Introduction
Receptor tyrosine kinases (RTKs) are transmembrane
proteins whose ligands induce receptor dimerization and activate
RTK downstream signaling pathways. RTKs are known to be involved in
cell proliferation, survival and differentiation pathways (1,2). As a
member of the TYRO3 protein tyrosine kinase, AXL RTK ligand (AXL)
and MER RTK (TAM) subfamily of RTKs, AXL has been reported to be a
promising oncogenic target owing to its overexpression in several
types of human cancer, including various types of leukemia and
solid tumors, as well as being an indicator of poor prognosis
(3–10). Furthermore, AXL serves a key role in
drug resistance (11–17). In addition, growth arrest-specific 6
(Gas6), the major ligand of AXL, has been identified as being
expressed in a number of types of human cancer, binding with AXL to
activate its phosphorylation and drive AXL/Gas6 signaling. This
signaling pathway has been revealed to be associated with the
activation of downstream signaling, including mitogen activated
protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/protein
kinase B (AKT), and focal adhesion kinase/steroid receptor
coactivator/nuclear factor κ-B signaling pathways, to drive tumor
cell metastasis, confer therapeutic resistance and promote disease
progression (11,18,19).
The crystal structure of a minimal human AXL/Gas6
complex revealed an assembly with 2:2 stoichiometry (20). Further analysis with structure-based
mutagenesis, protein binding assays, and receptor activation
experiments demonstrated that the major and minor Gas6 binding
sites are required for productive transmembrane signaling (20). In a previous study, several AXL
antagonists, including antibodies, small molecules and aptamers,
were reported to block signaling through the receptor; however,
current anti-AXL therapeutics either demonstrate modest antitumor
efficacy or induce substantial off-target effects (4,21–25).
Kariolis et al (26) examined
an AXL-decoy receptor, named MYD1, and revealed that this Fc fusion
protein possessed a high affinity to human Gas6. Furthermore, MYD1
could block the native AXL/Gas6 interaction and inhibit cancer cell
migration and invasion through the AXL signaling pathway; marked
effects were observed in an animal model.
Therefore, the present study aimed to effectively
and specifically disrupt the AXL/Gas6 signaling axis according to
its three-dimensional (3-D) complex structure. First, the
interaction mode of AXL/Gas6 was analyzed using computational
biology. Based on the theoretical analysis results, two types of
mutations were constructed, and the AXL mutants were added into
culture medium to capture free Gas6. The potential effects of these
mutations on the AXL/Gas6 signaling pathway were investigated in
human cancer cell lines.
Materials and methods
Reagents and antibodies
Recombinant Gas6 human protein (catalog no. 885-GSB)
and goat anti-AXL antibodies (catalog no. AF154) (all R&D
Systems, Inc., Minneapolis, MN, USA), Rabbit Anti-Goat IgG
(H&L) fluorescein isothiocyanate (catalog no. ab6737; Abcam,
Cambridge, UK), human full-length pCMV6-AXL plasmid (catalog no.
SC112559; OriGene Technologies, Inc., Rockville, MD, USA), TMB
Chromogen Solution (catalog no. 183657000; Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), RIPA (catalog no.
R0010; Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China), Giemsa (catalog no. G1010; Beijing Solarbio
Science & Technology Co., Ltd.), Taq Blend (catalog no.
BTQ-201; Toyobo Life Science, Osaka, Japan) and trypsin-EDTA
(0.25%; catalog no. 1967499; Thermo Fisher Scientific, Inc.) were
obtained. Lipofectamine® 3000 Transfection Reagent
(catalog no. L3000001; Invitrogen; Thermo Fisher Scientific, Inc.),
fetal bovine serum (FBS; catalog no. 1997802C; Gibco, Gaithersburg,
MD, USA), R428 inhibitor (catalog no. HY-15150; ChemCatch, CA,
USA), anti-AXL antibodies (catalog no. 4939), anti-phosphorylated
(phospho)-AXL (catalog no. 5724), and anti-GADPH antibodies
(catalog no. 51332) were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA), goat anti-human immunoglobulin G (IgG) was
from KPL, Inc., (catalog no. 01-10-06; Gaithersburg, MD, USA), and
horseradish peroxidase (HRP)-conjugated goat anti-human IgG was
from Thermo Fisher Scientific, Inc. (catalog no. A24494). The
proteins were purified using the ÄKTAprime® plus system
(catalog no. 11001313; GE Healthcare, Pittsburgh, PA, USA).
Cell culture
SKOV3 (catalog no. HTB-77), A549 (catalog no.
CCL-185), H1299 (catalog no. CRL-5803), 293T (catalog no. CRL-3216)
and MDA-MB-231 (catalog no. HTB-26) cells (all obtained from
American Type Culture Collection, Manassas, VA, USA) were
authenticated by Beijing ZhongYuan Company (Beijing, China;
http://www.sinozhongyuan.com) in 2014.
The cells were cultured in Dulbecco's modified Eagle's medium
(DMEM; catalog no. 8118210) and Mcoy's 5A medium (catalog no.
1835937) supplemented with 10% heat-inactivated FBS (catalog no.
1932594C) (all Gibco; Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin-streptomycin, and cultured in a cell incubator at 37°C
with 5% CO2.
Theoretical computational
analysis
All computational and theoretical analyses were
performed using InsightII 2000 software (MSI, San Diego, CA) in an
IBM Corp. workstation (Armonk, NY, USA). Based on the crystal
complex structures of AXL and Gas6 (20), the coordinates of the hydrogen atoms
were assigned under a consistent valence force field (CVFF), and
the whole complex structure was optimized using the steepest decent
and conjugate gradient method (InsightII 2000 software, Discovery
mode). With the optimized complex structure, the AXL/Gas6
interaction mode was evaluated using a computer graphics technique
and the distance geometry method (InsightII 2000 software, Standard
mode). Using Superimposition software (InsightII 2000 software,
Standard mode), the complex structure and the orientation of the
main-chain carbon atoms were identified, and the comparison of
their location was analyzed to determine the 3-D protein structures
of AXL and Gas6. Furthermore, using the interaction binding free
energy calculation method (InsightII 2000 software, Discovery
mode), the binding energy between Gas6 and AXL or its mutants was
calculated under the CVFF.
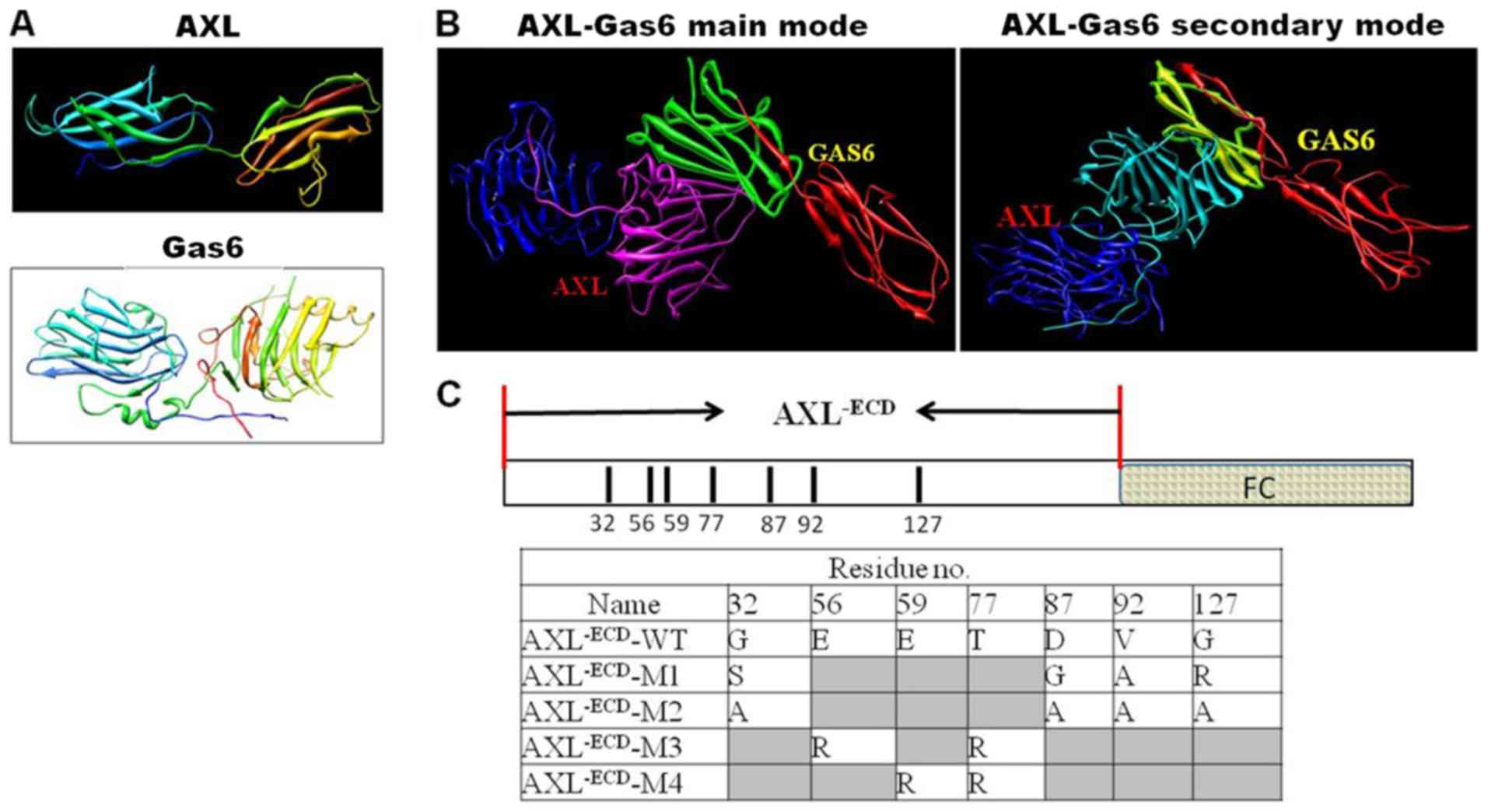
Construction and transfection
Using the human full-length pCMV6-AXL plasmid as the
template, polymerase chain reaction was performed using Taq Blend
kit to obtain AXL−ECD-Fc-wild-type (WT) and
AXL−ECD-Fc-M fragments, AXL−ECD-Fc-M1
(G32S, D87G, V92A and
G127R), AXL−ECD-Fc-M2 (G32A,
D87A, V92A and G127A),
AXL−ECD-Fc-M3 (E56R and T77R) and
AXL−ECD-Fc-M4 (E59R and T77R)
(M1/M2, high-affinity mutations; M3/M4, low-affinity mutations).
The thermocycling conditions for AXL-ECD-WT were as
follows: 94°C for 2 min; 94°C for 30 sec, 62°C for 30 sec, 72°C for
90 sec, 30 cycles; 72°C for 10 min. The target fragments were
amplified by p-up (forward)/p1 (reverse), p2 (forward)/p3
(reverse), p4 (forward)/p5 (reverse), p6 (forward)/p-down (reverse)
primers (Table I). The PCR
conditions were as follows: 94°C for 2 min, 94°C for 30 sec, 62°C
for 30 sec and 72°C for 30–90 sec (depending on the fragment
length) for 30 cycles; 72°C for 10 min. The recovered four
fragments were subjected to overlap PCR to amplify the full-length
fragment. Overlap PCR conditions were: No primer reaction: 94°C for
2 min; 94°C for 30 sec, 62°C for 30 sec, 72°C for 90 sec, 7 cycles;
72°C for 10 min. Subsequently, primers were added and a further
reaction was performed: 94°C for 30 sec, 62°C for 30 sec, 72°C for
90 sec, 23 cycles; 72°C for 10 min.
 | Table I.PCR primer sequences. |
Table I.
PCR primer sequences.
| Target | Sequence
(5′→3′) |
|---|
|
AXL−ECD-WT | p-up:
GCCAAGCTTACCACCATGGCGTGGCGGTGCCCCA |
|
| p-down: GCCCTCGAG
TGAAGGTTCCTTCACCAGCTGGTGGA |
|
AXL−ECD-M1 | p-up:
GCCAAGCTTACCACCATGGCGTGGCGGTGCCCCA |
|
| p1:
ATTCCCTGGGTTGGACACGAAGGGACTTTC |
|
| p2:
AGTCCCTTCGTGTCCAACCCAGGGAAT |
|
| p3:
GATTCTGAGCTGGCTGGCCACTATCCAGTCTCCCTGTTCATCCTC |
|
| p4:
GAGGATGAACAGGGAGACTGGATAGTGGCCAGCCAGCTCAGAATC |
|
|
p5:AGGCAAGCCCTCCAGCCGAACATAGCCAGGCTG |
|
| p6:
CAGCCTGGCTATGTTCGGCTGGAGGGCTTGCCTTA |
|
| p-down: GCCCTCGAG
TGAAGGTTCCTTCACCAGCTGGTGGA |
|
AXL−ECD-M2 | p-up:
GCCAAGCTTACCACCATGGCGTGGCGGTGCCCCA |
|
| p1:
ATATTCCCTGGGTTGGCCACGAAGGGACTT |
|
| p2:
AAGTCCCTTCGTGGCCAACCCAGGGAATAT |
|
| p3:
TCTGAGCTGGCTGGCCACTATCCAGTCAGCCTGTTCATCCTCACC |
|
| p4:
GGTGAGGATGAACAGGCTGACTGGATAGTGGCCAGCCAGCTCAGA |
|
| p5:
CAAGCCCTCCAGCGCAACATAGCCAGGCTGGG |
|
| p6:
CCCAGCCTGGCTATGTTGCGCTGGAGGGCTTG |
|
| p-down:
GCCCTCGAGTGAAGGTTCCTTCACCAGCTGGTGGA |
|
AXL−ECD-M3 | p-up:
GCCAAGCTTACCACCATGGCGTGGCGGTGCCCCA |
|
| p1:
TACCTCGGGGGGCCCTCCCTGAACCTGGAG |
|
| p2:
CTCCAGGTTCAGGGAGGGCCCCCCGAGGTA |
|
| p3:
CCCAGGGGCACCTGGCCCTGGGTGCTGTCC |
|
| p4:
GGACAGCACCCAGGGCCAGGTGCCCCTGGG |
|
| p-down:
GCCCTCGAGTGAAGGTTCCTTCACCAGCTGGTGGA |
|
AXL−ECD-M4 | p-up:
GCCAAGCTTACCACCATGGCGTGGCGGTGCCCCA |
|
| p1:
AAGCCAATGTACCCGGGGGGGCTCTCC |
|
| p2:
GGAGAGCCCCCCCGGGTACATTGGCTT |
|
| p3:
CCCAGGGGCACCTGCCGCTGGGTGCTGTCC |
|
| p4:
GACAGCACCCAGCGGCAGGTGCCCCTGGG |
|
| p-down:
GCCCTCGAGTGAAGGTTCCTTCACCAGCTGGTGGA |
The fragments were double-digested using HindIII
and NheI, and ligated to the optimized pCDNA®5
plasmid. For the Fc-fusion protein expression, the human Fc gene
was subcloned using HindIII/BamHI enzyme sites to obtain the target
protein with Fc fused in the C-terminus). Then, 5
AXL−ECD-WT/M fusion proteins were obtained using a
Lipofectamine® 3000 transfection system according to the
manufacturer's protocol, the 5 recombinant expression vectors were
transfected at 20 µg per well into 293T cells (7×106
cells/well). At 72 h, the purification of cell culture supernatants
was performed using ÄKTA prime plus instrument. SDS-PAGE was used
to determine the quality of the purified protein.
ELISA
ELISA plates were coated with 0.5 µg/ml Gas6 (100
µl/well) at 4°C overnight. A total of 10 serial 1:3 dilutions of
the AXL−ECD-Fc-WT/M fusions proteins were obtained (0–15
µg/ml) and added to the plates for 1 h at 37°C. Following three
washes, HRP-conjugated goat anti-human IgG (1:2,000) was added as
the secondary antibody, and plates were incubated for an additional
30 min at 37°C. Binding signals were visualized using TMB
substrate, and the light absorbance was measured using a SPECTRA
MAX 190 ELISA reader (Molecular Devices, LLC, Sunnyvale, CA,
USA) at 450 nm. Each ELISA experiment was repeated three times.
Binding kinetics assay
The binding kinetics of AXL−ECD-Fc-WT/M
fusion proteins to Gas6 were measured using a BIAcore 3000
instrument (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). The
assays were performed at 30°C in PBS. Sensor tips were pre-wetted
for 15 min in the same buffer immediately prior to use, and the
microplates were filled with 200 µl/well diluted Gas6 samples or
buffer and then agitated at 179 × g. Association (Kon)
and dissociation (Koff) rates were calculated using a
simple 1-to-1 Langmuir binding model (BIAcore Evaluation Software;
version 3.2; GE Healthcare Bio-Sciences). The equilibrium
dissociation constant (Kd) was determined using the
Koff/Kon ratio. The independent measurements
were performed three times.
Flow cytometry
Flow cytometry was used to detect AXL expression on
the cell surface as previously described (27). Briefly, the tumor cells were digested
and counted, and 1×106 cells per reaction were used. The
cells were stained with goat anti-AXL antibody (catalog no. AF154;
1:500; R&D Systems, Inc.) for 30 min at 4°C, washed three times
with buffer, and then incubated with secondary antibodies (1:1,000;
rabbit anti-goat IgG FITC; Abcam) for 30 min at 4°C. Once the assay
was complete, the cells were washed three times and the expression
of AXL was analyzed. To detect the expression of surface AXL, the
cell surface FITC intensity was analyzed using a BD FACSCalibur™
flow cytometer (ref. 342975; BD Biosciences, NY, USA) and FlowJo
software (version 7.6; BD Biosciences). The flow cytometry
experiments were repeated three times.
Migration assay
A549 and SKOV3 cells were serum-starved overnight.
Following trypsin digestion, the cells were counted and resuspended
in serum-free DMEM. Migration assays were performed by seeding
3×104 cells into BD-Falcon 24 Fluoroblock Transwell
inserts with 8-µm pores in the upper chamber. For Gas6-dependent
migration, 200 ng/ml Gas6 in the presence or absence of 100 µg/ml
AXL−ECD-Fc-WT/M proteins was added to the lower chamber
containing the migration medium (DMEM with 5% FBS). R428 (2 µM/ml)
+ Gas6 was used as a control. The AXL−ECD-Fc-WT/M
protein was added into culture systems to bind free Gas6 and block
it from binding with the cell surface AXL protein. Following 4 h of
migration, the upper chambers were fixed with 4% paraformaldehyde
for 30 min, stained with Giemsa solution for 10 min at room
temperature and counted by light microscopy (×10 magnification;
Nikon Corporation, Tokyo, Japan) using ImageJ software (version
18.0; National institutes of Health, MD, USA). The migration
experiments were repeated three times.
Western blot analysis
Gas6-induced phosphorylation of AXL in SKOV3 cells
was detected using western blot analysis. AXL-ECD-Fc WT and its
mutant were added to the cell culture as the decoy protein to bind
to free Gas6. SKOV3 cells were seeded in 6-well plates
(5×105 cells/well) for 12 h. The cells were placed in
serum-free medium and pre-treated for 8 h with or without AXL-WT/M
fusion proteins (10, 2, 0.4 or 0.08 µg/ml) at 37°C. Following the
addition of 200 ng/ml Gas6 for 30 min in 37°C, cells were collected
and lysed in ice-cold RIPA buffer (Beijing Solarbio Science &
Technology Co., Ltd.) supplemented with protease inhibitor cocktail
(Roche Diagnostics, Basel, Switzerland) for 30 min. Protein
concentrations were quantified using a BCA kit (Applygen
Technologies, Inc., Beijing, China). Lysates were separated and 20
µg protein was loaded per lane using 12% SDS-PAGE, transferred onto
a nitrocellulose filter membrane (EMD Millipore, Billerica, MA,
USA) and analyzed. Blots were incubated with the primary antibodies
anti-AXL, anti-pAXL and anti-GAPDH overnight at 4°C (all antibodies
were purchased from Cell Signaling Technology and diluted to
1:1,000). Subsequently, blots were incubated with HRP-conjugated
secondary antibodies (1:2,500) for 1 h at room temperature.
Immunoreactivity was detected and visualized using an enhanced
chemiluminescence detection system (SuperSignal West Pico Trial
kit; Pierce; Thermo Fisher Scientific, Inc.) and autoradiography.
GADPH was used as a loading control. Western blot experiments were
repeated three times.
Data analysis
Statistical analysis was performed using Prism
software v.5.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Dunnett's multiple comparisons test was used to compare all other
groups to the control group. P<0.05 was considered to indicate a
statistically significant difference. All measurements were within
95% confidence limits.
Results
Key amino acid residues in the
AXL/Gas6 interaction
Under CVFF and according to the 3-D crystal complex
structure of AXL/Gas6 (20), the
coordinates of hydrogen atoms were assigned and optimized using
molecular modeling. The theoretical 3-D structures of AXL, Gas6 and
the complex structure are presented in Fig. 1A and B. Using the Superimposition
software, the main-chain carbon atoms position of the proteins AXL
and Gas6 were determined and the orientation of the main-chain
carbon atoms was analyzed. The crystal and theoretical structures
were then compared. The root mean square deviation value of the
main-chain orientation was calculated to be 0.002 nm. The
coordinates of the heavy atoms were not altered under assignment of
the coordinates of hydrogen atoms; thus, the optimized method and
CVFF were suitable in the present study.
On the basis of the theoretical structures of AXL
and Gas6, the interaction mode between them was analyzed using a
computer graphics technique and the distance geometry method
(Fig. 1B). Through investigating the
van der Waals interactions and inter-molecular hydrogen bonds, the
key amino acid residues through which AXL interacted with Gas6 were
determined (Fig. 1C). The influence
of the single point mutants of AXL (G32, D87,
V92, G127, E56, E59 and
T77) was analyzed; the results revealed that it was not
significant and so 4 kinds of mutants were designed. The in
silico results of the present study revealed that the residues
E56 and E59 in AXL were bound to Gas6 through
electrostatic interactions. In addition, the residue T77
in AXL was bound to Gas6 through polar interactions. On the basis
of the analysis, the 4 mutants, AXL−ECD-Fc-M1
(G32S, D87G, V92A and
G127R), AXL−ECD-Fc-M2 (G32A,
D87A, V92A and G127A),
AXL−ECD-Fc-M3 (E56R and T77R) and
AXL−ECD-Fc-M4 (E59R and T77R),
were designed. In this analysis, the residues E56 and
E59 in AXL were bound to Gas6 through electrostatic
interactions, so E was replaced with R. In addition, the relative
binding energy of the 4 mutants to Gas6 compared with the parent
AXL was calculated. The results revealed that the relative binding
energies of AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2
were −18.58 and −15.62 kJ/mol, respectively, compared with that of
the WT. The relative binding energies of AXL−ECD-Fc-M3
and AXL−ECD-Fc-M4 were 5.38 and 6.16 kJ/mol,
respectively. The results demonstrated that the mutants
AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2 possessed
stronger binding affinity to Gas6 than the parent AXL; whereas the
mutants AXL−ECD-Fc-M3 and AXL−ECD-Fc-M4
exhibited lower affinity.
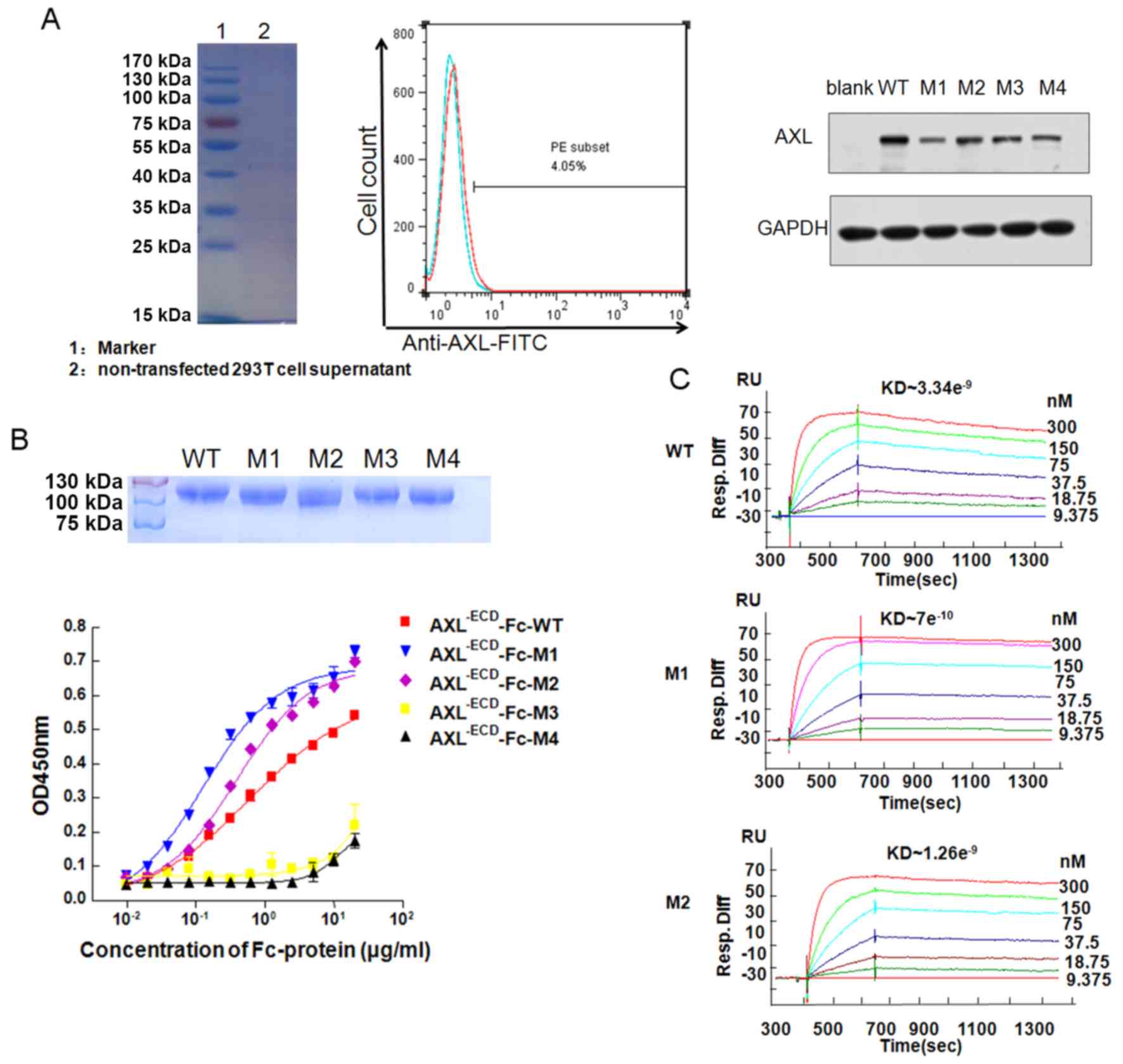
AXL−ECDFc fusion variants
bind with Gas6
293T cells did not express the AXL protein (Fig. 2A). The plasmids of
AXL−ECD-Fc WT and mutants transfected into the 293T
cells resulted in an increase in AXL Fc WT and mutant expression,
respectively (Fig. 2A). The 5
recombinant expression vectors were transfected into the 293T cells
and the proteins were purified from the supernatants. Molecular
weight of purified recombinants were verified using SDS-PAGE
(Fig. 2B). The ELISA demonstrated
that AXL−ECD-Fc-WT, AXL−ECD-Fc-M1 and
AXL−ECD-Fc-M2 could bind to Gas6 in a
concentration-dependent manner (Fig.
2B). In addition, the half maximal effective concentration
values of AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2
were 0.141 and 0.375 µg/ml, respectively, whereas that of
AXL−ECD-Fc-WT was 0.514 µg/ml, indicating that
AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2 possessed
stronger binding affinity than AXL−ECD-Fc-WT. In
contrast, AXL−ECD-Fc-M3 and AXL−ECD-Fc-M4 had
low binding affinity for Gas6. Additional BIAcore experiments
(Fig. 2C) yielded the same
results.
The Kd was determined using the
Koff/Kon ratio. As XL−ECD-Fc-M3
and AXL−ECD-Fc-M4 were low-affinity variants, they
exhibited markedly low binding to Gas6 (Fig. S1). In contrast, the Kd
values of AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2
were 7×10−10 and 1.48×10−9 M, respectively,
indicating higher affinity compared with that of
AXL−ECD-Fc-WT (Kd, 3.34×10−9
M).
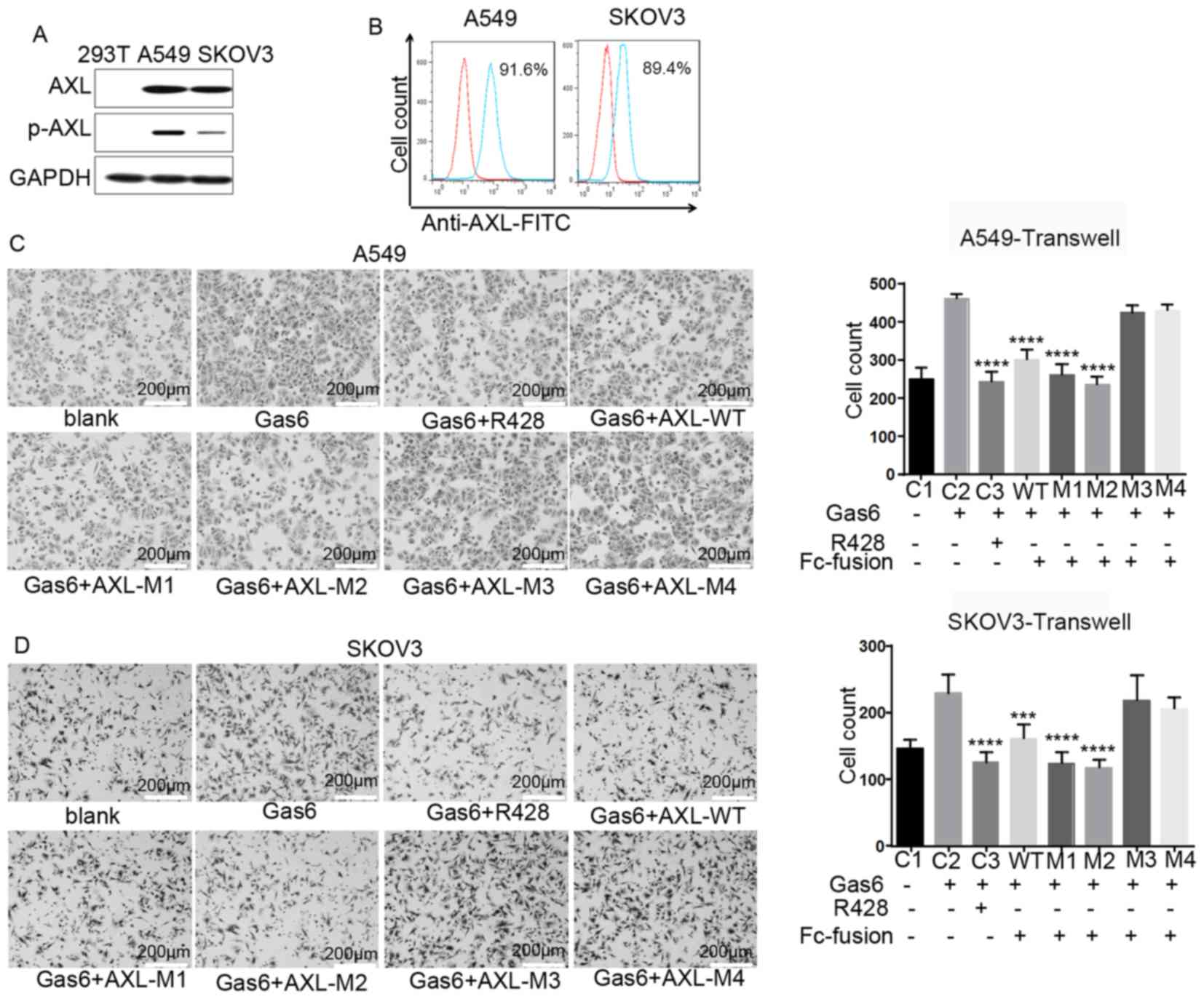
AXL−ECD-Fc fusion variants
inhibit cell migration
The western blot analysis revealed that AXL was
expressed in A549 and SKOV3 cells, but not in 293T cells (Fig. 3A), and the flow cytometry results
revealed that it was expressed on the cell surface (Fig. 3B). Expression of AXL was also
observed in other cell lines, such as H1299 and MDA-MB-231;
however, these cells had a strong autophosphorylation level of AXL,
so GAS6 was unable to trigger the phosphorylation of AXL, thus the
two cells lines was unsuitable as cell models in the current study
(Fig. S2). Therefore, A549 and
SKOV3 cells were selected for subsequent experimentation. To
investigate the potential function of AXL−ECD-Fc-WT/M
fusion proteins, a Transwell assay was performed. The purified Fc
fusion protein was added into culture systems to bind free Gas6 and
prevent it from binding to the cell surface AXL protein. The
results revealed that AXL−ECD-Fc-WT,
AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2 inhibited the
migration of A549 and SKOV3 cells induced by free Gas6 compared
with that observed with Gas6 alone, consistent with results
obtained with the AXL inhibitor R428 (Fig. 3C and D). In contrast, neither
AXL−ECD-Fc-M3 nor AXL−ECD-Fc-M4 inhibited
cell migration promoted by free Gas6. In summary,
AXL−ECD-Fc-WT/M1/M2 fusion proteins inhibited free
Gas6-induced cell migration, whereas the
AXL−ECD-Fc-M3/M4 variants demonstrated no such
inhibitory function.
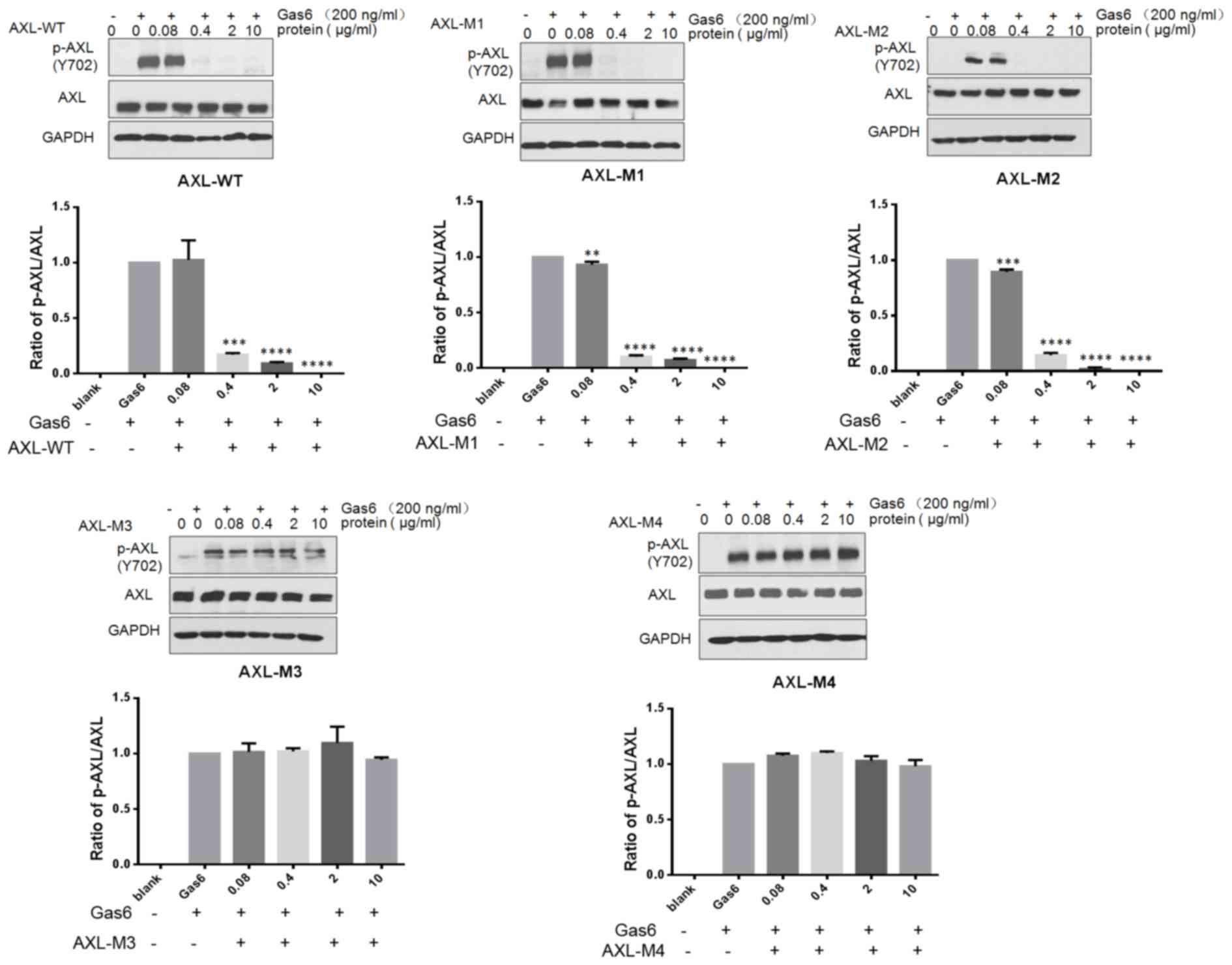
AXL−ECD-Fc fusion variants
block AXL phosphorylation in SKOV3 cells
Gas6 binds to the extracellular domain of AXL to
induce autophosphorylation of tyrosine residues on the
intracellular tyrosine kinase domain of AXL, subsequently
activating the MAPK/extracellular signal-regulated kinase and
PI3K/AKT signaling pathways. This results in the modulation of
numerous cellular activities, including cell survival,
proliferation, migration, invasion and drug resistance (17,28,29).
Therefore, the effects of AXL−ECD-Fc-WT/M fusion
proteins on Gas6-induced AXL signaling pathways were investigated
in SKOV3 cells through analyzing AXL phosphorylation by western
blotting (Fig. 4). Briefly, cells
were pretreated with the indicated concentrations of
AXL−ECD-Fc-WT/M fusion proteins for 8 h prior to
stimulation with Gas6. The results revealed that free Gas6
treatment induced AXL phosphorylation, whereas
AXL−ECD-Fc-WT/M1/M2 fusion protein pretreatment resulted
in a concentration-dependent decrease in phosphorylation.
Furthermore, the overall level of AXL expression was not changed by
AXL−ECD-Fc-WT/M fusion protein treatment. These results
suggest that AXL−ECD-Fc-WT/M1/M2 fusion proteins
neutralize the free Gas6 protein, preventing the interaction
between Gas6 and AXL in SKOV3 cells. This leads to the obstruction
of the Gas6-induced phosphorylation of AXL, and the downregulation
of the AXL/Gas6 downstream signaling pathway.
Discussion
In the present study, the key amino acid residues in
AXL that affect its interaction with Gas6 were identified
(E56, E59 and T77). Furthermore,
the inhibition of the AXL/Gas6 signaling pathway using
high-affinity mutants resulted in the inhibition of cancer cell
migration. AXL−ECD-Fc-M1 and -M2, which were identified
as high-affinity mutations, blocked the functions of AXL/Gas6 by
decreasing AXL phosphorylation. Cancer cell migration induced by
Gas6 was also inhibited. AXL−ECD-Fc-M3 and
AXL−ECD-Fc-M4 were characterized as low-affinity
mutations, supporting the notion that the amino acid T77
serves a key role in AXL binding to Gas6.
RTK activation is believed to result from
ligand-induced receptor dimerization, leading to the
autophosphorylation of multiple tyrosine residues in the cytosolic
domains and affecting downstream signaling (30). In cancer, the AXL signaling pathway
can be activated by Gas6 in an autocrine or paracrine manner
(5–8). The functional role of AXL/Gas6
signaling within the tumor microenvironment has previously been
reported to promote tumor progression, and nutrient deprivation
within the tumor microenvironment may contribute to activation of
the AXL/Gas6 signaling pathway (31). In addition, AXL has been identified
as a direct transcriptional target of hypoxia-inducible factor
(HIF)-1 and HIF-2 in tumor cells (31). In several studies, anti-AXL
antibodies were reported to block the AXL/Gas6 interaction and
inhibit AXL downstream signaling; however, tumor migration and
invasion were not altered, and tumor progression was not
effectively inhibited (25,32). Thus, although the roles of the AXL
signaling pathway in tumor migration, invasion and inflammation
have been extensively studied, the mechanisms mediating these
inhibitory effects have not yet been elucidated.
In the present study, the high-affinity mutants
AXL−ECD-Fc-M1 and AXL−ECD-Fc-M2 demonstrated
higher Gas6 binding ability than AXL−ECD-Fc-WT,
resulting in the inhibition of Gas6-induced cancer cell migration.
The low-affinity mutants AXL−ECD-Fc-M3 and
AXL−ECD-Fc-M4 could not block the AXL/Gas6 interaction
by binding with Gas6, and the inhibitory function was abolished,
indicating that the mutation sites of AXL−ECD-Fc-M3/M4
served key roles in the AXL/Gas6 interaction. Overall, the results
of the present study support that mutation of the amino acid
T77 in AXL inhibits the interaction between AXL and
Gas6. Further studies are required to determine the details of the
interactions between AXL and Gas6 using small molecule tyrosine
kinase inhibitors or antibodies. The present findings may provide
an important reference for designing such inhibitors or antibodies,
as well as critical insights into future preclinical and clinical
studies of AXL in cancer.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The National Natural Science Foundation of China
provided funding for the present study (grant no. 31771010).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
BS, ML and JF designed the study. YD, JW, HL and XL
performed the experiments. YD conducted the primary research. BH,
CQ, LL and TZ analyzed the data. YD wrote the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TAM
|
TYRO3 protein tyrosine kinase, AXL
receptor tyrosine kinase ligand and MER receptor tyrosine kinase
proteins
|
|
Gas6
|
growth arrest-specific 6
|
|
RTK
|
receptor tyrosine kinase
|
|
DMEM
|
Dulbecco's modified Eagle's
medium
|
|
CVFF
|
consistent valence force field
|
References
|
1
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Myers SH, Brunton VG and Unciti-Broceta A:
AXL inhibitors in cancer: A medicinal chemistry perspective. J Med
Chem. 59:3593–3608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Graham DK, DeRyckere D, Davies KD and Earp
HS: The TAM family: Phosphatidylserine sensing receptor tyrosine
kinases gone awry in cancer. Nat Rev Cancer. 14:769–785. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Holland SJ, Pan A, Franci C, Hu Y, Chang
B, Li W, Duan M, Torneros A, Yu J, Heckrodt TJ, et al: R428, a
selective small molecule inhibitor of Axl kinase, blocks tumor
spread and prolongs survival in models of metastatic breast cancer.
Cancer Res. 70:1544–1554. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paccez JD, Vogelsang M, Parker MI and
Zerbini LF: The receptor tyrosine kinase Axl in cancer: Biological
functions and therapeutic implications. Int J Cancer.
134:1024–1033. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheridan C: First Axl inhibitor enters
clinical trials. Nat Biotechnol. 31:775–776. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gjerdrum C, Tiron C, Høiby T, Stefansson
I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT,
et al: Axl is an essential epithelial-to-mesenchymal
transition-induced regulator of breast cancer metastasis and
patient survival. Proc Natl Acad Sci USA. 107:1124–1129. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koorstra JB, Karikari CA, Feldmann G,
Bisht S, Rojas PL, Offerhaus GJ, Alvarez H and Maitra A: The Axl
receptor tyrosine kinase confers an adverse prognostic influence in
pancreatic cancer and represents a new therapeutic target. Cancer
Biol Ther. 8:618–626. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu CW, Li AF, Chi CW, Lai CH, Huang CL, Lo
SS, Lui WY and Lin WC: Clinical significance of AXL kinase family
in gastric cancer. Anticancer Res. 22:1071–1080. 2012.
|
|
10
|
Sainaghi PP, Castello L, Bergamasco L,
Galletti M, Bellosta P and Avanzi GC: Gas6 induces proliferation in
prostate carcinoma cell lines expressing the Axl receptor. J Cell
Physiol. 204:36–44. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Konieczkowski DJ, Johannessen CM,
Abudayyeh O, Kim JW, Cooper ZA, Piris A, Frederick DT,
Barzily-Rokni M, Straussman R, Haq R, et al: A melanoma cell state
distinction influences sensitivity to MAPK pathway inhibitors.
Cancer Discov. 4:816–827. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong J, Peng D, Chen Z, Sehdev V and
Belkhiri A: ABL regulation by AXL promotes cisplatin resistance in
esophageal cancer. Cancer Res. 73:331–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hong CC, Lay JD, Huang JS, Cheng AL, Tang
JL, Lin MT, Lai GM and Chuang SE: Receptor tyrosine kinase AXL is
induced by chemotherapy drugs and overexpression of AXL confers
drug resistance in acute myeloid leukemia. Cancer Lett.
268:314–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Müller J, Krijgsman O, Tsoi J, Robert L,
Hugo W, Song C, Kong X, Possik PA, Cornelissen-Steijger PD, Geukes
Foppen MH, et al: Low MITF/AXL ratio predicts early resistance to
multiple targeted drugs in melanoma. Nat Commun. 5:57122014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou L, Liu XD, Sun M, Zhang X, German P,
Bai S, Ding Z, Tannir N, Wood CG, Matin SF, et al: Targeting MET
and AXL overcomes resistance to sunitinib therapy in renal cell
carcinoma. Oncogene. 35:2687–2697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Miller MA, Oudin MJ, Sullivan RJ, Wang SJ,
Meyer AS, Im H, Frederick DT, Tadros J, Griffith LG, Lee H, et al:
Reduced proteolytic shedding of receptor tyrosine kinases is a
post-translational mechanism of kinase inhibitor resistance. Cancer
Discov. 6:382–399. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagata K, Nakano T, Arita H, Zong C,
Hanafusa H and Mizuno K: Identification of the product of growth
arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer
receptor tyrosine kinases. J Biol Chem. 271:30022–30027. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Varnum BC, Young C, Elliott G, Garcia A,
Bartley TD, Fridell YW, Hunt RW, Trail G, Clogston C, Toso RJ, et
al: Axl receptor tyrosine kinase stimulated by the vitamin
K-dependent protein encoded by growth-arrest-specific gene 6.
Nature. 373:623–626. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sasaki T, Knyazev PG, Clout NJ, Cheburkin
Y, Göhring W, Ullrich A, Timpl R and Hohenester E: Structural basis
for Gas6-Axl signalling. EMBO J. 25:80–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan SB, Peek VL, Ajamie R, Buchanan SG,
Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, et al:
LY2801653 is an orally bioavailable multi-kinase inhibitor with
potent activity against MET, MST1R, and other oncoproteins, and
displays anti-tumor activities in mouse xenograft models. Invest
New Drugs. 31:833–844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leconet W, Larbouret C, Chardès T, Thomas
G, Neiveyans M, Busson M, Jarlier M, Radosevic-Robin N, Pugnière M,
Bernex F, et al: Preclinical validation of AXL receptor as a target
for antibody-based pancreatic cancer immunotherapy. Oncogene.
33:5405–5414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cerchia L, Esposito CL, Camorani S, Rienzo
A, Stasio L, Insabato L, Affuso A and de Franciscis V: Targeting
Axl with a high-affinity inhibitory aptamer. Mol Ther.
20:2291–2303. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Paolino M, Choidas A, Wallner S, Pranjic
B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa
JP, et al: The E3 ligase Cbl-b and TAM receptors regulate cancer
metastasis via natural killer cells. Nature. 507:508–512. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ye X, Li Y, Stawicki S, Couto S,
Eastham-Anderson J, Kallop D, Weimer R, Wu Y and Pei L: An anti-Axl
monoclonal antibody attenuates xenogra tumor growth and enhances
the effect of multiple anticancer therapies. Oncogene.
29:5254–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kariolis MS, Miao YR, Jones DS II, Kapur
S, Mathews II, Giaccia AJ and Cochran JR: An engineered Axl ‘decoy
receptor’ effectively silences the Gas6-Axl signaling axis. Nat
Chem Biol. 10:977–983. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boshuizen J, Koopman LA, Krijgsman O,
Shahrabi A, van den Heuvel EG, Ligtenberg MA, Vredevoogd DW, Kemper
K, Kuilman T, Song JY, et al: Cooperative targeting of melanoma
heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK
inhibitors. Nat Med. 24:203–212. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen PX, Li QY and Yang Z: Axl and
prostasin are biomarkers for prognosis of ovarian adenocarcinoma.
Ann Diagn Pathol. 17:425–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Darling RJ and Brault PA: Kinetic
exclusion assay technology: Characterization of molecular
interactions. Assay Drug Dev Technol. 2:647–657. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lew ED, Oh J, Burrola PG, Lax I, Zagórska
A, Través PG, Schlessinger J and Lemke G: Differential TAM receptor
ligand phospholipid interactions delimit differential TAM
bioactivities. Elife; 3. 2014
|
|
31
|
Rankin EB, Fuh KC, Castellini L,
Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A,
Lindgren D, et al: Direct regulation of GAS6/AXL signaling by HIF
promotes renal metastasis through SRC and MET. Proc Natl Acad Sci
USA. 111:13373–13378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J,
Kallop D, Ludlam MJ and Pei L: Axl as a potential therapeutic
target in cancer: Role of Axl in tumor growth, metastasis and
angiogenesis. Oncogene. 28:3442–3455. 2009. View Article : Google Scholar : PubMed/NCBI
|