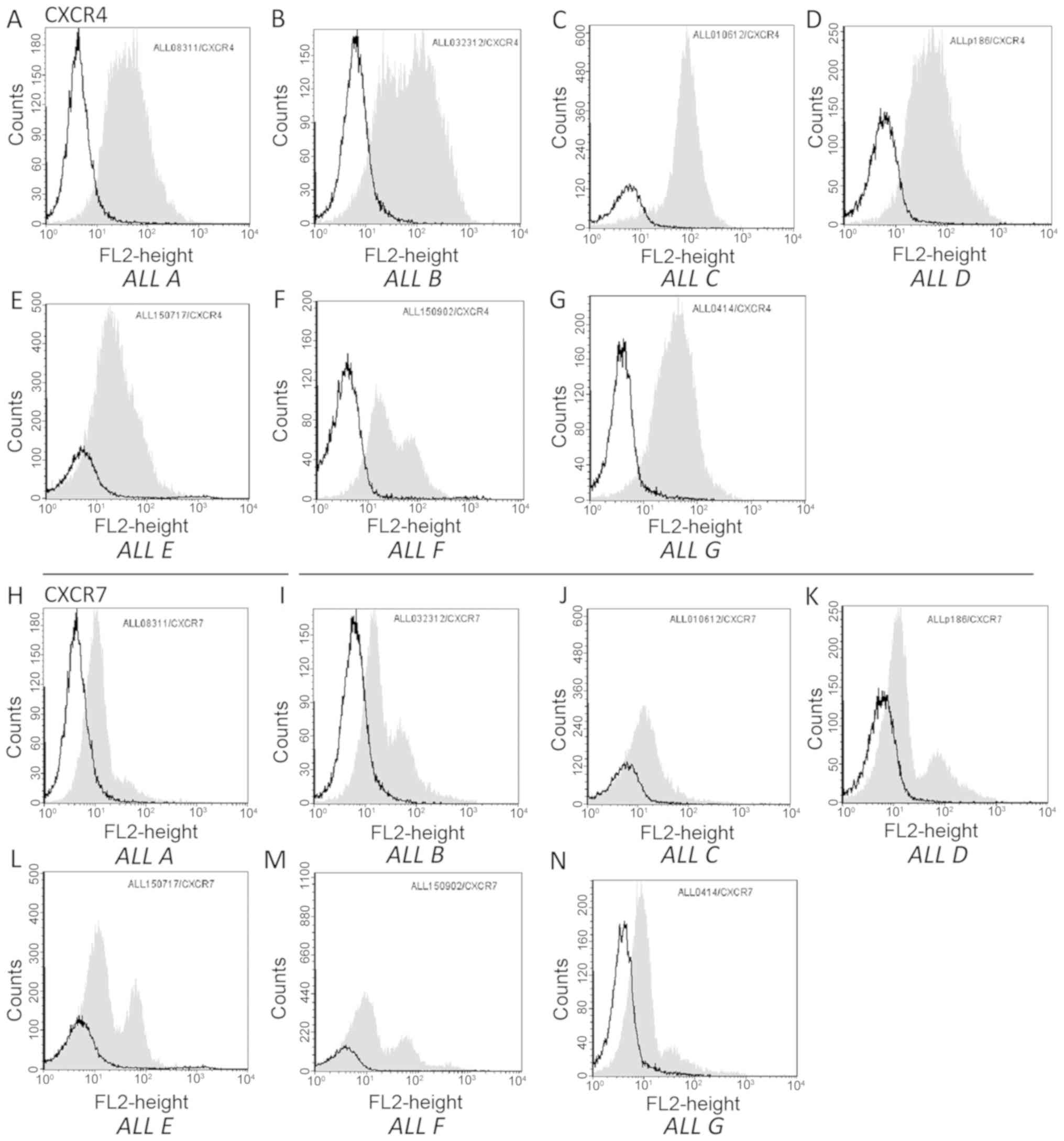
One possible explanation for the variability in the
effect of plerixafor could be differences in expression of
receptors for CXCL12 on leukemia cells. We performed flow cytometry
for both CXCR4 and CXCR7 (receptors for CXCL12) (15,16) on
the ALLs. We found similar levels of both receptors on both
plerixafor sensitive and insensitive leukemia cells (Fig. 3).
Effect of plerixafor on antileukemia
activity of commonly used leukemia drugs
We then explored whether interference of stromal
support with plerixafor would affect the antileukemia activity of
commonly used chemotherapy drugs (mercaptopurine, methotrexate,
dexamethasone and vincristine). Initially we performed
dose-response experiments with commonly used ALL chemotherapy
drugs. We then performed coculture assays in which we combined
plerixafor and conventional chemotherapy drugs. We used
concentrations of conventional chemotherapy drugs that were likely
to produce modest or minimal cytotoxicity alone because that would
allow us to detect either additive or synergistic activity with
plerixafor. We performed the assays with three unique ALLs, two of
which had shown significant sensitivity to plerixafor alone (ALL A
and ALL B) and one that had not (ALL C). Fig. 4 shows the results of these studies.
In general for ALL A and ALL B the combination of plerixafor and
chemotherapy drug produced greater antileukemia effect than
plerixafor or chemotherapy drug alone. We did not see similar
enhanced toxicity with ALL C.
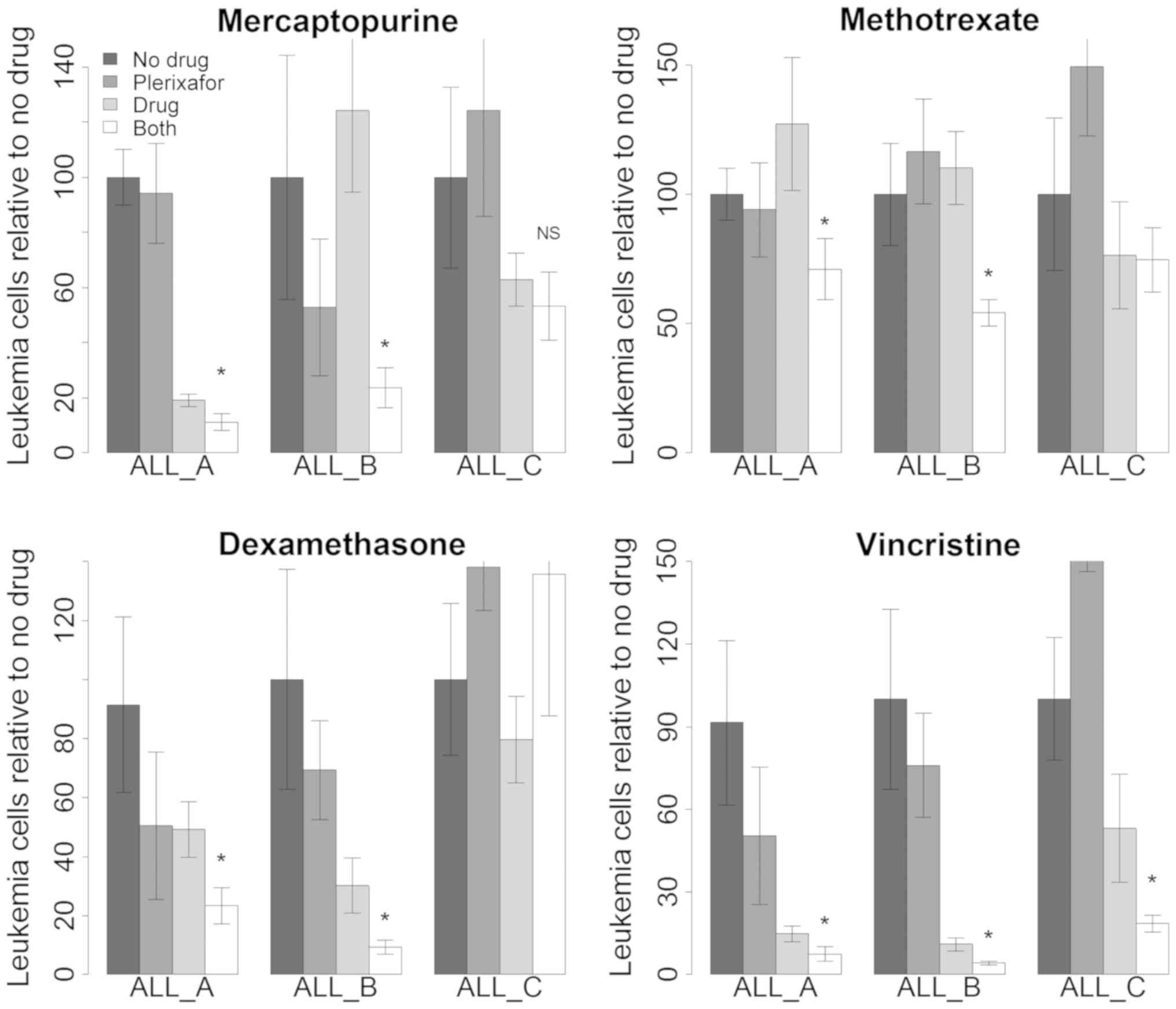 | Figure 4.Effect of plerixafor on antileukemia
activity of conventional chemotherapy drugs. Each panel represents
a different chemotherapy drug with or without plerixafor. For each
drug several ALLs were studied (ALLs A, B and C). Box and whisker
plots represent the data. Bars represent live the average number of
live ALL cells normalized to ‘no drug’. Error bars represent 95
percentile confidence intervals of the mean. From left to right the
bars represent no drug (black, n=8–12), plerixafor alone (dark
gray, n=8–12), chemotherapy drug alone (light gray, n=8–12), or the
combination of both (white, n=8–12). T tests were performed
comparing ‘drug alone’ to ‘both drug and plerixafor’. *P<0.05
vs. drug alone. NS, significant; ALL, acute lymphoblastic
leukemia. |
We analyzed the data for ALL A and B (in which we
consistently saw enhanced chemotherapy drug activity in the
presence of plerixafor) to determine if the effects were additive
or synergistic. For mercaptopurine and for methotrexate the
observed effect of combined treatment with plerixafor was greater
than the fractional product prediction which suggested that
synergistic interactions between the drugs were occurring (data not
shown).
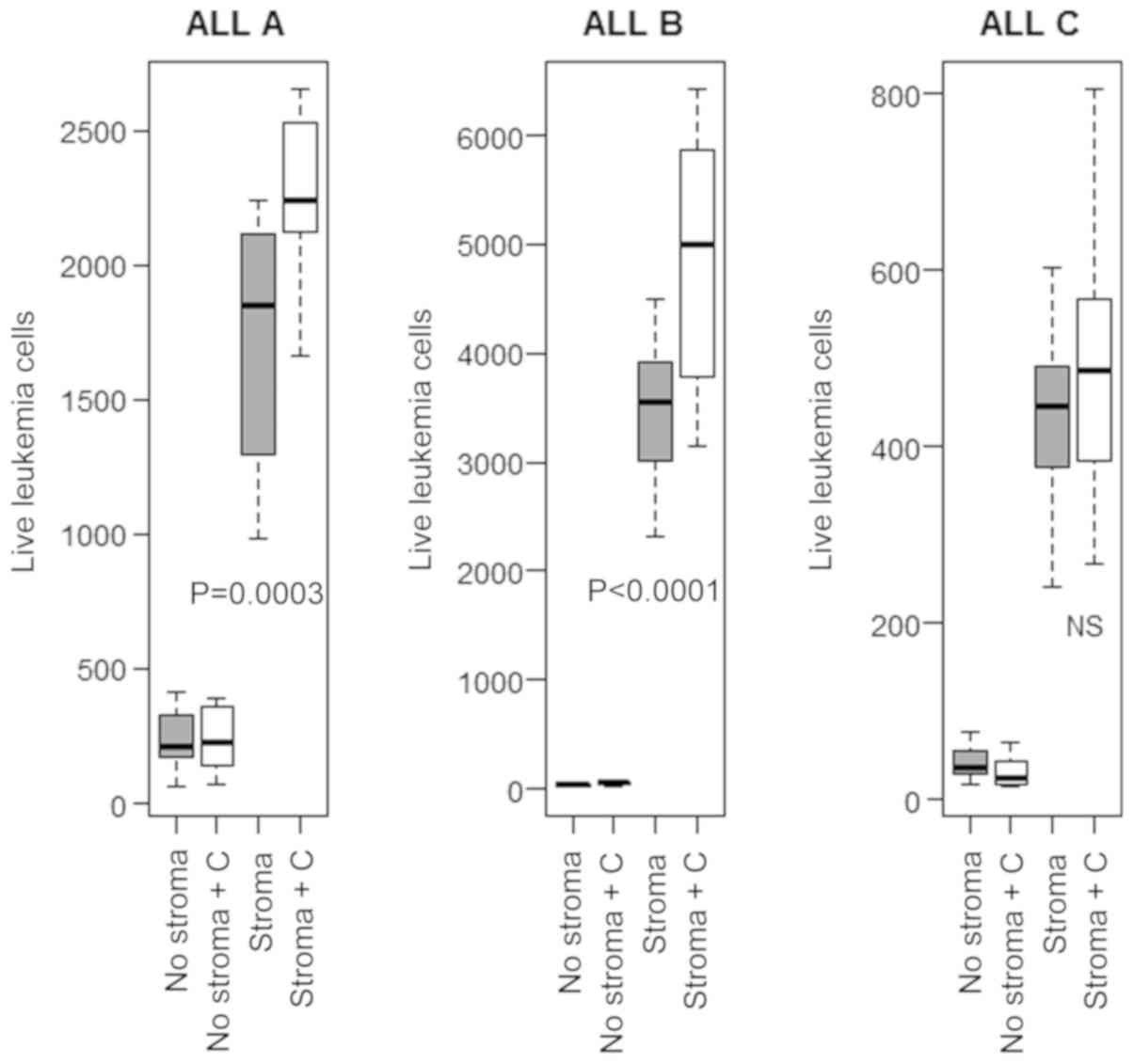
Recombinant CXCL12 cannot substitute
for stromal cell support of leukemia cells
The modest effects we saw with CXCL12 blockade
suggested that the mechanisms of stromal cell support were
multifactorial. To test this we performed three experiments with
three different leukemias in which recombinant CXCL12 was present
in the culture medium. For two of the leukemias CXCL12 in the
presence of stroma increased the number of surviving leukemia
cells. However, in the absence of stroma CXCL12 did not produce any
improvement of leukemia cell survival (Fig. 5).
Gene set enrichment analysis
The modest impact of interfering with CXCL12 is
consistent with the hypothesis that the mechanisms of stromal
support for leukemia involve many genes that contribute to the
overall effect. To gain some insight into this we performed gene
set enrichment analysis. In the first analysis we compared gene
expression profiles between cells that efficiently supported
leukemia cell survival and those cells that did not support
leukemia cells survival (see Table
II for the list of the cells in these groups). Within the set
of genes overrepresented among cells that support leukemia we
observed several related molecular functions (Table III). Not surprisingly we observed a
number of functions related to extracellular matrix. More
interestingly we saw enrichment of growth factor related genes
including platelet-derived growth factor and insulin-like growth
factor. We also unexpectedly observed enrichment of molecular
functions related to redox and energy metabolism.
| Table III.Gene set enrichment analysis. |
Table III.
Gene set enrichment analysis.
| A, Analysis of
genes differentially expressed in stromal cells compared to cells
that do not support ALL |
|---|
|
|---|
| Stromal cells that
support leukemia gene enrichment | Percentage of genes
involved | Multiple comparison
adjusted p-value |
|---|
| Extracellular
matrix | 14 |
5.10×10−25 |
| Cell surface | 11 |
2.60×10−09 |
| Extracellular
membrane bound organelle | 27 |
1.30×10−10 |
| Molecular
function |
|
|
| Growth factor
binding | 6 |
5.20×10−12 |
| PDGF binding | 2 |
1.60×10−06 |
| Receptor
binding | 15 |
2.60×10−06 |
| Biological
process |
|
|
| Extracellular
matrix organization | 14 |
7.70×10−25 |
| Biological
adhesion | 18 |
5.30×10−17 |
| Regulation of cell
death | 14 |
6.10×10−04 |
|
|---|
| B, Analysis of
genes differentially expressed in ALL cells after contact with
stromal cells |
|
|---|
| ALL leukemia
cell genes changed by contact with stroma gene enrichment | Percentage of
genes involved | Multiple
comparison adjusted p-value |
|
|---|
| Cellular
components |
|
|
| Extracellular
region | 35 |
1.60×10−12 |
| Extracellular
vesicle | 28 |
3.70×10−16 |
| Extracellular
membrane bound organelle | 27 |
3.70×10−16 |
| Molecular
function |
|
|
| Protein
binding | 63 |
2.30×10−11 |
| Purine nucleotide
binding | 16 |
3.10×10−05 |
| Small molecule
binding | 21 |
9.20×10−06 |
| Biological
process |
|
|
| Negative regulation
of biological process | 36 |
1.40×10−14 |
| Negative regulation
of apoptotic process | 9 |
3.30×10−05 |
| Cellular response
to cytokine stimulus | 7 |
3.40×10−05 |
|
| C, Analysis of
genes differentially expressed in stroma cells after contact with
ALL cells |
|
| Stromal genes
changed by contact with ALL leukemia gene enrichment | Percentage of
genes involved | Multiple
comparison adjusted p-value |
|
| Cellular
components |
|
|
| Extracellular
region | 31 |
3.30×10−09 |
| Extracellular
vesicle | 23 |
1.30×10−06 |
| Extracellular
organelle | 23 |
1.30×10−06 |
| Molecular
function |
|
|
|
Oxidoreductases | 4 |
1.30×10−04 |
| NAD/NADP/alcohol
metabolism | 2 |
1.30×10−04 |
| Daunorubicin
metabolism | 1 |
1.30×10−02 |
| Biological
process |
|
|
| Interferon
signaling | 5 |
5.70×10−19 |
| Regulation of cell
proliferation | 16 |
2.70×10−07 |
| Primary alcohol
metabolic process | 3 |
5.20×10−08 |
We performed a second analysis in which we compared
gene expression profiles before and after ALL A leukemia cells
physically interacted with stromal cells. After contact with
leukemia cells we observed 373 differentially expressed genes in
stromal cells. Genes related to redox reactions and energy
metabolism were prominently overrepresented, as were genes related
to growth factor and glycosaminoglycan binding (Table III). After contact with stromal
cells we observed 643 genes differentially expressed in the ALL A
leukemia cells. Again we saw enrichment of molecular functions
related to redox, to interactions with the extracellular matrix,
and to growth factor binding. We also saw overrepresentation of
genes related to TRAIL binding (Table
III).
Leukemia cells and stroma cells
exchange intracellular materials
The unexpected prominence of genes related to redox
reactions and energy metabolism in both stromal and leukemia cells,
coupled with our earlier observation that direct cell-cell contact
was essential for stromal support of leukemia cells, suggested the
possibility that the cells may be exchanging intracellular
materials related to or affecting metabolism. To test this
hypothesis we labeled cells with calcein AM which is a cell
permeant dye of molecular weight <1000 Daltons that in living
cells is converted to a fluorescent moiety. In these studies we
used the HS27 immortalized stromal cell line or primary bone marrow
stromal cells. We examined five of the leukemias (ALL A, ALL B, ALL
C, ALL F and ALL G). We then incubated either unlabeled leukemia
cells with labeled stromal cells, or unlabeled stromal cells with
labeled leukemia cells. 24 h later we performed flow cytometry to
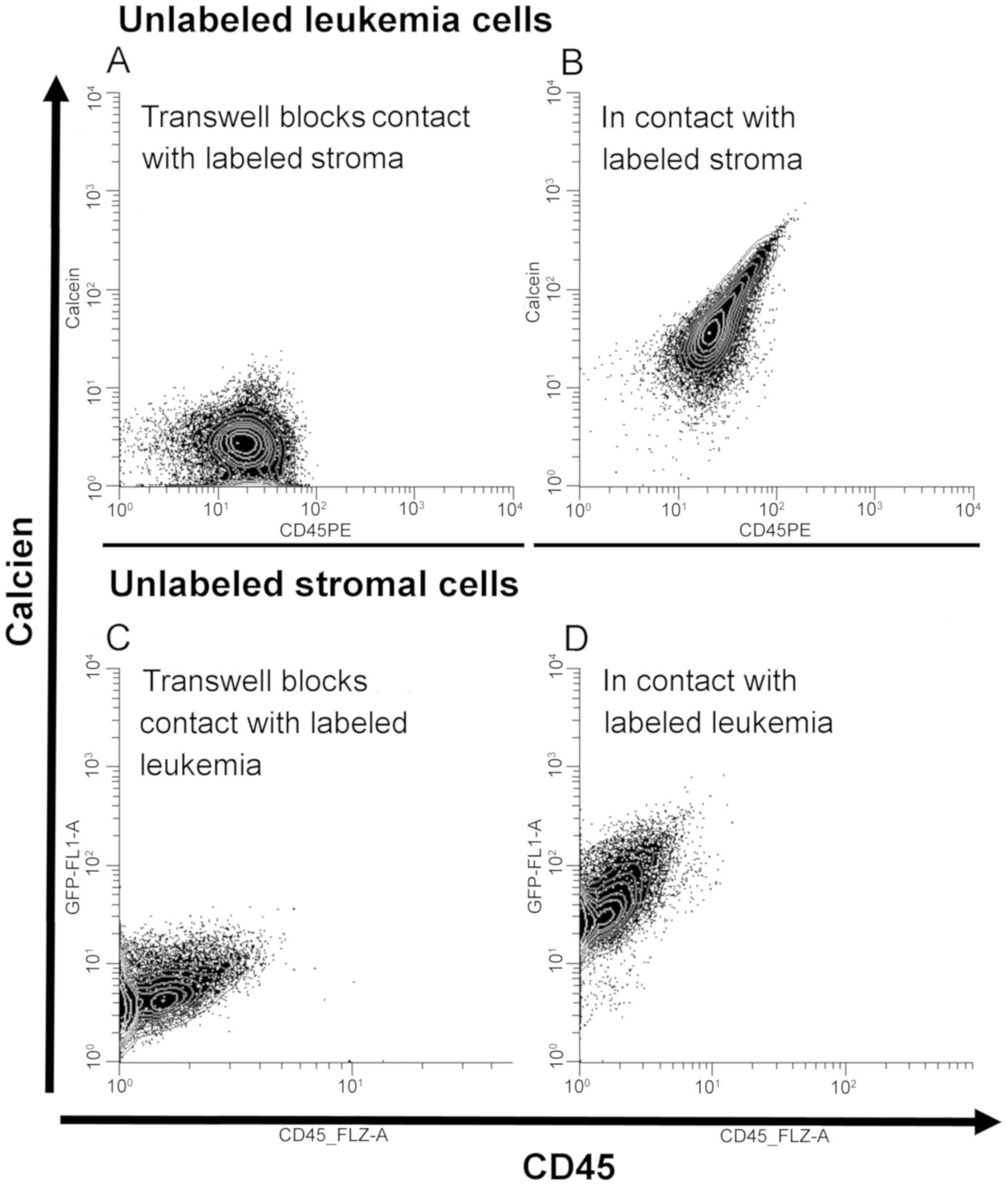
detect intracellular fluorescent calcein. Representative results
are presented in Fig. 6. We observed
transfer of calcein from leukemia cells to stromal cells (Fig. 6D), and also observed transfer of
label from stromal cells to leukemia cells (Fig. 6B). We repeated these experiments
using Transwell barriers in which leukemia cells and stromal cells
were in the same well but were unable to establish cell:cell
contact. When stromal cells and leukemia cells could were not in
physical contact we did not see transfer of calcein (Fig. 6A and 6C).
Leukemia cells and stroma cell
exchange mitochondria
Mitochondria play critical roles in cellular energy
metabolism. Since we had observed that genes related to energy
metabolism and redox reactions were associated with stromal cell
support of leukemia, and that cell-cell contact was critical for
leukemia cell survival, we hypothesized that mitochondria might
play a role. To assess this we treated either stromal cells or
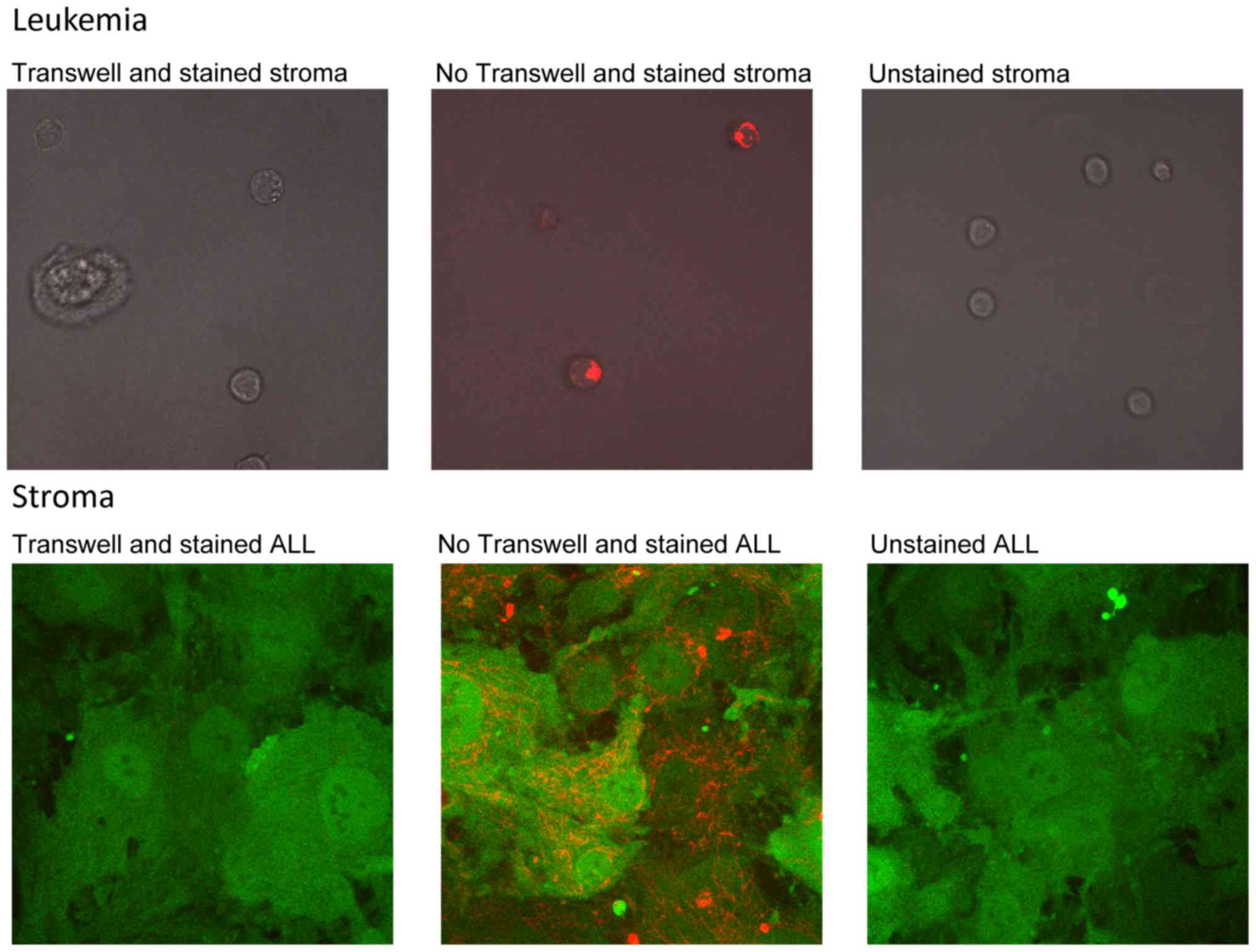
leukemia cells (ALL B or ALL F) with the vital dye MitoTracker Red
which accumulates in active mitochondria. Labeled cells were added
to unlabeled cells, and then examined by fluorescence microscopy
the next day. We observed transfer of mitochondria from labeled
leukemia cells to unlabeled stromal cell; we also observed transfer
of mitochondria from labeled stromal cells to unlabeled leukemia
cells (Fig. 7). Separation of cells
by a Transwell prevented mitochondrial transfer (data not
shown).
Discussion
Most cancers are mutationally diverse and the effort
to develop leukemia pathway-specific or leukemia mutation-specific
may lead to potent drugs that are active against only a fraction of
leukemias. Mutationally diverse leukemias appear to share a common
dependence on nonmalignant stroma. In principle, agents directed
against common mechanisms of microenvironmental support might
exhibit activity against mutationally diverse leukemias. Stromal
cell-derived CXCL12 has been identified as one potential mechanism.
Our study critically assessed the potential utility of targeting
CXCL12 in a panel of recently derived patient-derived ALL
xenografts in vitro. Our observations confirm observations
by others that CXCL12 may play a role in keeping ALL cells alive
(17,18). However, our findings extend these
observations by showing that the effect was not universal in ALL as
2 of the 7 leukemias were unaffected by plerixafor. Moreover, our
observations show that heterogeneity of the effect was not closely
associated with leukemia cell surface expression of either CXCR4 or
CXCR7, another receptor for CXCL12 (15,16).
We assessed the potential interaction of CXCL12
blockade by plerixafor with drugs commonly used in the treatment of
ALL. When used with leukemias more sensitive to plerixafor alone
(e.g., ALL A and ALL B) we saw synergistic antileukemia effects for
most conventional drugs with plerixafor. One should note that the
conventional drugs (mercaptopurine, methotrexate, dexamethasone and
vincristine) have different antileukemia mechanisms, but all
ultimately trigger apoptosis in damaged cells. This suggests the
hypothesis that the plerixafor interference with CXCL12 mediated
stromal support of leukemia may lower the apoptotic threshold for
drug-damaged cells. Our results confirm the observations of several
other groups that CXCR4 blockade can enhance the antileukemia
activity of conventional chemotherapy (17,19–23). Our
results extend these observations by demonstrating that the effect
of plerixafor may be a synergistic effect with conventional
chemotherapy. A recent phase I trial of plerixafor in combination
with cytarabine and etoposide reported biological responses but
modest clinical responses in a heavily pretreated group of patients
with ALL, acute myelogenous leukemia or myelodysplastic syndrome
(24).
The mechanisms of CXCL12 on hematopoietic or
leukemia cell are not fully elucidated. CXCL12 (also known as SDF1,
stromal cell-derived factor 1) has been extensively investigated
and is related to many cellular functions related to development,
immune responses and tumor growth and metastasis. In hematology it
is best known as a factor that contributes to angiogenesis and
regulates hematopoiesis and hematopoietic progenitor cell homing in
the marrow (25–27). One theme of these reports on the
mechanism is that CXL12 plays a role in the hematopoietic cell
homing and efficient binding of hematopoietic cells to stroma
through adhesion molecules (6–9).
One model of stromal cell-leukemia cell
microenvironmental interaction is that stromal cells present to
leukemia cells ligands or growth factors (such as CXCL12) on the
cell surface or in the intracellular region. The modest effect we
saw with plerixafor plus the biological themes that emerged from
the gene expression analyses led us to step back from this model.
To our surprise we observed changes in genes related to energy
metabolism and redox status, processes that are intracellular. This
led us to broaden the hypothesis that stromal cells provided
metabolic support to leukemia cells and that this metabolic support
contributed to leukemia cell survival. Central to this hypothesis
is the need to demonstrate some means by which stromal cells and
leukemia cells can exchange metabolites. Our experiments in which
we observed bidirectional transfer of calcien between stromal cells
and leukemia cells established that this is possible.
Given the prominence of pathways related to energy
metabolism and redox reactions in the gene set enrichment analyses
we also hypothesized that intercellular mitochondria exchange may
be involved. Intercellular transport of organelles between cells
though tunneling nanotubes was described >10 years ago (28). Exchange of functioning mitochondria
and improved aerobic respiration in recipient cells has also been
observed (29). Exchange of
mitochondria between malignant cells and nonmalignant cells has
also been observed (30–34). Our observation of bidirectional
transfer between nonmalignant stromal cells and ALL cells is
consistent with these observations in other systems.
There are limitations in this approach to dissecting
the mechanisms by which nonmalignant stromal cells provide support
to ALL cells. The first is the simplicity of our coculture system.
In the living marrow environment there are many types of cells
interacting including a variety of cells derived from mesenchymal
stem cells, cells derived from normal hematopoietic stem cells and
leukemia cells. Our in vitro model is very simple and does
not fully recreate the in vivo microenvironment. The second
is that the stromal cells and leukemia cells are from different
humans. It is possible there are polymorphisms in a number of
functions that may obscure critical interdependencies. The third is
the lack of knowledge of the genetic abnormalities in the ALLs
studied. If one wished to apply this or a similar system to define
novel therapeutic targets, such knowledge would be very helpful in
defining which ALL's might be successfully targeted.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
St. Baldrick's Foundation (Monrovia, CA, USA; to CAM) and Alex's
Lemonade Stand Foundation for Childhood Cancer (Wynnewood, PA, USA;
to CAM).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SU designed and performed experiments, interpreted
data and wrote an initial draft of the manuscript. US, OT and LN
designed, performed and interpreted experiments. JCS assisted with
data interpretation and manuscript editing. CAM was responsible for
the overall project, designed experiments, interpreted data and
wrote the final version of the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The Research Subjects Review Board of the University
of Rochester approved the use of the human tissues in the present
study (RSRB00046358, approval dates 2/28/2013 to present). The
University Committee on Animal Resources approved the use of mice
used in these studies (UCAR 102081/UCAR-2003-237E, approval dates
2003 to present).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ALL
|
acute lymphoblastic leukemia
|
|
GFP
|
green fluorescent protein
|
References
|
1
|
Shahrabi S, Rezaeeyan H, Ahmadzadeh A,
Shahjahani M and Saki N: Bone marrow blood vessels: Normal and
neoplastic niche. Oncol Rev. 10:3062016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ramasamy SK: Structure and Functions of
Blood Vessels and Vascular Niches in Bone. Stem Cells Int.
2017:50469532017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Manabe A, Coustan-Smith E, Behm FG,
Raimondi SC and Campana D: Bone marrow-derived stromal cells
prevent apoptotic cell death in B-lineage acute lymphoblastic
leukemia. Blood. 79:2370–2377. 1992.PubMed/NCBI
|
|
4
|
Manabe A, Murti KG, Coustan-Smith E,
Kumagai M, Behm FG, Raimondi SC and Campana D: Adhesion-dependent
survival of normal and leukemic human B lymphoblasts on bone marrow
stromal cells. Blood. 83:758–766. 1994.PubMed/NCBI
|
|
5
|
Dominici M, Le Blanc K, Mueller I,
Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A,
Prockop DJ and Horwitz E: Minimal criteria for defining multipotent
mesenchymal stromal cells. The International Society for Cellular
Therapy position statement. Cytotherapy. 8:315–317. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glodek AM, Le Y, Dykxhoorn DM, Park SY,
Mostoslavsky G, Mulligan R, Lieberman J, Beggs HE, Honczarenko M
and Silberstein LE: Focal adhesion kinase is required for
CXCL12-induced chemotactic and pro-adhesive responses in
hematopoietic precursor cells. Leukemia. 21:1723–1732. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Acharya M, Edkins AL, Ozanne BW and
Cushley W: SDF-1 and PDGF enhance alphavbeta5-mediated ERK
activation and adhesion-independent growth of human pre-B cell
lines. Leukemia. 23:1807–1817. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Arnaud MP, Vallée A, Robert G, Bonneau J,
Leroy C, Varin-Blank N, Rio AG, Troadec MB, Galibert MD and
Gandemer V: CD9, a key actor in the dissemination of lymphoblastic
leukemia, modulating CXCR4-mediated migration via RAC1 signaling.
Blood. 126:1802–1812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shen N, Ffrench P, Guyotat D, Ffrench M,
Fiere D, Bryon PA and Dechavanne M: Expression of adhesion
molecules in endothelial cells during allogeneic bone marrow
transplantation. Eur J Haematol. 52:296–301. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mihara K, Imai C, Coustan-Smith E, Dome
JS, Dominici M, Vanin E and Campana D: Development and functional
characterization of human bone marrow mesenchymal cells
immortalized by enforced expression of telomerase. Br J Haematol.
120:846–849. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roecklein BA and Torok-Storb B:
Functionally distinct human marrow stromal cell lines immortalized
by transduction with the human papilloma virus E6/E7 genes. Blood.
85:997–1005. 1995.PubMed/NCBI
|
|
12
|
Greco WR, Bravo G and Parsons JC: The
search for synergy: A critical review from a response surface
perspective. Pharmacol Rev. 47:331–385. 1995.PubMed/NCBI
|
|
13
|
To LB, Levesque JP and Herbert KE: How I
treat patients who mobilize hematopoietic stem cells poorly. Blood.
118:4530–4540. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fricker SP: Physiology and pharmacology of
plerixafor. Transfus Med Hemother. 40:237–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sánchez-Martín L, Sánchez-Mateos P and
Cabañas C: CXCR7 impact on CXCL12 biology and disease. Trends Mol
Med. 19:12–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Asri A, Sabour J, Atashi A and Soleimani
M: Homing in hematopoietic stem cells: Focus on regulatory role of
CXCR7 on SDF1a/CXCR4 axis. EXCLI J. 15:134–143. 2016.PubMed/NCBI
|
|
17
|
Sison EA, Rau RE, McIntyre E, Li L, Small
D and Brown P: MLL-rearranged acute lymphoblastic leukaemia stem
cell interactions with bone marrow stroma promote survival and
therapeutic resistance that can be overcome with CXCR4 antagonism.
Br J Haematol. 160:785–797. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishii K, Katayama N, Miwa H, Shikami M,
Masuya M, Shiku H and Kita K: Survival of human leukaemic B-cell
precursors is supported by stromal cells and cytokines: Association
with the expression of bcl-2 protein. Br J Haematol. 105:701–710.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Juarez J, Bradstock KF, Gottlieb DJ and
Bendall LJ: Effects of inhibitors of the chemokine receptor CXCR4
on acute lymphoblastic leukemia cells in vitro. Leukemia.
17:1294–1300. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Welschinger R, Liedtke F, Basnett J, Dela
Pena A, Juarez JG, Bradstock KF and Bendall LJ: Plerixafor
(AMD3100) induces prolonged mobilization of acute lymphoblastic
leukemia cells and increases the proportion of cycling cells in the
blood in mice. Exp Hematol. 41:293–302, e291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sison EA, Magoon D, Li L, Annesley CE, Rau
RE, Small D and Brown P: Plerixafor as a chemosensitizing agent in
pediatric acute lymphoblastic leukemia: Efficacy and potential
mechanisms of resistance to CXCR4 inhibition. Oncotarget.
5:8947–8958. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sison EA, Magoon D, Li L, Annesley CE,
Romagnoli B, Douglas GJ, Tuffin G, Zimmermann J and Brown P:
POL5551, a novel and potent CXCR4 antagonist, enhances sensitivity
to chemotherapy in pediatric ALL. Oncotarget. 6:30902–30918. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Randhawa S, Cho BS, Ghosh D, Sivina M,
Koehrer S, Müschen M, Peled A, Davis RE, Konopleva M and Burger JA:
Effects of pharmacological and genetic disruption of CXCR4
chemokine receptor function in B-cell acute lymphoblastic
leukaemia. Br J Haematol. 174:425–436. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cooper TM, Sison EAR, Baker SD, Li L,
Ahmed A, Trippett T, Gore L, Macy ME, Narendran A, August K, et al:
A phase 1 study of the CXCR4 antagonist plerixafor in combination
with high-dose cytarabine and etoposide in children with relapsed
or refractory acute leukemias or myelodysplastic syndrome: A
Pediatric Oncology Experimental Therapeutics Investigators'
Consortium study (POE 10-03). Pediatr Blood Cancer.
doi.10.1002/pbc.26414.
|
|
25
|
Anthony BA and Link DC: Regulation of
hematopoietic stem cells by bone marrow stromal cells. Trends
Immunol. 35:32–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Karpova D and Bonig H: Concise Review:
CXCR4/CXCL12 signaling in immature hematopoiesis-lessons from
pharmacological and genetic models. Stem Cells. 33:2391–2399. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gomes AC and Gomes MS: Hematopoietic
niches, erythropoiesis and anemia of chronic infection. Exp
Hematol. 44:85–91. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rustom A, Saffrich R, Markovic I, Walther
P and Gerdes HH: Nanotubular highways for intercellular organelle
transport. Science. 303:1007–1010. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Spees JL, Olson SD, Whitney MJ and Prockop
DJ: Mitochondrial transfer between cells can rescue aerobic
respiration. Proc Natl Acad Sci USA. 103:1283–1288. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X and Gerdes HH: Transfer of
mitochondria via tunneling nanotubes rescues apoptotic PC12 cells.
Cell Death Differ. 22:1181–1191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lou E, Fujisawa S, Morozov A, Barlas A,
Romin Y, Dogan Y, Gholami S, Moreira AL, Manova-Todorova K and
Moore MA: Tunneling nanotubes provide a unique conduit for
intercellular transfer of cellular contents in human malignant
pleural mesothelioma. PLoS One. 7:e330932012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pasquier J, Guerrouahen BS, Al Thawadi H,
Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L,
Rafii S, et al: Preferential transfer of mitochondria from
endothelial to cancer cells through tunneling nanotubes modulates
chemoresistance. J Transl Med. 11:942013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moschoi R, Imbert V, Nebout M, Chiche J,
Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Y, et
al: Protective mitochondrial transfer from bone marrow stromal
cells to acute myeloid leukemic cells during chemotherapy. Blood.
128:253–264. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marlein CR, Zaitseva L, Piddock RE,
Robinson SD, Edwards DR, Shafat MS, Zhou Z, Lawes M, Bowles KM and
Rushworth SA: NADPH oxidase-2 derived superoxide drives
mitochondrial transfer from bone marrow stromal cells to leukemic
blasts. Blood. 130:1649–1660. 2017.PubMed/NCBI
|