Introduction
Melanoma is the most lethal skin malignancy, and the
estimated number of new cases with melanoma of the skin based on
conventional cancer incidence rates is 96,480 and the estimated
number of cases of mortality is 7,230 for 2019 (1–3).
Cutaneous melanoma is the fifth most common type of cancer, with
its incidence rate increasing continuously during the past decades
(4,5). Despite high diagnostic accuracy and
appropriate treatment due to medical advances, its pathogenesis and
natural course remain unclear.
Melanoma is a malignant type of tumor, which may be
composed of epidermal melanocytes, nevocytes or dermal melanocytes.
Epidemiology studies revealed that melanoma is likely to be caused
by hereditary factors and environmental risk factors, including
sex, age, ethnicity and geographic location (6,7). This is
in line with the most vital and potentially reversible risk factor
of malignant melanoma arising from an interaction between
environmental exposure and genetic susceptibility. Gandini et
al (8) revealed the association
between melanoma and ultraviolet (UV)-irradiation, and concluded
that intermittent sunlight may be a vital determinant of risk.
Additionally, other types of UV ray exposure, including sunburn and
artificial UV-irradiation, may cause melanoma development (9,10). Out
of all melanoma cases, ~25% occur together with pre-existing moles,
suggesting that melanocytes are benign accumulations of themselves
and nevus cells (11). A family
history may be a strong risk factor for the disease, and provides a
direction for elucidating the genetic basis of melanoma (12,13). In
addition, melanoma has a high degree of malignancy, and is prone to
early lymphoid and blood tract metastasis, which are associated
with poor prognosis; therefore, it is important to identify
susceptibility genes. Genomic profiling and sequencing will form
the basis for molecular taxonomy for more accurate subgrouping of
patients with melanoma in the future.
Identification of associated genes is a central
challenge of modern genetics aiming to modulate signal pathways and
biological processes for a number of well-known diseases, including
various types of cancer. Melanoma is highly complicated; therefore,
research strategies are limited. Computational methods provide
important complementary tools for this problem. Over the past
decades, as biotechnology has advanced, a large amount of gene
expression data has been collected and submitted to several
large-scale databases. These have been extensively used to explore
the molecular mechanism of tumorigenesis with bioinformatics
analysis methods (14,15). However, a previous study (16) based on sequence- and network-based
methods has been conducted to identify melanoma-associated
genes.
The purpose of the present study was to establish a
comprehensive method to identify melanoma-associated biological
mechanisms and to predict key genes associated with melanoma using
the random walk method. Gene Ontology (GO) terms and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were
analyzed to illuminate the biological pathways and processes which
may be altered in patients with melanoma. Additionally, network and
Kaplan-Meier analyses identified several potential therapeutic
targets for melanoma. The results of the present study may provide
novel strategies for diagnosis and treatment of melanoma and
uncover the probable mechanism of tumorigenesis.
Materials and methods
Data collection/data set
The present study searched for genes associated with
melanoma using the Online Mendelian Inheritance in Man (OMIM)
database (https://www.omim.org/), which is a
comprehensive, authoritative and timely research resource, and
includes descriptions of human genes, phenotypes and their
associations (17). A total of 74
melanoma-associated genes were identified. These were used as seed
genes for prioritization of melanoma candidate genes in the present
study.
The Search Tool for the Retrieval of Interacting
Genes/Proteins (STRING) database (http://string-db.org) provides a comprehensive
catalogue of protein-protein interactions along with their
confidence score according to means obtaining them. In the present
study, the full list of gene pairs as background was obtained and
the melanoma-associated network was extracted by retaining only the
direct neighborhoods of the 74 seed genes, which resulted in a
network of 11,181 genes and 39,043 interaction pairs.
Prioritization of melanoma candidate
genes
Random walk on heterogeneous networks is an emerging
approach for effective disease gene prioritization.
Melanoma-associated genes obtained from OMIM and candidate genes in
the melanoma-associated network were used as inputs. The random
walk algorithm produced a ranked list of the genes, according to
their strength of association with melanoma. The risk score of each
candidate gene was calculated as follows:
pt+1=(1-r)Qpt+rp0
Where pt represents the melanoma risk vector at walk
step t, the parameter r ϵ (0, 1) is the restart
probability, and Q is the risk probability transfer
matrix.
According to the Perron-Frobenius theorem (18), the eigenvalues of stochastic matrix
Q are in the range of [-1, 1] and r=0.1 was finally
obtained as the optimal parameter. Generally, the transition
probability from gene qi to phenotype qj is defined
as:
qij=wij∑k∈neighbor(i)wkj
These probabilities can reach a steady state
following a sufficiently large number of iterations. The iterations
were conducted until the difference between ps
and ps+1 (measured by the L1 norm)
fell below 10−6.
Functional enrichment analysis
The GO database (http://geneontology.org), which unifies genetic and
gene product characteristics of all species, is an effective and
efficient tool to screen genes. In the present study, GO functional
enrichment analysis of melanoma candidate genes was performed to
interpret their biological significance using the WEB-based Gene
SeT AnaLysis Toolkit (http://www.webgestalt.org) (19), with a cut-off criterion of
P<0.05.
The full list of KEGG pathways was downloaded from
the KEGG database (https://www.genome.jp/kegg/), and pathway enrichment
analysis for melanoma candidate genes was conducted using the
following formula:
p=1-∑j=0m-1(mj)(N-Mn-j)(Nn)
Where N is the total number of genes in the KEGG
pathway database, n is the number of melanoma candidate genes
annotated by the KEGG pathway database, m is the number of melanoma
candidate genes annotated by a specific KEGG pathway and M is the
total number of genes involved in a specific pathway. KEGG pathways
were considered as statistically significant if P<0.05 following
correction using the Benjamini-Hochberg method (20).
Pathway crosstalk analysis
Associations among significantly enriched pathways
were explored through crosstalk analysis using the overlap
coefficient (OC) and the Jaccard coefficient (JC), which were
defined as follows:
OC=|A∩B|min(|A|,|B|)
JC=|A∩BA∪B|
Where A and B represent the melanoma candidate gene
numbers contained in pathway A and pathway B, respectively. Pathway
A and pathway B were considered to be connected if OC>0.5 and
JC>0.25. Furthermore, Cytoscape 3.0 software (http://cytoscape.org/) was used to visualize
connections among KEGG pathways.
Melanoma-specific network
analysis
The melanoma candidate genes were uploaded to the
STRING database to obtain a melanoma-specific network by using the
threshold of confidence score >0.4. Additionally, gene degree
and betweenness were applied for evaluating gene importance in the
network. Khuri and Wuchty (21)
hypothesized that MDSets could provide a novel method of evaluating
the core proteins of an organism. The present study considered
genes contained in the MDSet with a high degree and betweenness as
important biomarkers for melanoma progression.
Survival analysis
To evaluate the association among
melanoma-associated biomarkers and survival rates, the Gene
Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn) database was used to
perform survival analysis. Melanoma samples were classified into
high or low expression groups according to the median expression
value of specific respective genes. The Kaplan-Meier method and a
two-side log-rank test were used for overall survival comparison
between two groups. P<0.05 was considered to indicate a
statistically significant difference.
Statistical analysis
All statistical analyses were conducted in R version
3.4.1 (https://www.r-project.org/). For
functional analysis, the enrichment method was used. For survival
analysis, the Kaplan-Meier method was adopted to plot survival
curves, followed by a log-rank test to compare the differences
between the survival curves. Data was presented as the mean ±
standard deviation with at least three repeats in every group.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Melanoma candidate gene
prioritization
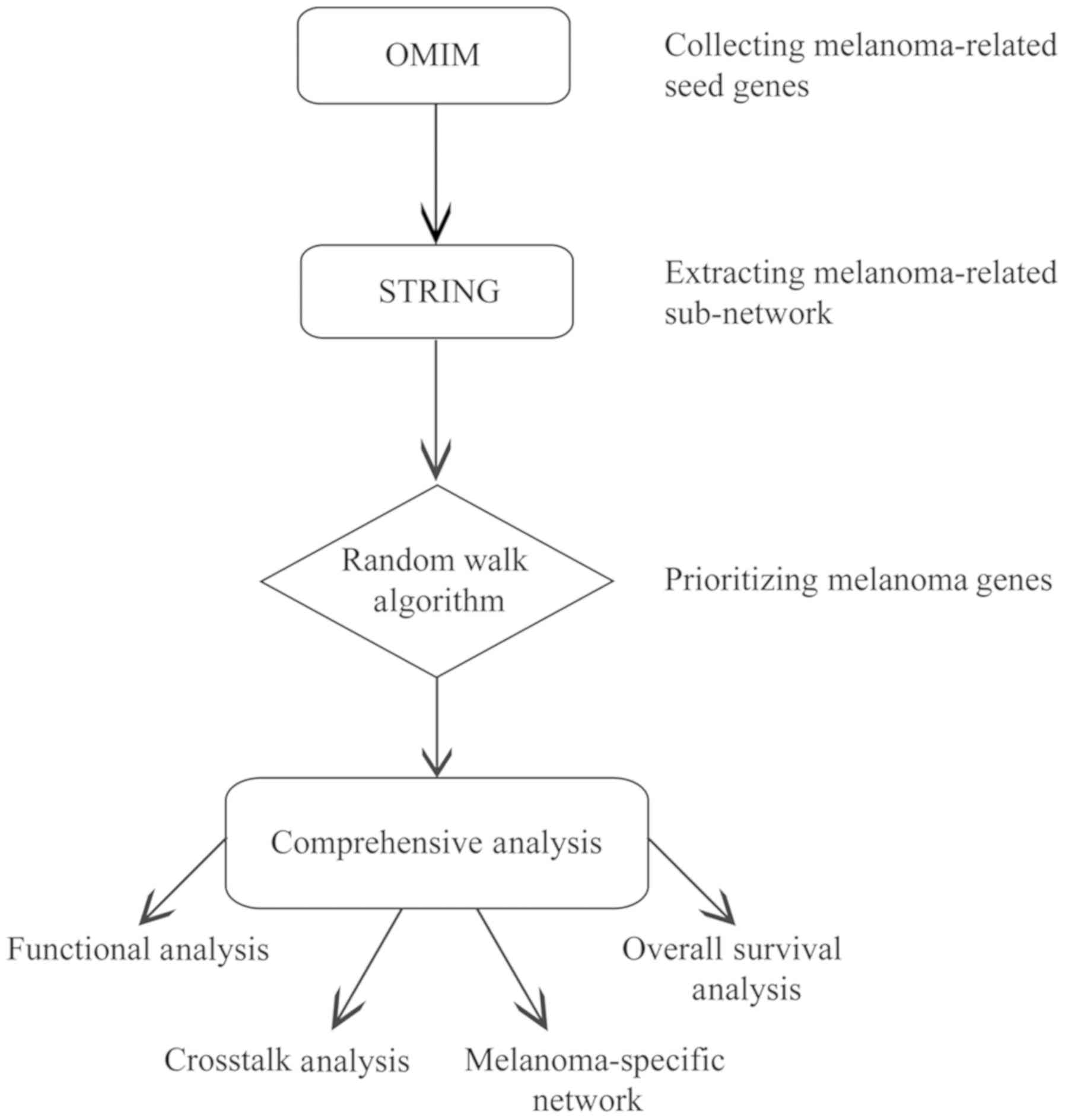
Fig. 1 shows the
workflow of the present study. A total of 74 genes were reported to
be associated with melanoma in the OMIM database, and used as seed
genes for following analyses. Additionally, 39,043 interaction
pairs among 11,181 genes were obtained from the STRING database by
extracting the direct interactions of the 74 seed genes, which was
considered as the melanoma-associated network. By applying the
random walk algorithm to the 74 seed genes and 11,181 candidate
genes, a score was assigned to every candidate gene and the top 1%
of genes (n=111) with the highest scores were considered as
melanoma-associated genes.
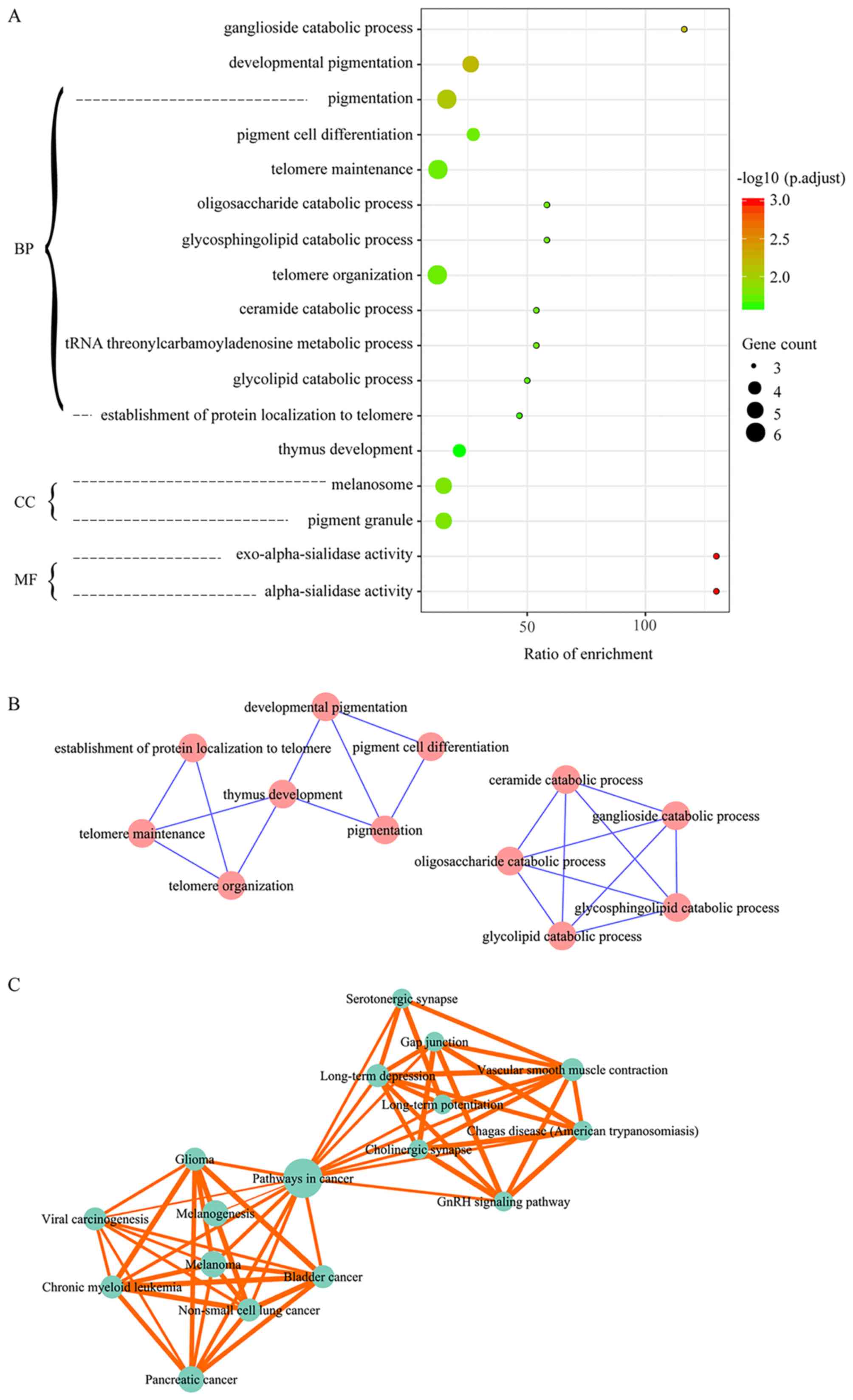
Significantly enriched functions
The 111 melanoma-associated genes were identified to
be significantly enriched in 13 GO biological process (BP) terms,
two cellular component (CC) terms and two molecular function terms
(Fig. 2A). The BP terms mainly
involved in the process of pigmentation, catabolism, telomeres and
thymus development. The CC terms melanosome and pigment granule
were also considerably enriched. These are involved in
pigmentation.
Enrichment Map (http://www.baderlab.org/Software/EnrichmentMap) was
used to explore the associations among significantly enriched GO
terms, which resulted in two clusters (Fig. 2B). One cluster included biological
processes associated with telomeres and pigmentation, including
‘telomere maintenance’ and ‘pigment cell differentiation’, and the
other cluster included biological processes associated with
catabolism.
There were 30 pathways that were significantly
enriched in association with the 111 melanoma-associated genes
(Table I), and 12 of them were
involved in cancer-associated processes. In addition, endocrine
system pathways (melanogenesis and gonadotropin-releasing hormone
signaling pathway) and pathways in the translation process
[epidermal growth factor receptor signaling pathway and mechanistic
target of rapamycin (mTOR) signaling pathway] were also included in
the results. The immune system (T cell receptor signaling pathway,
NOD-like receptor signaling pathway and natural killer cell
mediated cytotoxicity) was identified to be overrepresented in
melanoma-associated genes. Pathway crosstalk analysis obtained two
pathway clusters that were associated with cancer development and
the neuroendocrine system, respectively, which were connected by
pathways in cancer (Fig. 2C).
 | Table I.Significantly enriched Kyoto
Encyclopedia of Genes and Genomes pathways of the 111
melanoma-associated genes. |
Table I.
Significantly enriched Kyoto
Encyclopedia of Genes and Genomes pathways of the 111
melanoma-associated genes.
| Pathway | P-value | PBH-value | Melanoma-associated
genes included in the pathway |
|---|
| Melanoma |
9.45×10−7 |
1.30×10−4 | MITF; CDK4; BRAF;
MAPK3; MAPK1; CDKN2A |
| Bladder cancer |
1.32×10−6 |
1.30×10−4 | CDK4; BRAF; MAPK3;
MAPK1; CDKN2A |
| Pancreatic
cancer |
6.10×10−7 |
1.30×10−4 | CDK4; BRCA2; BRAF;
MAPK3; MAPK1; CDKN2A |
| Non-small cell lung
cancer |
6.37×10−6 |
4.21×10−4 | CDK4; BRAF; MAPK3;
MAPK1; CDKN2A |
| Melanogenesis |
7.12×10−6 |
4.21×10−4 | GNAQ; MITF; TYR;
MC1R; MAPK3; MAPK1 |
| Long-term
depression |
8.99×10−6 |
4.43×10−4 | GNAQ; GNA11; BRAF;
MAPK3; MAPK1 |
| Glioma |
1.34×10−5 |
4.94×10−4 | CDK4; BRAF; MAPK3;
MAPK1; CDKN2A |
| Pathways in
cancer |
1.18×10−5 |
4.94×10−4 | GNAQ; MITF; CDK4;
BRCA2; GNA11; BIRC7; BRAF; MAPK3; MAPK1; CDKN2A |
| Chronic myeloid
leukemia |
2.36×10−5 |
7.77×10−4 | CDK4; BRAF; MAPK3;
MAPK1; CDKN2A |
| Other glycan
degradation |
8.13×10−5 |
2.41×10−3 | NEU4; NEU2;
NEU3 |
| Vascular smooth
muscle contraction |
2.55×10−4 |
6.67×10−3 | GNAQ; GNA11; BRAF;
MAPK3; MAPK1 |
| Long-term
potentiation |
2.70×10−4 |
6.67×10−3 | GNAQ; BRAF; MAPK3;
MAPK1 |
| Thyroid cancer |
3.51×10−4 |
7.99×10−3 | BRAF; MAPK3;
MAPK1 |
| Gap junction |
8.11×10−4 |
1.60×10−2 | GNAQ; GNA11; MAPK3;
MAPK1 |
| ErbB signaling
pathway |
7.77×10−4 |
1.60×10−2 | NCK1; BRAF; MAPK3;
MAPK1 |
| GnRH signaling
pathway |
9.20×10−4 |
1.70×10−2 | GNAQ; GNA11; MAPK3;
MAPK1 |
| Sphingolipid
metabolism |
1.47×10−3 |
2.36×10−2 | NEU4; NEU2;
NEU3 |
| Chagas disease
(American trypanosomiasis) |
1.51×10−3 |
2.36×10−2 | GNAQ; GNA11; MAPK3;
MAPK1 |
| T cell receptor
signaling pathway |
1.51×10−3 |
2.36×10−2 | NCK1; CDK4; MAPK3;
MAPK1 |
| Serotonergic
synapse |
1.99×10−3 |
2.68×10−2 | GNAQ; BRAF; MAPK3;
MAPK1 |
| Endometrial
cancer |
1.97×10−3 |
2.68×10−2 | BRAF; MAPK3;
MAPK1 |
| Cholinergic
synapse |
1.93×10−3 |
2.68×10−2 | GNAQ; GNA11; MAPK3;
MAPK1 |
| Acute myeloid
leukemia |
2.56×10−3 |
3.03×10−2 | BRAF; MAPK3;
MAPK1 |
| NOD-like receptor
signaling pathway |
2.56×10−3 |
3.03×10−2 | CXCL1; MAPK3;
MAPK1 |
| Neurotrophin
signaling pathway |
2.56×10−3 |
3.03×10−2 | MAGED1; BRAF;
MAPK3; MAPK1 |
| mTOR signaling
pathway |
2.97×10−3 |
3.25×10−2 | BRAF; MAPK3;
MAPK1 |
| Viral
carcinogenesis |
2.86×10−3 |
3.25×10−2 | CDK4; HLA-E; MAPK3;
MAPK1; CDKN2A |
| Colorectal
cancer |
3.26×10−3 |
3.44×10−2 | BRAF; MAPK3;
MAPK1 |
| Natural killer cell
mediated cytotoxicity |
3.92×10−3 |
3.87×10−2 | BRAF; HLA-E; MAPK3;
MAPK1 |
| Renal cell
carcinoma |
3.89×10−3 |
3.87×10−2 | BRAF; MAPK3;
MAPK1 |
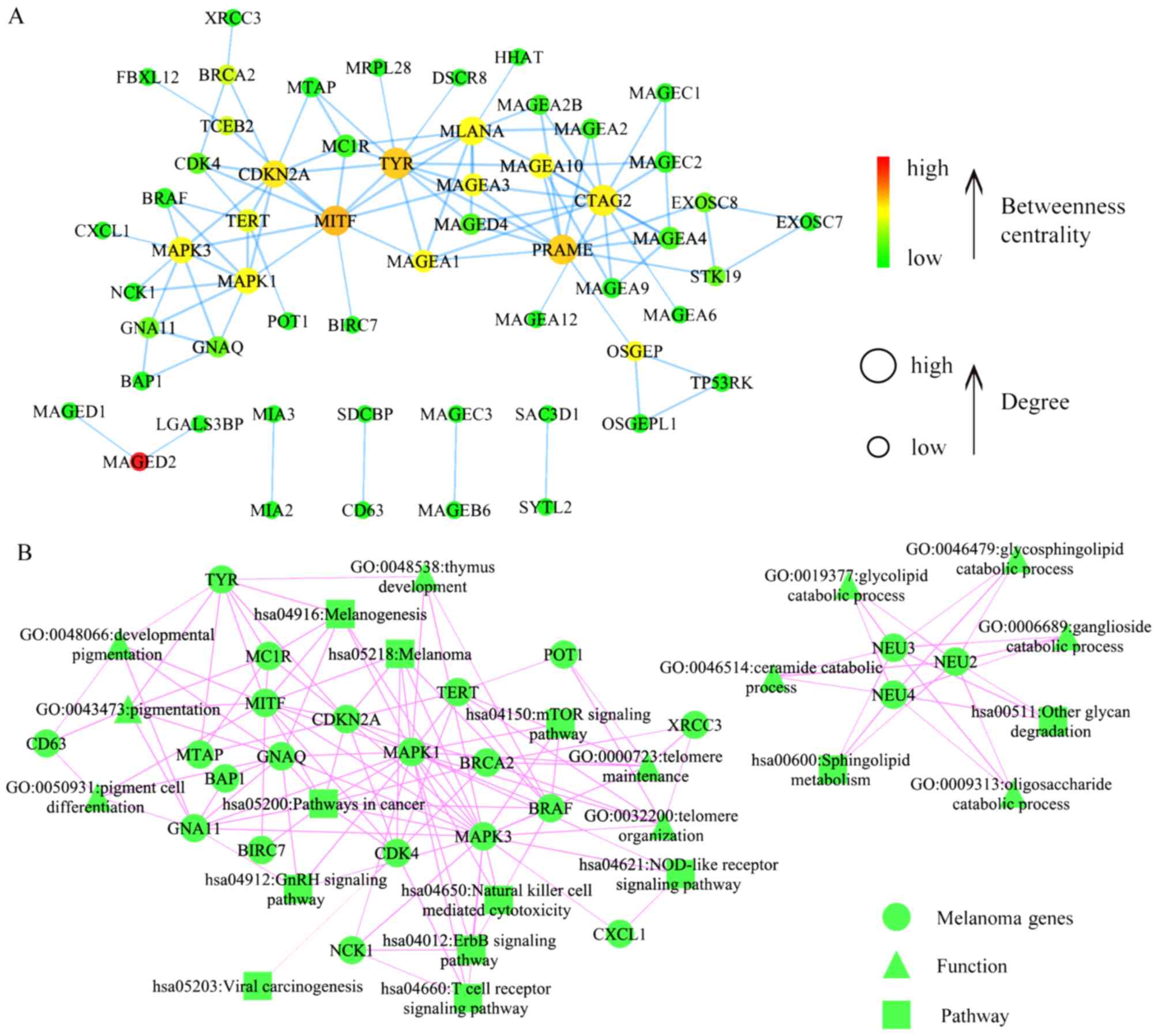
Melanoma-specific network
Fig. 3A illustrates
the melanoma-specific network that contains 56 melanoma-associated
genes and 98 gene pairs. The network MDSet, obtained by using the
binary integer linear method, contained 16 genes. Three genes,
including tyrosinase (TYR), mitogen-activated protein kinase (MAPK)
3 and cancer/testis antigen 2 (CTAG2), intersected with the top 16
genes with highest betweenness or degree and were considered as
important melanoma progression biomarkers. Fig. 3B illustrates associations of genes in
the network with significantly enriched GO terms and KEGG
pathways.
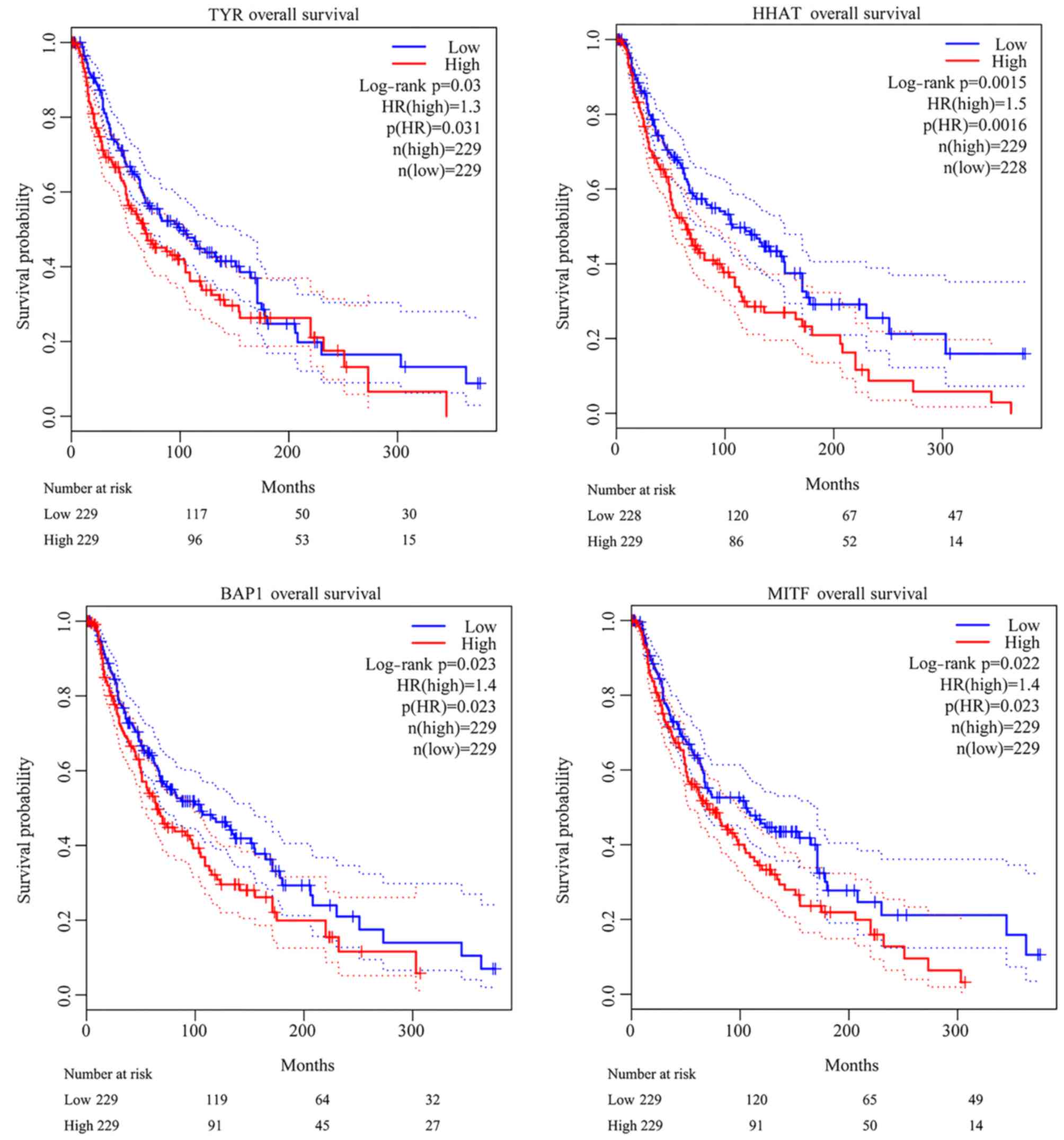
Survival analysis
Associations among genes contained in the network
and overall survival rates of patients with melanoma were evaluated
using the GEPIA database. As a result, two genes contained in the
MDSet, hedgehog acyltransferase (HHAT) and BRCA1-associated protein
1 (BAP1), and another two genes, TYR and microphthalmia-associated
transcription factor (MITF), were identified to be significantly
associated with the overall survival rates of patients with
melanoma. Elevated expression levels of the four genes were all
closely associated with poor melanoma prognosis (Fig. 4).
Discussion
The purpose of the present study was to explore the
underlying mechanism of melanoma and potential gene biomarkers
using comprehensive bioinformatics methods. The random walk method
identified 111 potential melanoma-associated genes, and functional
enrichment analysis identified several biological processes and
pathways which may contribute to melanoma progression. A
melanoma-specific network in combination with survival analysis
identified more reliable biomarkers for melanoma diagnosis and
treatment.
GO enrichment analysis indicated that ‘developmental
pigmentation’, ‘pigmentation’ and ‘pigment cell differentiation’
were closely associated with melanoma-associated genes.
Pigmentation is determined by the proportion of several melanin
species, for instance the alkali soluble yellow-red pheomelanin and
insoluble brown-black eumelanin (22). Synthesis of the two types of melanin
starts with the tyrosine oxidation of TYR, which produces
L-3,4-dihydroxyphenylalanine, which then produces dopaquinone and
eventually the common precursor. The range of individual
pigmentation phenotypes is the result of the ratio of true melanin
to brown melanin. Individuals with darker hair or skin possess
higher ratios of true melanin to brown melanin than those with
lighter hair or skin. Several studies have demonstrated that skin
which contains brown melanin provides weak UV protection,
suggesting that brown melanin may actually be a prooxidant and
photosensitive to DNA damage, while eumelanin may act as a
light-protective antioxidant (23,24).
Reactive oxygen species (ROS) may also be produced as byproducts
during pigment synthesis (24). It
has been considered that the role of melanocyte organelles is to
provide protection against ROS-induced injury by containment
(25). Leakage of toxic substances
from the melanosome can be damaging to melanocytes. Additionally,
pheomelanin synthesis stimulates melanoma genesis via an
UV-independent pathway that induces ROS-mediated DNA damage, which
may lead to melanoma carcinogenesis (26).
The deubiquitinating enzyme BAP1 is a tumor
suppressor, which is inactivated in a variety of types of cancer
(27). BAP1 inactivation is expected
to affect transcription regulation, either through direct gene
expression dysregulation or chromatin structure perturbation
(28,29). It has been reported that BAP1 affects
ROS homeostasis and serves a major role in regulating cell
morphology, cell migration and mitochondrial respiration in
mesothelioma (30). In uveal
melanoma, BAP1 inactivation is associated with metastasis
development and poor prognosis (31). Therefore, BAP1 may promote tumor cell
motility and invasiveness, and stimulates metastasis formation
in vivo, which is consistent with the results of the study
by Joseph et al (32).
MITF is the main regulator of melanocytes and serves
a vital role in the pigmentation pathway. It also has antioxidant
and prooxidant components (33).
There are at least nine isoforms of MITF, which exhibit
tissue-specific expression patterns, and the M isoform of MITF is
selectively expressed in melanocytes (34). Melanoma is considered to arise from
melanocytes, the pigment-producing cells of the skin, hair and
eyes. In addition to its vital roles in normal melanocytes, MITF
may also serve vital roles in melanoma, where it has been revealed
to be a genetically-specific survival oncogene that is amplified in
~20% of human types of melanoma (35). Previously, MITF has been demonstrated
to be involved in regulating metabolism and oxidative stress by
directly regulating the expression of the main mitochondrial
regulator; peroxisome proliferator-activated receptor γ coactivator
1 α (36).
The TYR gene, which encodes melanogenesis enzymes,
has been associated with melanoma (37). Subsequent to identifying that
variants of TYR affect pigmentation features, these same variants
were revealed to be associated with the risk of skin cancer, even
when accounting for available assessments of pigmentation (38). The association of TYR and its
variants is robust in regulating pigmentation effects.
The hedgehog family of secreted proteins act as
morphogens to control embryonic patterning and development in a
variety of organ systems. HHAT mutation occurs in the conserved
membrane bound O-acyltransferase domain. HHAT has been demonstrated
to be expressed in the somatic cells of XX and XY gonads at the
time of sex determination, and HHAT loss of function recapitulates
the majority of the testicular, skeletal, neuronal and growth
defects observed in humans (39). In
the developing testis, HHAT is not required for Sertoli cell
commitment but serves a role in proper testis cord formation and
the differentiation of fetal Leydig cells.
Optimal BP terms were consistent with expectations,
and several catabolism-associated terms were among them. Optimal
features contained the terms ganglioside catabolic process,
oligosaccharide catabolic process, glycosphingolipid catabolic
process, ceramide catabolic process, transfer RNA
threonylcarbamoyladenosine metabolic process and glycolipid
catabolic process. A previous study demonstrated that numerous
melanoma genes control cell metabolism, suggesting that melanoma
may be a metabolic disease (40).
Melanoma patterns in normal cells depend on the availability of
substrates and oxygen. The regulation of metabolic pathways in
cancer cells and the regulation of metabolic pathways in normal
cells are significantly different. A number of studies have
revealed fundamental differences in metabolic characteristics
between normal melanocytes and melanoma cells in vitro
(41–43). The metabolic characteristics of the
two cell types were further characterized by the use of stable
isotopes as tracers to quantify cell metabolic flux (44). According to the Warburg effect, the
relative contribution of mitochondrial respiration to energy
production in melanoma cells is generally low due to its higher
glycolytic rate. In addition, melanoma cells exhibit high levels of
glutaminase, which is the reverse flux of the tricarboxylic acid
cycle and provides carbon for synthesis of fatty acids (44).
A total of 40–60% of cutaneous types of melanoma
harbor B-Raf proto-oncogene (BRAF)V600 mutations
(45). BRAF mutations are
known to constitutively activate the MAPK signaling pathway
(46,47). MAPK3 belongs to the MAPK family,
which includes extracellular signal regulation kinases (48). When activated by upstream kinases,
MAPK phosphorylates several transcription factors and serves a
vital role in regulating cancer cell proliferation, differentiation
and other cellular activities directly associated with cancer. A
previous study revealed that the expression levels of members of
the MAPK signaling pathway were altered in patients with metastatic
melanoma, which may activate this signaling pathway (49).
The telomere-associated terms, including ‘telomere
maintenance’, ‘telomere organization’ and ‘establishment of protein
localization to telomere’, may also be associated with melanoma.
Telomere length was considered to be a major risk factor for
melanoma, increasing melanoma risk in a study of 557 cases
(50), and increasing nevus number
(51,52). In addition, high penetrative melanoma
mutations were reported in genes encoding the protection of
telomeres 1 (POT1) components, which are essential for telomere
maintenance and signal transduction functions (53). POT1 mutations result in longer
telomeres (54). A previous study
investigated the mechanism of melanoma, and hypothesized that
longer telomeres increase the duration of cell proliferation in a
melanocytic nevus (52). If the
process of aging is delayed in melanocytes, it could lead to
further mutations, which increases the probability of developing a
malignant tumor (50).
The thymus development process is also associated
with melanoma. Interleukin-32 (IL-32) is a novel cytokine, involved
in cancer development, and expressed in numerous human tissues and
organs, including the thymus. Nicholl et al (55) reported that exogenous addition of
IL-32 can effectively inhibit the proliferation of human melanoma
cell lines, and that it is associated with increased expression of
p21, p53 and tumor necrosis factor receptor superfamily member 10a.
In previous studies, IL-32 has been reported to inhibit the growth
and metastasis of cancer cells by regulating nuclear factor κB
(NF-κB) signaling (56,57). NF-κB is a transcription factor that
regulates the expression of cytokines, growth factors and apoptosis
inhibiting factors. Additionally, IL-32 is associated with signal
transducer and activator of transcription 3 (STAT3) signaling in
cancer cell growth (58). Numerous
studies have reported that STAT3 signaling promotes cancer
development, while loss of STAT3 inhibits cancer development
(59–61).
In KEGG pathway analysis, certain types of cancer,
including pancreatic and bladder cancer, were significantly
enriched, which may indicate that melanoma has a similar phenotype,
mechanism or co-morbidity with these types of cancer. Pathways,
including chronic myeloid leukemia and acute myeloid leukemia, are
directly associated with cancer and potential oncogenes. As a
result, the abnormal expression of oncogenes may lead to cell cycle
disturbances, continued cell proliferation, reduced apoptosis and
tumor metastasis (62). The mTOR
signaling pathway is involved in regulating cell growth, division,
survival and transcription (63).
Overactivation of mTOR can promote proliferation and reduce
apoptosis. In addition, mTOR pathway dysfunction has been observed
in several types of tumor (64,65). A
previous study suggested that CTAG2 is closely associated with
cancer/testis antigen 1B (CTAG1B) (66). CTAG2 and CTAG1B are overexpressed in
numerous types of tumor, including melanoma, sarcoma and multiple
myeloma, as well as certain types of carcinoma, including lung,
head and neck, and ovarian carcinoma (66,67).
In the present study, TYR, HHAT, BAP1 and MITF were
identified as potential therapeutic targets for melanoma. As the
occurrence and development of melanoma is a complex event, the
specific roles of these genes in the regulatory network remain to
be determined by further experiments. Furthermore, in addition to
the present study, multiple biomarkers have been identified to be
associated with melanoma progression, proliferation, immune
response, oncogenesis and other aspects by microarray studies
(68,69). As the public databases used,
including OMIM and STRING, did not provide patient characteristics
information, the association between the expression levels of
candidate genes and clinicopathological parameters could not be
assessed. Further analysis will be conducted in a future study with
information from The Cancer Genome Atlas and Gene Expression
Omnibus data sets. Additionally, it remains a great challenge to
apply this information in a therapeutic manner.
In conclusion, in the present study, prioritization
and comprehensive analysis of melanoma candidate genes were
conducted to obtain several potential biomarkers and biological
processes which may contribute to melanoma progression. This should
be helpful in understanding the underlying mechanisms of melanoma,
as well as for its diagnosis and treatment. However, further
experimental validation of the results of the present study is
required.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
CF and MB made substantial contributions to the
conception and design of the study. CF, HZ and AZ participated in
the acquisition, analysis and interpretation of data. MB, HZ and WZ
have been involved in drafting the manuscript or revising it
critically for important intellectual content. WZ participated in
the data analysis. CF and MB gave final approval of the version to
be published. CF and MB have agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
MacKie RM, Hauschild A and Eggermont AM:
Epidemiology of invasive cutaneous melanoma. Ann Oncol. 20 (Suppl
6):vi1–7. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2019. CA Cancer J Clin. 69:7–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Caini S, Gandini S, Sera F, Raimondi S,
Fargnoli MC, Boniol M and Armstrong BK: Meta-analysis of risk
factors for cutaneous melanoma according to anatomical site and
clinico-pathological variant. Eur J Cancer. 45:3054–3063. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ingraffea A: Melanoma. Facial Plast Surg
Clin North Am. 21:33–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Owens B: Melanoma. Nature. 515:S1092014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Markovic SN, Erickson LA, Rao RD, Weenig
RH, Pockaj BA, Bardia A, Vachon CM, Schild SE, McWilliams RR, Hand
JL, et al: Malignant melanoma in the 21st century, part 1:
Epidemiology, risk factors, screening, prevention, and diagnosis.
Mayo Clin Proc. 82:364–380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rigel DS: Epidemiology of melanoma. Semin
Cutan Med Surg. 29:204–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gandini S, Sera F, Cattaruzza MS, Pasquini
P, Picconi O, Boyle P and Melchi CF: Meta-analysis of risk factors
for cutaneous melanoma: II. Sun exposure. Eur J Cancer. 41:45–60.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Elwood JM and Jopson J: Melanoma and sun
exposure: An overview of published studies. Int J Cancer.
73:198–203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stern RS; PUVA Follow up Study, : The risk
of melanoma in association with long-term exposure to PUVA. J Am
Acad Dermatol. 44:755–761. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bevona C, Goggins W, Quinn T, Fullerton J
and Tsao H: Cutaneous melanomas associated with nevi. Arch
Dermatol. 139:1620–1624. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tsao H and Niendorf K: Genetic testing in
hereditary melanoma. J Am Acad Dermatol. 51:803–808. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stam-Posthuma JJ, van Duinen C, Scheffer
E, Vink J and Bergman W: Multiple primary melanomas. J Am Acad
Dermatol. 44:22–27. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hanauer DA, Rhodes DR, Sinha-Kumar C and
Chinnaiyan AM: Bioinformatics approaches in the study of cancer.
Curr Mol Med. 7:133–141. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cho A, Shim JE, Kim E, Supek F, Lehner B
and Lee I: MUFFINN: Cancer gene discovery via network analysis of
somatic mutation data. Genome Biol. 17:1292016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ding KF, Finlay D, Yin H, Hendricks WPD,
Sereduk C, Kiefer J, Sekulic A, LoRusso PM, Vuori K, Trent JM and
Schork NJ: Network rewiring in cancer: Applications to melanoma
cell lines and the cancer genome atlas patients. Front Genet.
9:2282018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amberger JS, Bocchini CA, Schiettecatte F,
Scott AF and Hamosh A: OMIM.org: Online mendelian inheritance in
man (OMIM(R)), an online catalog of human genes and genetic
disorders. Nucleic Acids Res. 43:D789–D798. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dembélé D: Analysis of high-throughput
biological data using their rank values. Stat Methods Med Res.
96228021876418. 2018.(Epub ahead of print). View Article : Google Scholar
|
|
19
|
Wang J, Duncan D, Shi Z and Zhang B:
WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013.
Nucleic Acids Res. 41:W77–W83. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abbas A, Kong XB, Liu Z, Jing BY and Gao
X: Automatic peak selection by a Benjamini-Hochberg-based
algorithm. PLoS One. 8:e531122013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Khuri S and Wuchty S: Essentiality and
centrality in protein interaction networks revisited. BMC
Bioinformatics. 16:1092015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito S and Wakamatsu K: Chemistry of mixed
melanogenesis-pivotal roles of dopaquinone. Photochem Photobiol.
84:582–592. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Samokhvalov A, Hong L, Liu Y, Garguilo J,
Nemanich RJ, Edwards GS and Simon JD: Oxidation potentials of human
eumelanosomes and pheomelanosomes. Photochem Photobiol. 81:145–148.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Meredith P and Sarna T: The physical and
chemical properties of eumelanin. Pigment Cell Res. 19:572–594.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen KG, Valencia JC, Gillet JP, Hearing
VJ and Gottesman MM: Involvement of ABC transporters in
melanogenesis and the development of multidrug resistance of
melanoma. Pigment Cell Melanoma Res. 22:740–749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Montano SP, Pigli YZ and Rice PA: The mu
transpososome structure sheds light on DDE recombinase evolution.
Nature. 491:413–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wiesner T, Murali R, Fried I, Cerroni L,
Busam K, Kutzner H and Bastian BC: A distinct subset of atypical
Spitz tumors is characterized by BRAF mutation and loss of BAP1
expression. Am J Surg Pathol. 36:818–830. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dey A, Seshasayee D, Noubade R, French DM,
Liu J, Chaurushiya MS, Kirkpatrick DS, Pham VC, Lill JR, Bakalarski
CE, et al: Loss of the tumor suppressor BAP1 causes myeloid
transformation. Science. 337:1541–1546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baymaz HI, Fournier A, Laget S, Ji Z,
Jansen PW, Smits AH, Ferry L, Mensinga A, Poser I, Sharrocks A, et
al: MBD5 and MBD6 interact with the human PR-DUB complex through
their methyl-CpG-binding domain. Proteomics. 14:2179–2189. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hebert L, Bellanger D, Guillas C, Campagne
A, Dingli F, Loew D, Fievet A, Jacquemin V, Popova T, Jean D, et
al: Modulating BAP1 expression affects ROS homeostasis, cell
motility and mitochondrial function. Oncotarget. 8:72513–72527.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harbour JW, Onken MD, Roberson ED, Duan S,
Cao L, Worley LA, Council ML, Matatall KA, Helms C and Bowcock AM:
Frequent mutation of BAP1 in metastasizing uveal melanomas.
Science. 330:1410–1413. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Joseph RW, Kapur P, Serie DJ, Eckel-Passow
JE, Parasramka M, Ho T, Cheville JC, Frenkel E, Rakheja D,
Brugarolas J and Parker A: Loss of BAP1 protein expression is an
independent marker of poor prognosis in patients with low-risk
clear cell renal cell carcinoma. Cancer. 120:1059–1067. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hershey CL and Fisher DE: Genomic analysis
of the microphthalmia locus and identification of the MITF-J/Mitf-J
isoform. Gene. 347:73–82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fuse N, Yasumoto K, Suzuki H, Takahashi K
and Shibahara S: Identification of a melanocyte-type promoter of
the microphthalmia-associated transcription factor gene. Biochem
Biophys Res Commun. 219:702–707. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Garraway LA, Widlund HR, Rubin MA, Getz G,
Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J,
et al: Integrative genomic analyses identify MITF as a lineage
survival oncogene amplified in malignant melanoma. Nature.
436:117–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Haq R, Shoag J, Andreu-Perez P, Yokoyama
S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL,
et al: Oncogenic BRAF regulates oxidative metabolism via PGC1α and
MITF. Cancer Cell. 23:302–315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gudbjartsson DF, Sulem P, Stacey SN,
Goldstein AM, Rafnar T, Sigurgeirsson B, Benediktsdottir KR,
Thorisdottir K, Ragnarsson R, Sveinsdottir SG, et al: ASIP and TYR
pigmentation variants associate with cutaneous melanoma and basal
cell carcinoma. Nat Genet. 40:886–891. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Saternus R, Pilz S, Gräber S, Kleber M,
März W, Vogt T and Reichrath J: A closer look at evolution:
Variants (SNPs) of genes involved in skin pigmentation, including
EXOC2, TYR, TYRP1, and DCT, are associated with 25(OH)D serum
concentration. Endocrinology. 156:39–47. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Callier P, Calvel P, Matevossian A,
Makrythanasis P, Bernard P, Kurosaka H, Vannier A, Thauvin-Robinet
C, Borel C, Mazaud-Guittot S, et al: A Loss of Function mutation in
the palmitoyl-transferase HHAT Leads to Syndromic 46,XY disorder of
sex development by impeding hedgehog protein palmitoylation and
signaling. PLoS Genet. 10:e10043402014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Abildgaard C and Guldberg P: Molecular
drivers of cellular metabolic reprogramming in melanoma. Trends Mol
Med. 21:164–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Abildgaard C, Dahl C, Basse AL, Ma T and
Guldberg P: Bioenergetic modulation with dichloroacetate reduces
the growth of melanoma cells and potentiates their response to
BRAFV600E inhibition. J Transl Med. 12:2472014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Barbi de Moura M, Vincent G, Fayewicz SL,
Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C,
et al: Mitochondrial respiration-an important therapeutic target in
melanoma. PLoS One. 7:e406902012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hall A, Meyle KD, Lange MK, Klima M,
Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen
PB, et al: Dysfunctional oxidative phosphorylation makes malignant
melanoma cells addicted to glycolysis driven by the (V600E)BRAF
oncogene. Oncotarget. 4:584–599. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Scott DA, Richardson AD, Filipp FV,
Knutzen CA, Chiang GG, Ronai ZA, Osterman AL and Smith JW:
Comparative metabolic flux profiling of melanoma cell lines: Beyond
the Warburg effect. J Biol Chem. 286:42626–42634. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dong J, Phelps RG, Qiao R, Yao S, Benard
O, Ronai Z and Aaronson SA: BRAF oncogenic mutations correlate with
progression rather than initiation of human melanoma. Cancer Res.
63:3883–5. 2003.PubMed/NCBI
|
|
46
|
Pflugfelder A, Kochs C, Blum A, Capellaro
M, Czeschik C, Dettenborn T, Dill D, Dippel E, Eigentler T, Feyer
P, et al: Malignant melanoma S3-guideline ‘diagnosis, therapy and
follow-up of melanoma’. J Dtsch Dermatol Ges. 11 (Suppl 6):S1–S126.
2013. View Article : Google Scholar
|
|
47
|
Stadler S, Weina K, Gebhardt C and Utikal
J: New therapeutic options for advanced non-resectable malignant
melanoma. Adv Med Sc. 60:83–88. 2015. View Article : Google Scholar
|
|
48
|
Garcia F, Zalba G, Paez G, Encio I and de
Miguel C: Molecular cloning and characterization of the human p44
mitogen-activated protein kinase gene. Genomics. 50:69–78. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Orouji E, Orouji A, Gaiser T, Larribere L,
Gebhardt C and Utikal J: MAP kinase pathway gene copy alterations
in NRAS/BRAF wild-type advanced melanoma. Int J Cancer.
138:2257–2262. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nan H, Du M, De Vivo I, Manson JE, Liu S,
McTiernan A, Curb JD, Lessin LS, Bonner MR, Guo Q, et al: Shorter
telomeres associate with a reduced risk of melanoma development.
Cancer Res. 71:6758–6763. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Han J, Qureshi AA, Prescott J, Guo Q, Ye
L, Hunter DJ and De Vivo I: A prospective study of telomere length
and the risk of skin cancer. J Invest Dermatol. 129:415–421. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bataille V, Kato BS, Falchi M, Gardner J,
Kimura M, Lens M, Perks U, Valdes AM, Bennett DC, Aviv A and
Spector TD: Nevus size and number are associated with telomere
length and represent potential markers of a decreased senescence in
vivo. Cancer Epidemiol Biomarkers Prev. 16:1499–1502. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Horn S, Figl A, Rachakonda PS, Fischer C,
Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al:
TERT promoter mutations in familial and sporadic melanoma. Science.
339:959–961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Robles-Espinoza CD, Harland M, Ramsay AJ,
Aoude LG, Quesada V, Ding Z1, Pooley KA, Pritchard AL, Tiffen JC,
Petljak M, et al: POT1 loss-of-function variants predispose to
familial melanoma. Nat Genet. 46:478–481. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nicholl MB, Chen X, Qin C, Bai Q, Zhu Z,
Davis MR and Fang Y: IL-32α has differential effects on
proliferation and apoptosis of human melanoma cell lines. J Surg
Oncol. 113:364–369. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng
WL, Tseng YH, Chen CY, Lin CD, Wu JI, Wang LH and Lin KH:
Interleukin-32 increases human gastric cancer cell invasion
associated with tumor progression and metastasis. Clin Cancer Res.
20:2276–2288. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yun HM, Oh JH, Shim JH, Ban JO, Park KR,
Kim JH, Lee DH, Kang JW, Park YH, Yu D, et al: Antitumor activity
of IL-32β through the activation of lymphocytes, and the
inactivation of NF-κB and STAT3 signals. Cell Death Dis.
4:e6402013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Oh JH, Cho MC, Kim JH, Lee SY, Kim HJ,
Park ES, Ban JO, Kang JW, Lee DH, Shim JH, et al: IL-32γ inhibits
cancer cell growth through inactivation of NF-κB and STAT3 signals.
Oncogene. 30:3345–3359. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chan KS, Sano S, Kiguchi K, Anders J,
Komazawa N, Takeda J and DiGiovanni J: Disruption of Stat3 reveals
a critical role in both the initiation and the promotion stages of
epithelial carcinogenesis. J Clin Invest. 114:720–728. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jenkins BJ, Roberts AW, Najdovska M, Grail
D and Ernst M: The threshold of gp130-dependent STAT3 signaling is
critical for normal regulation of hematopoiesis. Blood.
105:3512–3520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Lee H, Herrmann A, Deng JH, Kujawski M,
Niu G, Li Z, Forman S, Jove R, Pardoll DM and Yu H: Persistently
activated Stat3 maintains constitutive NF-kappaB activity in
tumors. Cancer Cell. 15:283–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Arvanitis C and Felsher DW: Conditional
transgenic models define how MYC initiates and maintains
tumorigenesis. Semin Cancer Biol. 16:313–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hay N and Sonenberg N: Upstream and
downstream of mTOR. Genes Dev. 18:1926–1945. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beevers CS, Li F, Liu L and Huang S:
Curcumin inhibits the mammalian target of rapamycin-mediated
signaling pathways in cancer cells. Int J Cancer. 119:757–764.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Matsubara S, Ding Q, Miyazaki Y, Kuwahata
T, Tsukasa K and Takao S: mTOR plays critical roles in pancreatic
cancer stem cells through specific and stemness-related functions.
Sci Rep. 3:32302013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lethe B, Lucas S, Michaux L, De Smet C,
Godelaine D, Serrano A, De Plaen E and Boon T: LAGE-1, a new gene
with tumor specificity. Int J Cancer. 76:903–908. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nicholaou T, Ebert L, Davis ID, Robson N,
Klein O, Maraskovsky E, Chen W and Cebon J: Directions in the
immune targeting of cancer: Lessons learned from the cancer-testis
Ag NY-ESO-1. Immunol Cell Biol. 84:303–317. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Vizkeleti L, Kiss T, Koroknai V, Ecsedi S,
Papp O, Szasz I, Adany R and Balazs M: Altered integrin expression
patterns shown by microarray in human cutaneous melanoma. Melanoma
Res. 27:180–188. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wozniak M, Peczek L, Czernek L and Düchler
M: Analysis of the miRNA profiles of melanoma exosomes derived
under normoxic and hypoxic culture conditions. Anticancer Res.
37:6779–6789. 2017.PubMed/NCBI
|