Introduction
Cutaneous squamous cell carcinoma (cSCC), also known
as squamous-cell skin cancer, is a malignancy originating from
keratinocytes in the epidermis or epidermal appendages (1). cSCC represents the second most common
type of non-melanoma skin cancer following basal cell carcinoma and
accounts for ~20% of all cutaneous malignancies (2). Exposure to chronic ultraviolet
radiation is considered to be a risk factor for the development of
cSCC, which is associated with a notable alteration in EFGR
expression (3,4). EGFR, a receptor tyrosine kinase, serves
an important regulatory role in the Ras/mitogen-activated protein
kinase, phosphoinositide 3-kinase/protein kinase B and
phospholipase C pathways in squamous cells (5), which are involved in cell apoptosis,
proliferation, invasion, metastasis and angiogenesis (6). However, the deregulation of EGFR
activation has been associated with the development and progression
of cSCC (7). Thus, an increasing
number of studies have investigated EGFR-targeted therapies in
recent years, including monoclonal antibodies (mAbs) and small
molecule tyrosine kinase inhibitors (TKIs) (8–10). mAbs,
which include cetuximab, panitumumab, nimotuzumab and zalutunumab,
target the extracellular portion of the receptor; however, TKIs,
including gefitinib, erlotinib, lapatinib and afatinib, block the
intracellular downstream signalling pathway (11). In the present study, the cytotoxic
and apoptotic effects exhibited by A431 cells treated with
gefitinib were investigated.
Cells and tumours overexpressing EGFR have been
demonstrated to exhibit dysregulated autophagy (12), resulting in cells degrading and
recycling cellular constituents (13). The exact role of autophagy is
unknown. It has been suggested that autophagy represents an
alternative tumour-suppressing mechanism and is associated with
genomic instability, suppression of cell growth and degradation of
important cellular components (14).
Recycled proteins and energy contribute to the maintenance of
cellular homeostasis and increase the survival of tumour cells
under stress conditions (15).
However, it has been reported that autophagy represents a survival
strategy exhibited by skin cancer cells in response to cisplatin,
an adjuvant chemotherapy used for the treatment of patients with
invasive cSCC (16). Recently,
numerous studies have demonstrated that autophagy represents an
important mechanism associated with resistance to TKIs (17,18). It
has also been revealed that inhibition of autophagy enhances the
anti-cancer effect of EGFR inhibitors in human bladder cancer cells
(19). Furthermore, targeting
autophagy in triple negative breast cancer cells is an effective
treatment for the enhancement of sensitivity to EGFR inhibitors
(20). However, to the best of our
knowledge, the role of autophagy associated with the administration
of gefitinib as a neoadjuvant treatment followed by surgery and/or
radiotherapy for the treatment of patients with aggressive cSCC has
not been clearly determined.
To determine the effects of autophagy on the
cytoprotection of gefitinib-treated A431 cells, chloroquine
diphosphate (CQ), an inhibitor of autophagolysosome formation was
used in the present study to inhibit autophagy. The results
demonstrated that gefitinib induced caspase-dependent apoptosis and
activated the autophagic response in A431 cells. In addition, the
role of autophagy in sSCC cell survival, was examined by assessing
the anti-proliferative effect following co-treatment with CQ and
gefitinib.
Materials and methods
Cell culture
The cSCC cell line A431 (derived from an 85-year-old
female patient suffering from vulvar squamous cell carcinoma; China
Centre for Type Culture Collection and Cell Bank of the Chinese
Academy of Sciences, Shanghai, China) was cultured in Dulbecco's
modified Eagle medium (DMEM) supplemented with 10% foetal bovine
serum, 100 units/ml penicillin and streptomycin (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), and maintained at 37°C
with 5% CO2 in a humidified atmosphere.
Reagents and antibodies
Gefitinib (cat. no. S1025) was purchased from
Selleck Chemicals (Houston, TX, USA) and CQ (cat. no. A506569) was
purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
Gefitinib and CQ were dissolved in DMSO and DMEM, respectively, and
subsequently stored at a stock concentration of 100 mM at −20°C.
The following primary antibodies and dilutions were used in the
present study: Microtubule associated protein 1 light chain 3-B
(LC3B; cat. no. 3868S; Cell Signalling Technology, Inc., Danvers,
MA, USA; 1:1,000), caspase-3 (cat. no. sc-7272; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; 1:1,000), poly-(ADP-ribose)
polymerase (PARP; cat. no. 9532S, Cell Signalling Technology, Inc.;
1:5,000), β-actin (cat. no. sc-47778; Santa Cruz Biotechnology,
Inc., Inc.; 1:1,000) and α-tubulin (cat. no. sc-5286; Santa Cruz
Biotechnology; Inc.; 1:1,000). Secondary antibodies used in the
present study were horseradish peroxidase (HRP)-tagged anti-mouse
IgG (cat. no. 31430; Invitrogen; Thermo Fisher Scientific, Inc.;
1:5,000) and HRP-tagged anti-rabbit IgG (cat. no. 31460;
Invitrogen; Thermo Fisher Scientific, Inc.; 1:5,000).
Cytotoxicity assay
Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was used to perform
cytotoxicity assays. Cells were plated in triplicate in 96-well
plates at a density of 8×103 cells/well and cultured
overnight. Media was then removed via suction with an aspirator and
replaced with 0.1 ml fresh DMEM containing different concentrations
of gefitinib (0, 10, 20, 30, 40 or 50 µM) or CQ (0, 50, 100, 150,
200, 250 or 300 µM). Control cells were treated with the same
volumes of DMSO or DMEM as the experimental groups. Following this,
the plates were incubated at 37°C for 12 h. Each well was
subsequently incubated with 100 µl DMEM medium containing 10 µl
CCK-8 for 2 h. The absorbance was measured at 450 nm using a
microplate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and half-maximal inhibitory concentration (IC50) values
were calculated based on log values using GraphPad Prism version 5
software (GraphPad Software, Inc., La Jolla, CA, USA).
Drug combination analysis
Gefitinib and CQ were added separately and together
in a constant ratio, as calculated from a dose-effect curve.
Inhibition effect was scored from 0 to 1, where a score of 0
represented no effect and a score of 1 represented 100% effect.
CompuSyn software (version 1.0; T. C. Chou and N. Martin, Memorial
Sloan-Kettering Cancer Centre, New York) was used to calculate the
combination index (CI) and an isobologram was established to
quantitatively determine the effect of drug interactions.
Investigation of apoptotic morphology
via fluorescent microscopy
Following the treatment of A431 cells with either
gefitinib (20 µM), CQ (188 µM) or gefitinib (20 µM) + CQ (188 µM)
at 37°C for 12 h, morphological observations of apoptosis and cell
death were investigated using acridine orange/ethidium bromide
staining (Beijing Solarbio Science & Technology Co., Ltd.,
Beijing, China). Following incubation, cells were washed with PBS
and subsequently fixed with 4% formaldehyde at room temperature for
15 min. Fixed cells were again washed with PBS and stained with
acridine orange/ethidium bromide at room temperature for 5 min.
Stained cells were subsequently observed and imaged under a
Ti-Eclipse inverted fluorescent microscope (Nikon Corporation,
Tokyo, Japan; magnification, ×10).
Annexin V/propidium iodide (PI)
staining assay for apoptosis
Following treatment with either gefitinib (20 µM),
CQ (188 µM) or gefitinib (20 µM) + CQ (188 µM) at 37°C for 12 h,
A431 cells were collected and washed three times using ice-cold
PBS. Cells were then resuspended in 400 µl binding buffer and
subsequently incubated with 5 µl Annexin V-FITC and 5 µl PI at room
temperature for 15 min in the dark. Following this, flow cytometric
analysis was immediately performed and data was analysed using
Cell-Quest software (version 5.1; BD Biosciences, San Jose, CA,
USA).
Monodansylcadaverine (MDC) staining
for the identification of autophagic vacuoles
Autophagic vacuoles were stained as previously
described (21,22). A431 cells were seeded in 24
well-plate at a density of 3×104 cells/well. Following a
12 h incubation with gefitinib (20 µM) and CQ (188 µM), either
alone or in combination at 37°C, cells were cultured in 50 µM MDC
for 15 min at 37°C. Cells were then washed with PBS (pH 7.4), and
levels of fluorescence were subsequently measured and imaged using
an inverted fluorescence microscope (Nikon Eclipse Ti; Nikon
Corporation; magnification, ×20). All experiments were repeated at
least three times.
Western blotting
Cells were treated with CQ (188 µM) and/or Ge (0–40
µM) for 0–12 h at 37°C. Plates were subsequently washed twice with
ice-cold PBS and the cells were lysed using
radioimmunoprecipitation assay buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology, Haimen, China). Protein concentrations
were quantified using Bradford reagent. Denatured proteins (20
µg/well) were separated by 12% SDS-PAGE and subsequently
transferred onto polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). Following this, membranes were
blocked at room temperature for 1 h in blocking buffer containing
5% dried skimmed milk that was diluted with TBST containing 0.1%
Tween 20. Membranes were then incubated with primary antibodies
against LC3-II, PARP, caspase-3, and α-tubulin at 4°C overnight.
Following this, membranes were washed with TBST and then incubated
with HRP-conjugated secondary antibodies (1:5,000 in 0.1% TBST) for
90 min at room temperature. Membranes were again washed with TBST
and immune complexes were then detected using enhanced
chemiluminescence reagents (cat. no. WBLUF0500; Merck KGaA,
Darmstadt, Germany). Densitometry of the western blot bands was
performed using ImageJ software (v1.52i; National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
Differences between groups were compared using
two-tailed Student's t tests or one-way analysis of variance
followed by Student-Newman-Keuls-q post hoc test. Data were
analysed using SPSS 13.0 software (SPSS, Inc., Chicago, IL, USA)
and presented as the means ± standard error of the mean. P<0.05
was considered to indicate a statistically significant difference.
All experiments were repeated at least three times.
Results
Gefitinib and CQ induce cytotoxic
effects in A431 cells
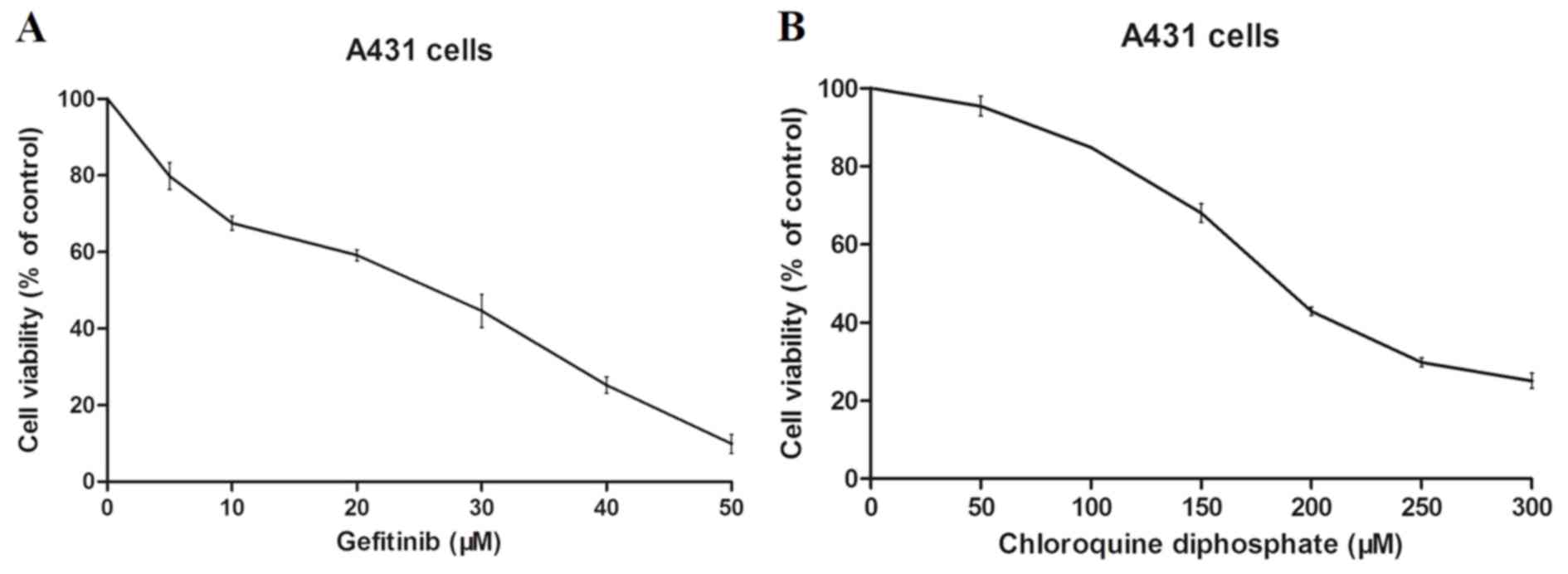
To investigate the cytotoxicity of gefitinib and CQ,
A431 cells were treated with various doses of gefitinib or CQ for
12 h. The results revealed that gefitinib and CQ both induce
cytotoxic effects in A431 cells in a dose-dependent manner
(Fig. 1A and B). Following 12 h of
treatment, the IC50 values of gefitinib and CQ in A431
cells were demonstrated to be 19.77±1.76 and 189.1±3.29 Μm,
respectively.
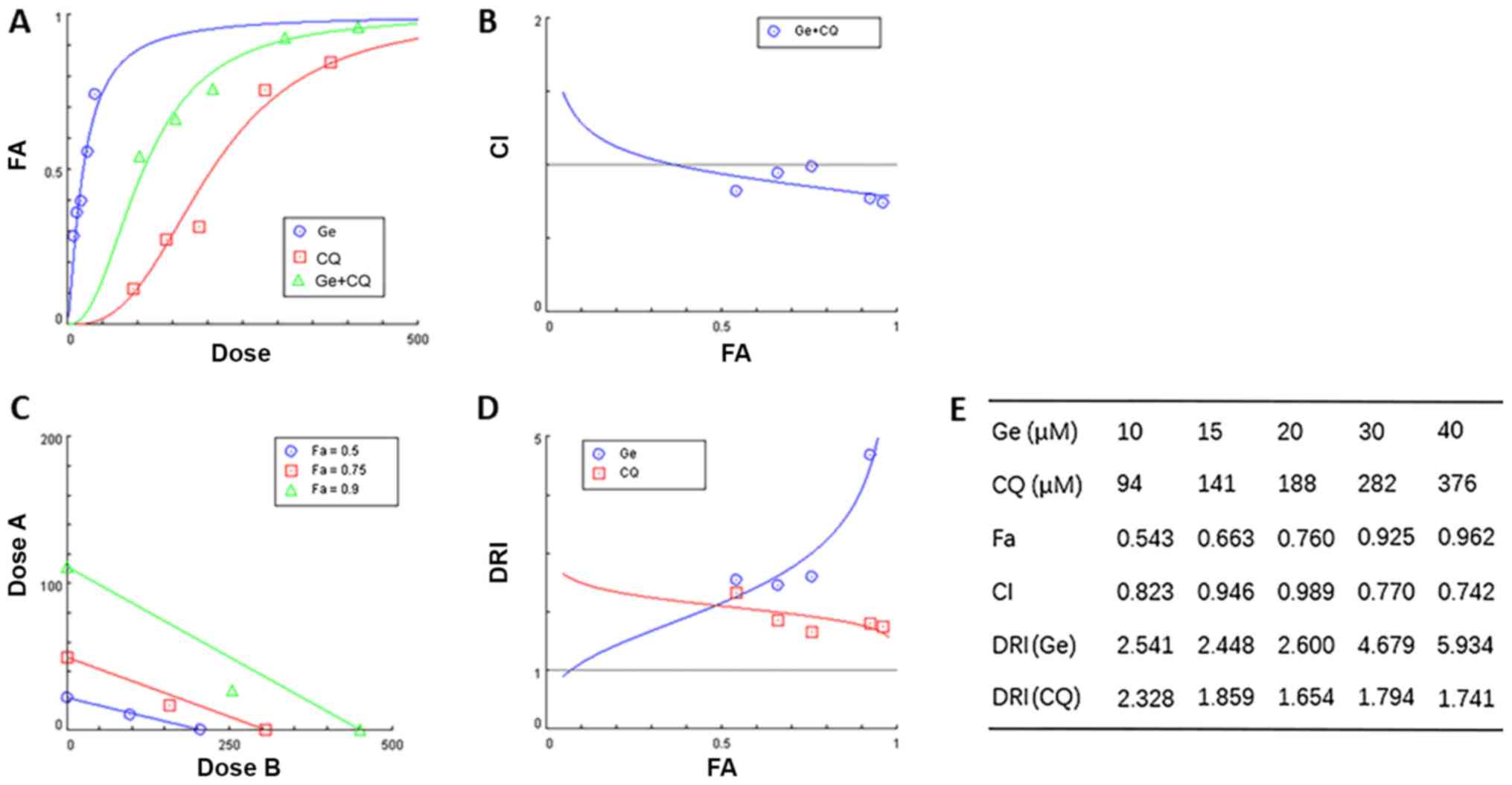
Gefitinib and CQ synergistically
inhibit the proliferation of A431 cells
Combinatory administration of gefitinib and CQ was
investigated using CompuSyn software. Concentrations of 10, 15, 20,
30 and 40 µM, and 94, 141, 188, 282 and 376 µM were used to
establish a dose-effect curve for gefitinib and CQ, respectively. A
constant ratio (20/188=5:47) was used to establish the doses used
in combinatory treatment groups (10+94, 15+141, 20+188, 30+282 and
40+376 Μm). Gefitinib and CQ exhibited a synergistic effect
(Fig. 2A-E). The dose-response
effects of gefitinib, CQ and gefitinib + CQ are presented in
Fig. 2A. CI values, a quantitative
definition for synergism, were revealed to be <1 in A431 cells,
which indicated that combinatory treatment with gefitinib and CQ
exhibited synergistic cytotoxic effects in A431 cells (Fig. 2B). Isobolograms, representing
equipotent combinations of two drugs administered at different
dosages, were also established via CompuSyn analysis. The dosages
of drug combinations revealed by the isobologram also suggested
that gefitinib and CQ exhibited synergistic effects in A431 cells
(Fig. 2C). Dose reduction index
(DRI) values of each drug in combination treatment, which measures
the number of folds by which single drug doses can be reduced by
when used in combination, were revealed to be >1, thus
indicating a favourable drug combination (Fig. 2D). Data obtained via CompuSyn
analyses are presented in Fig.
2E.
Gefitinib and CQ induce apoptosis via
the caspase-dependent apoptosis pathway
To determine the mechanism of cell death induced in
A431 cells following combinatory treatment with gefitinib and CQ,
acridine orange/ethidium bromide staining assays were performed.
The results demonstrated that the number of apoptotic cells (early
apoptotic cells with yellow-green fluorescence and late apoptotic
cells with orange fluorescence) in gefitinib + CQ treatment groups
were markedly increased compared with cells treated with gefitinib
or CQ alone (Fig. 3A). To determine
the apoptotic rates of A431 cells following treatment with
gefitinib and/or CQ, flow cytometry with Annexin V/PI staining was
performed. Significantly increased levels of Annexin V-positive
A431 cells were identified in the combinatory treatment group
compared with cells treated with gefitinib or CQ alone (Fig. 3B and C).
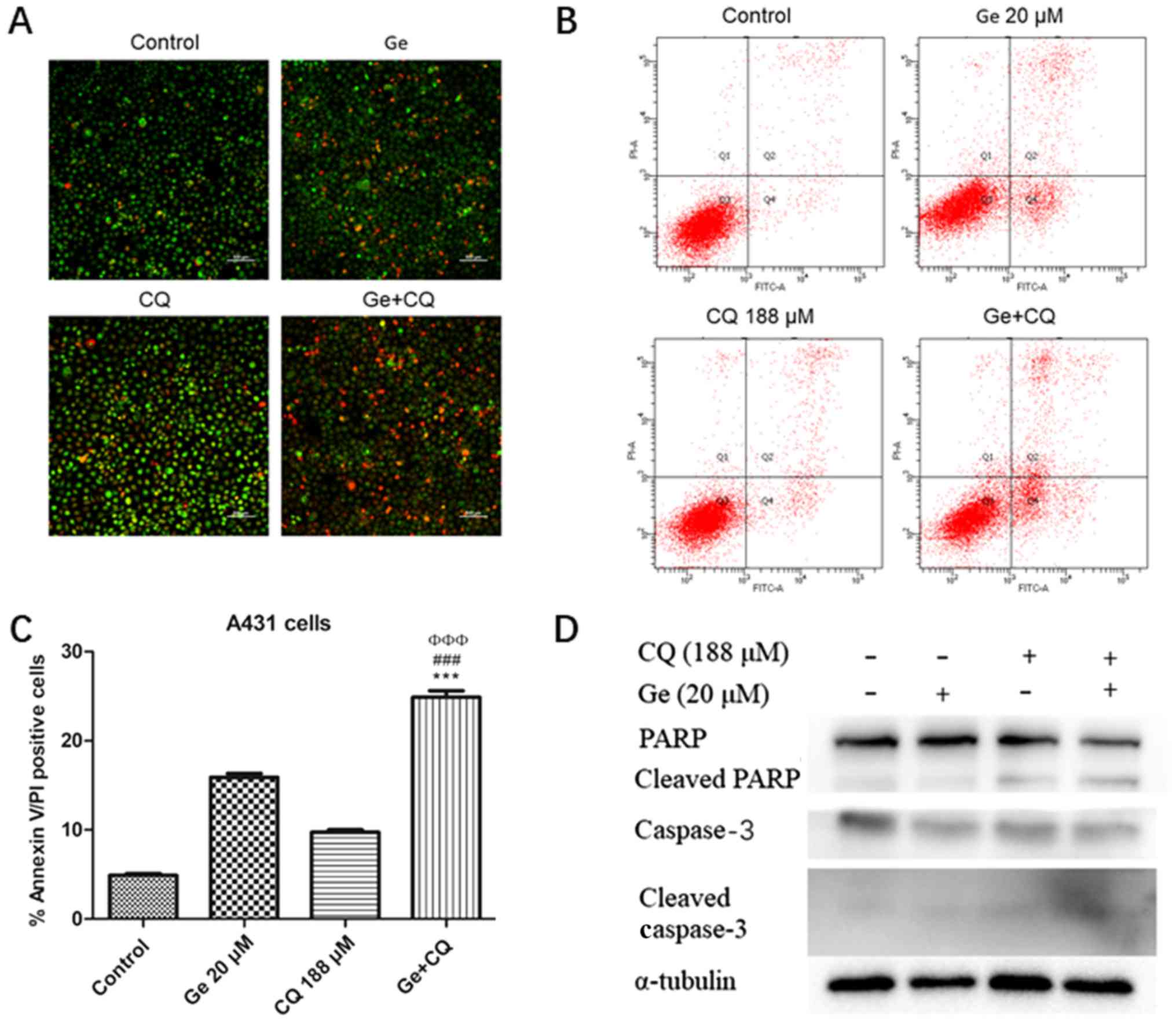 | Figure 3.Ge and CQ induce apoptosis via the
caspase-dependent apoptosis pathway. (A) A431 cells were stained
with acridine orange/ethidium bromide to determine levels of
apoptosis and then observed under a fluorescence microscope.
Magnification, ×10. Scale bar, 500 µm. (B) Following treatment with
20 µM Ge and 188 µM CQ for 12 h, levels of apoptosis were analysed
in A431 cells. (C) Statistical analysis of apoptosis levels in A431
cells in untreated, single drug treatment and combination treatment
groups. Data are expressed as the mean ± standard deviation (n=3).
***P<0.001 vs. control group. ΦΦΦP<0.001 vs. Ge treatment
group. ###P<0.001 vs. CQ treatment group. (D) Protein expression
levels of caspase-3, cleaved caspase-3, PARP and cleaved PARP. CQ,
chloroquine diphosphate; Ge, gefitinib; PARP, poly-(ADP-ribose)
polymerase; PI, propidium iodide; FITC, fluorescein
isothiocyanate. |
Furthermore, whether caspase-3 and PARP proteins
serve important roles in the gefitinib + CQ-induced apoptosis of
A431 cells was investigated via western blot analysis. Compared
with the negative control, levels of the cleaved subunits of
caspase-3 as well as cleaved PARP protein levels were increased in
all treatment groups. In particular, the combination group
(gefitinib + CQ) exhibited enhanced protein levels of cleaved PARP
and cleaved caspase-3 (Fig. 3D).
These results suggest that apoptosis in A431 cells is induced by
co-treatment with CQ and gefitinib via caspase-dependent
pathways.
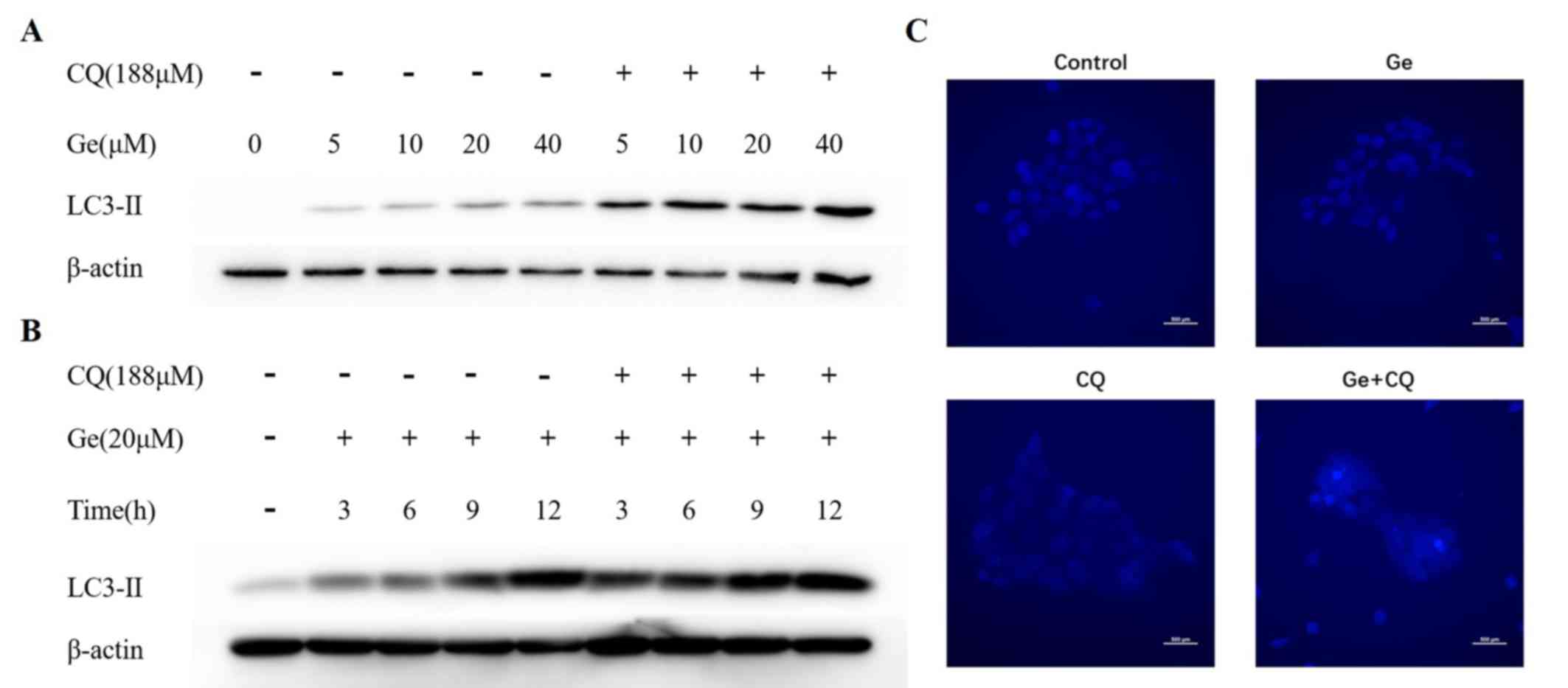
CQ suppresses autophagy via inhibition
of autophagosome degradation
Following pretreatment with CQ for 1 h, increased
levels of LC3-II protein were observed in A431 cells treated with
increasing concentrations of gefitinib for 12 h (Fig. 4A). LC3-II protein levels were
increased in a time-independent manner, which suggested that CQ
increased the number of autophagosomes by preventing fusion of
lysosomes and autophagosomes, which can lead to autophagy
inhibition (Fig. 4B) (21). To further investigate the effects of
CQ on autophagic activity in A431 cells, basic autophagy activities
exhibited by A431 cells were determined via MDC staining. Compared
with the negative control, A431 cells treated with either gefitinib
or CQ demonstrated weak fluorescence intensity in the cytosol;
however, a number of bright foci were visualised in cells belonging
to the combination group (Fig. 4C).
These results suggest that gefitinib activates the autophagy
response in A431 cells and CQ blocks autophagy via inhibition of
autophagosome degradation.
Discussion
In the present study, the pro-apoptotic role of
gefitinib in cSCC cells was investigated and autophagy induced by
treatment with gefitinib was revealed to represent a survival
mechanism in cSCC cells. In addition, the results revealed that
pro-survival autophagic flux may be blocked via treatment with CQ,
which interferes with the fusion of autophagosomes with lysosomes.
The results of the present study suggested that combinatorial usage
of gefitinib with CQ may represent an effective therapeutic
strategy for the treatment of patients with cSCC.
Preclinical data have demonstrated that EGFR has an
important role in the carcinogenesis of cSCC (23), which resulted in the development of
EGFR-targeting antibodies and TKIs, including gefitinib. When used
as a neoadjuvant therapy, gefitinib has a 45.5% response rate and
is well tolerated in patients with aggressive cSCC (24,25).
Gefitinib has a therapeutic effect on patients suffering from
non-small cell lung cancer (NSCLC) with EGFR mutations (26); however, the majority of patients
exhibiting a response eventually develop acquired resistance to
EGFR-TKIs (27). It has been well
established that the therapeutic benefits of EGFR-targeting therapy
may be suppressed by the requirement of autophagy for growth,
survival and therapy resistance (28). The present study investigated the
potential of autophagy inhibition, induced by CQ, for the
enhancement of anti-proliferative effects of gefitinib in A431
cells.
The results of the present study demonstrated that
gefitinib and CQ inhibited the proliferation of A431 cells in a
dose-dependent manner. Analysis performed using CompuSyn software
revealed that combinatory treatment with gefitinib and CQ inhibited
cell growth and enhanced synergistic drug interaction. Such drug
combination methods allow for quantitative determination of drug
interactions by determining CI values, in which CI<1, =1 and
>1 indicate synergism, additive effect and antagonism,
respectively (29). Combinatory
treatment with gefitinib and CQ exhibited moderate synergistic
effects in A431 cells, with CI values ranging from 0.742–0.989 for
fraction affected (Fa)=0.543–0.962. Fa is
commonly used to assess cell mortality following drug treatment,
although this value does not demonstrate synergistic effects, which
was evaluated by CI value (28).
Drug synergism was also investigated using an isobologram, the
results of which were previously revealed to be in agreement with
Fa-CI plots (30).
Synergistic effects were demonstrated by different dosages of drug
combinations below their respective Fa isobole. DRI
values are used to determine the effects of combinatory drug
treatments (31). The DRI value of
combinatory treatment with gefitinib and CQ was revealed to be
>1 (1.654–2.328) in A431 cells, thus suggesting drug synergism.
Furthermore, flow cytometric analysis and acridine orange/ethidium
bromide staining revealed that CQ enhanced gefitinib-induced
apoptosis, which was further demonstrated by increased expression
levels of cleaved PARP and cleaved caspase-3 protein. Collectively,
these results suggested that combinatory treatment with gefitinib
and CQ synergistically induced apoptosis in A431 cells via the
caspase-dependent apoptosis pathway.
The sensitivity of EGFR-targeting therapy is
increased by inhibition of autophagy in NSCLC cells (32). The results of the present study
demonstrated that suppressed levels of autophagy enhanced the
levels of apoptosis in A431 cells. In conclusion, these results
suggest that autophagy has a self-protective role in cell survival
and contributes to drug resistance (33). LC3 is a marker for autophagy, and
contains LC3-I and LC3-II (34).
Cytosolic LC3-I is converted into membrane-bound LC3-II during the
initiation of autophagy and thus LC3-II levels are associated with
the number of autophagosomes (35).
In the present study, gefitinib-induced autophagy in A431 cells was
indicated by markedly increased levels of LC3-II in a
dose-dependent manner. Furthermore, pre-treatment with CQ prior to
treatment with gefitinib further increased LC3-II, which indicated
autophagy was blocked by CQ. The results of the MDC staining
demonstrated this effect. Therefore, the results of the present
study suggested that autophagy induced by gefitinib regulates
cytoprotective effects in A431 cells, and could be inhibited by CQ.
However, the possible mechanisms of autophagy associated with this
effect require further investigation. Numerous studies have
demonstrated that increased levels of cytotoxicity associated with
autophagy inhibition are exhibited by glioblastoma cells induced by
vandetanib (36), by lung cancer
cells induced by gefitinib and erlotinib (37), and by breast cells induced by
gefitinib (38). However, limited
studies have demonstrated the synergistic effect between autophagy
inhibition and EGFR-targeting therapy, which was investigated in
the present study. The results of the present study are notable, as
the Chou-Talalay method regarding drug combination was used in the
present study to quantitatively determine synergistic effects,
which revealed that combinatory therapy of EGFR-targeting and
autophagy-inhibition may represent a therapeutic strategy for
patients with cSCC.
In conclusion, combinatory treatment using gefitinib
and CQ on A431 cells exhibited a synergistic effect regarding
increased levels of apoptosis. Autophagy, a cytoprotective effect
associated with drug administration, was revealed to be inhibited
by CQ, which subsequently enhanced gefitinib-mediated apoptosis via
caspase-dependent pathways. Therefore, combinatory treatment using
gefitinib and CQ may present a potential novel therapeutic strategy
for the treatment of patients with cSCC. To further confirm the
results of the present study, future studies should determine the
underlying mechanisms associated with such effects. CQ represents a
promising adjuvant approach for improving the efficacy of gefitinib
for the treatment of patients with cSCC; however, this should be
investigated further using in vivo preclinical models.
Acknowledgements
Not applicable.
Funding
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81472922).
Availability of data and materials
All data generated or analysed during the present
study are included in this published article.
Authors' contributions
JW, CW, XH, ZD and MZ conceived and designed the
study. JW, CW and XH drafted the manuscript. JW, CW, XH and CY
participated in implementation of the study. CW, XH and LZ assisted
in collecting the data. JW, CW, XH and CY performed the statistical
analysis. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
cSCC
|
cutaneous squamous cell carcinoma
|
|
EGFR
|
epidermal growth factor receptor
|
|
CQ
|
chloroquine diphosphate
|
|
IC50
|
half-maximal inhibitory
concentration
|
|
CI
|
combination index
|
|
DRI
|
dose reduction index
|
|
mAbs
|
monoclonal antibodies
|
|
TKIs
|
tyrosine kinase inhibitors
|
|
DMEM
|
Dulbecco's modified Eagle's medium
|
|
CCK-8
|
Cell Counting Kit-8
|
|
MDC
|
monodansylcadaverine
|
|
TBST
|
Tris-buffered saline-Tween 20
|
|
NSCLC
|
non-small cell lung cancer
|
References
|
1
|
Stratigos A, Garbe C, Lebbe C, Malvehy J,
del Marmol V, Pehamberger H, Peris K, Becker JC, Zalaudek I, Saiag
P, et al: Diagnosis and treatment of invasive squamous cell
carcinoma of the skin: European consensus-based interdisciplinary
guideline. Eur J Cancer. 51:1989–2007. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rogers HW, Weinstock MA, Harris AR,
Hinckley MR, Feldman SR, Fleischer AB and Coldiron BM: Incidence
estimate of nonmelanoma skin cancer in the United States, 2006.
Arch Dermatol. 146:283–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
de Vries E, Trakatelli M, Kalabalikis D,
Ferrandiz L, Ruiz-de-Casas A, Moreno-Ramirez D, Sotiriadis D,
Ioannides D, Aquilina S, Apap C, et al: Known and potential new
risk factors for skin cancer in European populations: A multicentre
case-control study. Br J Dermatol. 167 (Suppl 2):S1–S13. 2012.
View Article : Google Scholar
|
|
4
|
El-Abaseri TB, Fuhrman J, Trempus C,
Shendrik I, Tennant RW and Hansen LA: Chemoprevention of UV
light-induced skin tumorigenesis by inhibition of the epidermal
growth factor receptor. Cancer Res. 65:3958–3965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hynes NE and Lane HA: ERBB receptors and
cancer: The complexity of targeted inhibitors. Nat Rev Cancer.
5:341–354. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sahu N and Grandis JR: New advances in
molecular approaches to head and neck squamous cell carcinoma.
Anticancer Drugs. 22:656–664. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cañueto J, Cardeñoso E, García JL,
Santos-Briz Á, Castellanos-Martín A, Fernández-López E, Blanco
Gómez A, Pérez-Losada J and Román-Curto C: Epidermal growth factor
receptor expression is associated with poor outcome in cutaneous
squamous cell carcinoma. Br J Dermatol. 176:1279–1287. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng Y, Su C, Zhao L and Shi Y: mAb
MDR1-modified chitosan nanoparticles overcome acquired EGFR-TKI
resistance through two potential therapeutic targets modulation of
MDR1 and autophagy. J Nanobiotechnology. 15:662017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang S, Armstrong EA, Benavente S,
Chinnaiyan P and Harari PM: Dual-agent molecular targeting of the
epidermal growth factor receptor (EGFR): Combining anti-EGFR
antibody with tyrosine kinase inhibitor. Cancer Res. 64:5355–5362.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Corkery B, Crown J, Clynes M and O'Donovan
N: Epidermal growth factor receptor as a potential therapeutic
target in triple-negative breast cancer. Ann Oncol. 20:862–867.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
El Guerrab A, Bamdad M, Kwiatkowski F,
Bignon YJ, Penault-Llorca F and Aubel C: Anti-EGFR monoclonal
antibodies and EGFR tyrosine kinase inhibitors as combination
therapy for triple-negative breast cancer. Oncotarget.
7:73618–73637. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tan X, Thapa N, Sun Y and Anderson RA: A
kinase-independent role for EGF receptor in autophagy initiation.
Cell. 160:145–160. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N: The pleiotropic role of
autophagy: From protein metabolism to bactericide. Cell Death
Differ. 12 (Suppl 2):S1535–S1541. 2005. View Article : Google Scholar
|
|
14
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Degenhardt K, Mathew R, Beaudoin B, Bray
K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Claerhout S, Verschooten L, Van Kelst S,
De Vos R, Proby C, Agostinis P and Garmyn M: Concomitant inhibition
of AKT and autophagy is required for efficient cisplatin-induced
apoptosis of metastatic skin carcinoma. Int J Cancer.
127:2790–2803. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fung C, Chen X, Grandis JR and Duvvuri U:
EGFR tyrosine kinase inhibition induces autophagy in cancer cells.
Cancer Biol Ther. 13:1417–1424. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jutten B, Keulers TG, Schaaf MB,
Savelkouls K, Theys J, Span PN, Vooijs MA, Bussink J and Rouschop
KM: EGFR overexpressing cells and tumors are dependent on autophagy
for growth and survival. Radiother Oncol. 108:479–483. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang M, Lee KH, Lee HS, Jeong CW, Kwak C,
Kim HH and Ku JH: Concurrent autophagy inhibition overcomes the
resistance of epidermal growth factor receptor tyrosine kinase
inhibitors in human bladder cancer cells. Int J Mol Sci. 18:2017.
View Article : Google Scholar
|
|
20
|
Liu Z, He K, Ma Q, Yu Q, Liu C, Ndege I,
Wang X and Yu Z: Autophagy inhibitor facilitates gefitinib
sensitivity in vitro and in vivo by activating mitochondrial
apoptosis in triple negative breast cancer. PLoS One.
12:e01776942017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Munafó DB and Colombo MI: A novel assay to
study autophagy: Regulation of autophagosome vacuole size by amino
acid deprivation. J Cell Sci. 114:3619–3629. 2001.PubMed/NCBI
|
|
23
|
Uribe P and Gonzalez S: Epidermal growth
factor receptor (EGFR) and squamous cell carcinoma of the skin:
Molecular bases for EGFR-targeted therapy. Pathol Res Pract.
207:337–342. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lewis CM, Glisson BS, Feng L, Wan F, Tang
X, Wistuba II, El-Naggar AK, Rosenthal DI, Chambers MS, Lustig RA
and Weber RS: A phase II study of gefitinib for aggressive
cutaneous squamous cell carcinoma of the head and neck. Clin Cancer
Res. 18:1435–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
William WN Jr, Feng L, Ferrarotto R,
Ginsberg L, Kies M, Lippman S, Glisson B and Kim ES: Gefitinib for
patients with incurable cutaneous squamous cell carcinoma: A
single-arm phase II clinical trial. J Am Acad Dermatol.
77:1110–1113.e2. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang MH, Lee JH, Chang YJ, Tsai HH, Lin
YL, Lin AM and Yang JC: MEK inhibitors reverse resistance in
epidermal growth factor receptor mutation lung cancer cells with
acquired resistance to gefitinib. Mol Oncol. 7:112–120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jutten B and Rouschop KM: EGFR signaling
and autophagy dependence for growth, survival, and therapy
resistance. Cell Cycle. 13:42–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chou TC: Drug combination studies and
their synergy quantification using the Chou-Talalay method. Cancer
Res. 70:440–446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chou TC and Talalay P: Generalized
equations for the analysis of inhibitions of Michaelis-Menten and
higher-order kinetic systems with two or more mutually exclusive
and nonexclusive inhibitors. Eur J Biochem. 115:207–216. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li YY, Lam SK, Mak JC, Zheng CY and Ho JC:
Erlotinib-induced autophagy in epidermal growth factor receptor
mutated non-small cell lung cancer. Lung Cancer. 81:354–361. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen J, Zheng H, Ruan J, Fang W, Li A,
Tian G, Niu X, Luo S and Zhao P: Autophagy inhibition induces
enhanced proapoptotic effects of ZD6474 in glioblastoma. Br J
Cancer. 109:164–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J,
Ge W, Feng L, Lin X, Wang X, et al: EGFR tyrosine kinase inhibitors
activate autophagy as a cytoprotective response in human lung
cancer cells. PLoS One. 6:e186912011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dragowska WH, Weppler SA, Wang JC, Wong
LY, Kapanen AI, Rawji JS, Warburton C, Qadir MA, Donohue E, Roberge
M, et al: Induction of autophagy is an early response to gefitinib
and a potential therapeutic target in breast cancer. PLoS One.
8:e765032013. View Article : Google Scholar : PubMed/NCBI
|