Introduction
Cervical cancer and melanoma are aggressive cancers
with increasing incidence worldwide (1). Whereas cervical cancer ranks fourth
cancer for both incidence and mortality, malignant melanoma is the
most serious type of skin cancer and accounts for the majority of
skin cancer-associated mortalities (1,2).
Cervical cancer is commonly treated with a combination of
radiotherapy and platinum-based chemotherapy, which may also damage
normal cells (3). At present, there
is no globally accepted standard treatment that offers a
significant survival benefit for patients with advanced-stage
melanoma (2). Until 2011, the
chemotherapeutic drugs dacarbazine, temozolomide and fotemustine
were most commonly used for metastatic melanoma treatment (4); however, only a low percentage of
patients who received these compounds exhibited a significant
response. These treatments have been mostly replaced by the
immune-checkpoint inhibitors, including cytotoxic
T-lymphocyte-associated protein-4 (CTLA-4), B-Raf proto-oncogene
(BRAF) and mitogen-activated protein kinase kinase (MEK) inhibitors
(5). However, these therapeutic
alternatives display adverse events, including secondary cutaneous
squamous cell carcinomas and keratoacanthomas, which occur in ~20%
of patients treated with BRAF-inhibitor (6). A previous study reported that MEK
inhibitors have more serious adverse effects and a lower efficacy
compared with BRAF inhibitors (5).
The development of novel effective agents for the treatment of
these cancers is therefore crucial.
Imidazopyridines possess a wide range of biological
activities. In particular, the imidazo[1,2-a]pyridine moieties of
imidazopyridine have recently gained significant interest as
potential anticancer agents due to their potent inhibitory role of
cancer cell growth, which is commonly due to survival kinases
inhibition (7,8). Various drugs containing
imidazo[1,2-a]pyridine moieties are currently used to treat cardiac
disorders, insomnia, antianxiety, ulcers and HIV infections
(9). Although these compounds have
numerous medicinal applications, none of them have been accepted as
an anti-cancer drug. However, previous studies have reported that
imidazo[1,2-a]pyridines have some anti-cancer abilities. For
example, Goel et al (9)
recently reported that 3-{1-[(4-fluorophenyl)sulfonyl]-
1H-pyrazol- 3-yl}-2-methylimimoredazo[1,2-a]pyridine may be
a novel PIK3CA inhibitor with an half maximal inhibitory
concentration (IC50) of 0.67 µM. Further optimization of
the substituents resulted in thiazole groups substituted by
imidazo[1,2-a]pyridines, which are more potent inhibitor of
phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit
alpha (PI3KCA) with an IC50 of 0.0028 µM (10). In addition, this compound exerts a
promising anti-proliferation effect against melanoma (A375) and
cervical (HeLa) cancer cells, with IC50 values of 0.14
and 0.21 µM, respectively. Notably, 50 mg/kg of this compound
significantly inhibits HeLa human cervical tumor xenografts growth
in mice. Additional series of imidazo[1,2-a]pyridine derivatives
were designed and synthesized as PI3Kα inhibitors (6). One of these compounds, comprising the
bioisosteric 1,2,4-oxadiazole group as a substituent, exhibits
potent PI3Kα inhibition with an IC50 of 2 nM. In
addition, this compound inhibits various types of breast cancer
cell line proliferation with an IC50 of >10 µM.
Furthermore, Annexin V results demonstrated that it significantly
increases T47D breast cancer cell apoptosis. At a molecular level,
these effects are associated with PI3K signaling inhibition.
Notably, this compound has some anti-angiogenic effects through
VEGF expression inhibition. In addition, ethyl 6-(5-(phenyl
sulfonamide)pyridin-3-yl)imidazo[1,2-a]pyridine-3-carboxylate was
also reported (11). This compound
has significant anti-cancer activity in non-small cell lung cancer
cells. Cell treatment with this compound inhibits the phospho
(p)-protein kinase B (PI3K)-protein kinase B (Akt)-mechanistic
target of rapamycin (mTOR) pathway-induced intrinsic apoptosis
(11). A recent study reported that
selenylated imidazo[1,2-a]pyridines inhibits breast cancer cell
proliferation by inducing DNA damage and apoptosis (12). Additional anti-cancer properties of
imidazo[1,2-a]pyridines, including inhibition of nicotinamide
phosphoribosyltransferase (13),
cyclin-dependent kinases (14) and
insulin-like growth factor 1 receptor tyrosine kinase (8) were also reported.
The present study aimed to investigated the
anticancer activities of three imidazo[1,2-a]pyridines, the
compounds 5–7, in melanoma and cervical cancer cells through
analyses of the Akt/mTOR pathway, the cell cycle and apoptosis.
Materials and methods
Chemicals and reagents
Chemicals and solvents, radioimmunoprecipitation
assay (RIPA) lysis buffer (cat. no. R0278) were used without
further purification and were supplied by Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany), unless otherwise stated. Media and cell
culture reagents were from Biological Industries (Kibbutz Beit
Haemek, Israel). Western blotting reagents were obtained from
Bio-Rad Laboratories Inc. (Hercules, CA, USA). DAPI and primary
antibodies against poly(ADP-ribose) polymerase 1/2 (PARP1/2; cat.
no. sc-7150), p53 (cat. no. sc-126), p21 (cat. no. sc-756), BCL2
associated X protein (BAX; cat. no. sc-7480), cyclin B (cat. no.
sc-sc-53236), AKT (cat. no. sc-514032) and tubulin (cat. no.
sc-5286) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Primary antibodies against B-cell lymphoma 2
(BCL2; cat. no. 2876) and caspase-9 (cat. no. 9502) were provided
by Cell Signaling Technology, Inc. (Danvers, MA, USA). Horseradish
peroxidase-conjugated secondary antibodies were provided by Bio-Rad
Laboratories, Inc. (Hercules, CA, USA) and electrochemiluminescence
reaction (ECL) detection system was purchased from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA).
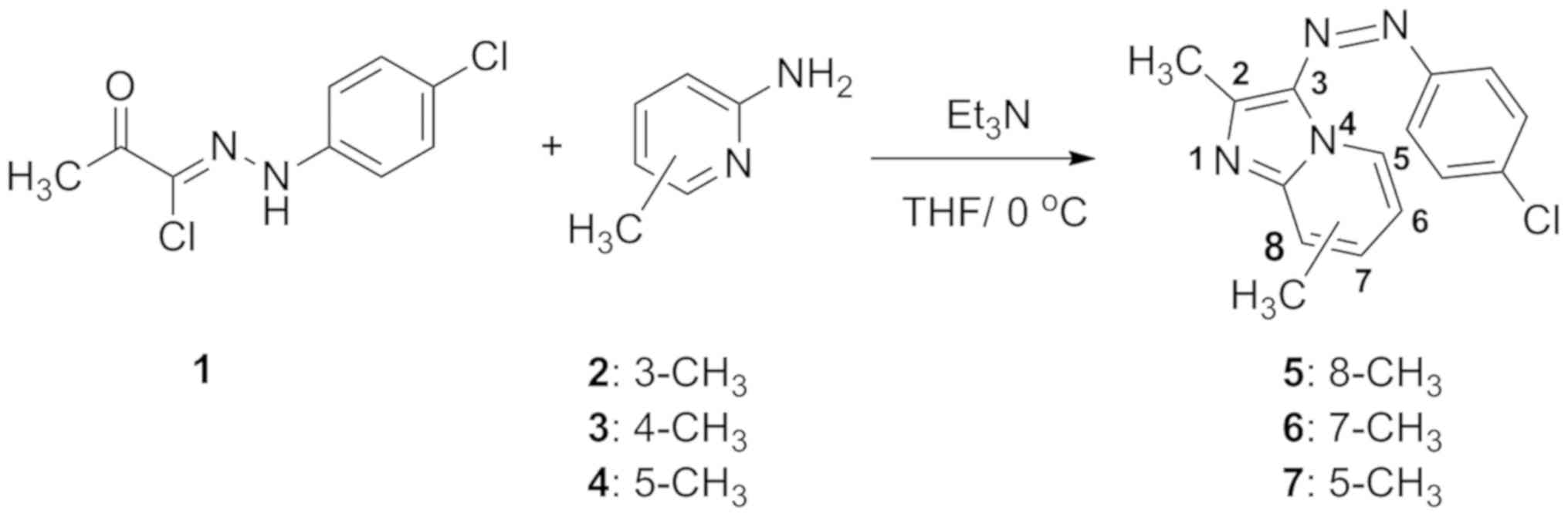
Synthesis of compounds 5–7
Triethylamine (0.6 g, 0.006 mol) diluted in 10 ml
dry tetrahydrofuran (THF) was dropwise added to a stirring solution
of hydrazonoyl chloride 1 (0.005 mol) and substituted picolines 2–4
(0.65 g, 0.006 mol) in 25 ml THF at room temperature. Stirring was
continued overnight, and the solvent was evaporated in
vacuo. The residual solid was washed with water to remove the
triethylammonium salt, and the crude products were recrystallized
from the appropriate solvents to obtain the compounds 5–7. The
physical data of these compounds are available in the original
publication (15). The chemical
formulas of the three compounds were as follows:
3-[(4-Chlorophenyl)diazenyl]-2,8- dimethylimidazo[1,2- a]pyridine
(compound 5), 3-[(4-Chlorophenyl)diazenyl]- 2,7-
dimethylimidazo[1,2-a]pyridine (compound 6) and
3-[(4-Chlorophenyl)diazenyl]-2,5-dimethylimidazo[1,2- a]pyridine
(compound 7).
Cell culture and treatments
The human melanoma cell lines A375 and WM115 and the
cervical cancer cell line HeLa, sourced from the Department of
Human Biology, University of Cape Town (Cape Town, South Africa),
were maintained in Dulbecco's modified Eagle's medium supplemented
with 10% fetal bovine serum and placed at 37°C in a humidified
incubator containing 5% CO2. Compounds 5–7 were
dissolved in dimethyl sulfoxide (DMSO) to obtain 10 mM stock
solutions that were stored at room temperature for a maximum of 10
days. Control cells were treated with equivalent concentrations of
DMSO (vehicle).
Small interfering RNA (siRNA)
Suppression of p53 expression was achieved by siRNA
that specifically targeted p53 mRNA. Cells at 70% confluence were
transfected with 50 nM anti-p53 siRNA (cat. no. sc-756) or a
scrambled control RNA (cat. no. sc-37007) from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA) using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. The transfection
reagent and siRNA complex were added drop-wise to the cells and
incubated for 30 h at 37°C. The subsequent experiments were
performed 30 h after transfection.
Cytotoxicity assays
Cells were seeded in 96-well plates at
4–5×103 cells per well and allowed to settle for 48 h.
Cells were treated with increasing concentrations of compounds 5–7
(0–100 µM) or vehicle for 48 h. Cytotoxicity was assessed using MTT
assay (Roche Diagnostics GmbH, Mannheim, Germany) (16) according to the manufacturer's
instructions. Briefly, 10 µl MTT solution was added to each well
and cells were incubated at 37°C for 4 h. The solubilization buffer
(100 µl, 10% SDS in 0.01 M hydrochloric acid) was added on cells
for 16 h at 37°C. Absorbance was determined at 585 nm with a
microplate reader, and the mean cell viability was calculated as a
percentage of the mean vehicle control.
Cell cycle analysis
The effect of compound 6 on the cell cycle profile
of cancer cells was determined according to a previous protocol
(17). Briefly, cells were seeded at
3–4×105 cells per 6-cm dish and allowed to settle for 24
h. Log-phase cultures were exposed to compounds 5–7 (0 to 100 µM)
or vehicle for 48 h. Cells were trypsinized, washed with PBS and
fixed overnight in 95% ethanol at 4°C, and subjected to RNase
treatment and stained for 30 min at room temperature with propidium
iodide (PI; 500 µg/ml; Sigma-Aldrich; Merck KGaA). Cellular DNA
content was determined by flow cytometry with a FACSCalibur flow
cytometer with a 488 nm Coherent laser (BD Biosciences, Franklin
Lakes, NJ, USA). The CellQuest Pro version 5.2.1 software (BD
Biosciences) was used for data acquisition and analyses were
performed using Modfit version 2.0 software (BD Biosciences).
Detection of apoptosis
Log-phase cultures (60–70% confluence) were treated
with compound 6 or vehicle for 48 h. Adherent and floating cells
were collected and double-labelled with Annexin V-Fluorescein
isothiocyanate (FITC) and PI (Sigma-Aldrich; Merck KGaA) for 10 min
at room temperature. Annexin V-FITC was used to determine apoptotic
cells percentage whereas PI stained all dead cells. Cells were
analyzed by flow cytometry with a 488 nm Coherent laser equipped
with FACStation running CellQuest software (BD Biosciences).
Nuclear fragmentation
Melanoma and cervical cancer cells were
treated with compound 6 (10 and 35 µM, respectively) for 24 h at
37°C and stained with DAPI (10 µg/ml) nuclear stain for 10 min at
room temperature. Cells were observed by fluorescence microscopy
(Zeiss GmbH, Jena, Germany).
Western blotting
Cells were harvested with RIPA lysis buffer for 30
min on ice. The lysates were collected, and the protein
concentrations were determined using Bradford's reagent (Bio-Rad
Laboratories, Inc.), according to the manufacturer's instructions
and using albumin as a standard. A total of 20 µg protein was
separated by 8–15% SDS-PAGE, and transferred to a Hybond ECL
nitrocellulose membrane (GE Healthcare, Chicago, IL, USA) (17). Following blocking for 1 h at room
temperature with PBS containing 0.05% Tween-20 and 5% powdered skim
milk, membranes were incubated overnight with primary antibodies
(1:1,000) at 4°C. After primary antibody incubation, membranes were
incubated with appropriate HRP-conjugated secondary antibodies
(1:5,000) for 1 h at room temperature. Bands were detected using
enhanced chemiluminescence detection system (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) (18). Relative expression level of the
proteins was analyzed by UN-SCAN-IT gel 6.1 software by Silk
Scientific Corporation (Orem, UT, USA) and normalized to the
loading controls.
Statistical analysis
Results are presented as the mean ± standard error
of the mean (SEM) of the three independent experiments. Statistical
analysis of data was performed using the two-sample t-test in
Microsoft Excel 2013 (Microsoft Corporation, Redmond, WA, USA) or a
one-way ANOVA with Tukey's post hoc test in Graph Pad Prism
(version 5; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Imidazo[1,2-a]pyridines induces
cytotoxicity and cell cycle arrest
Imidazo[1,2-a]pyridines 5–7 were synthesized
according to our published method (15). As presented in Fig. 1, the reaction of hydrazonoyl chloride
1 with the appropriate substituted methyl-2-aminopicolines 2–4 in
the presence of triethylamine as a base at 0°C gave the compounds
5–7 (Fig. 1).
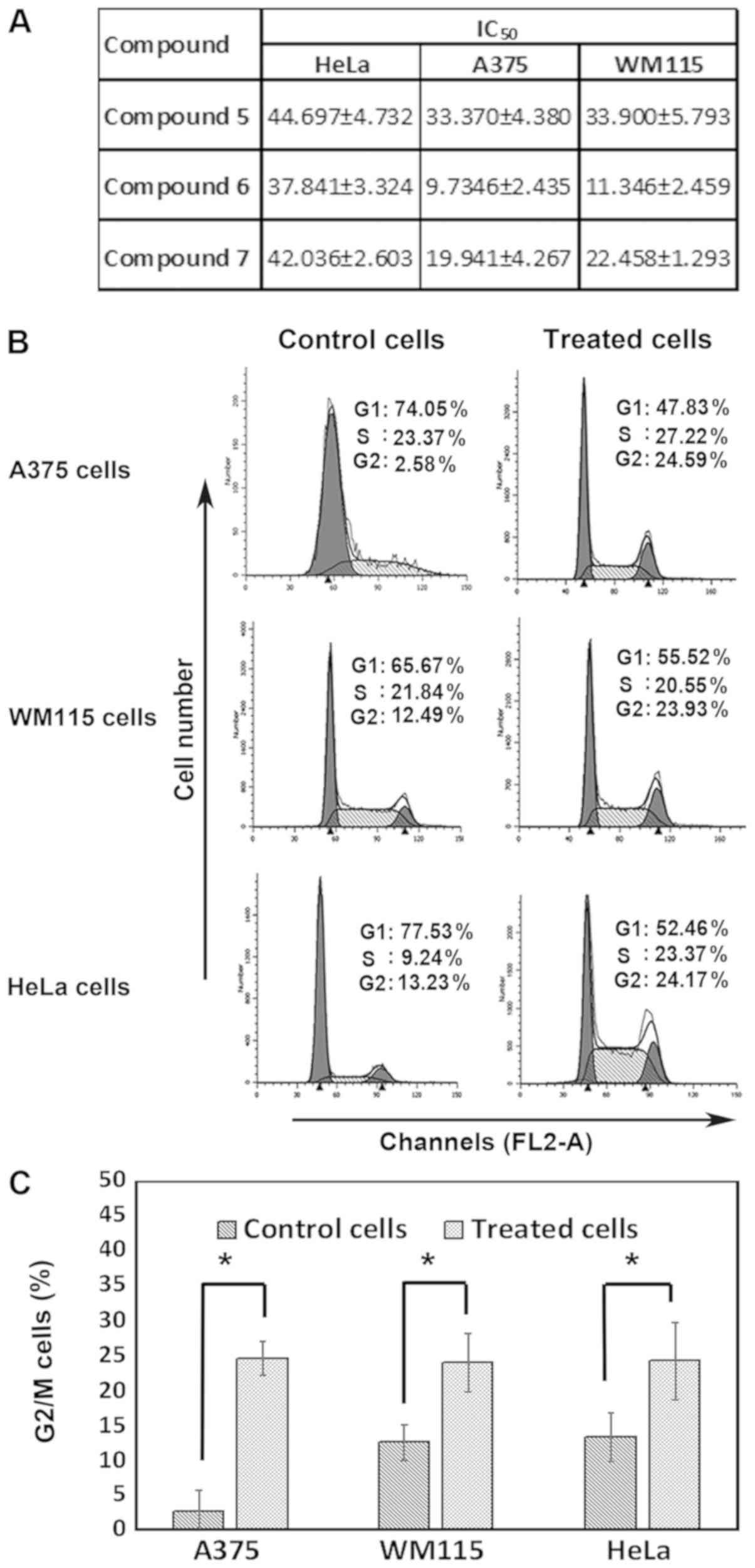
The cytotoxic effects of compounds 5–7 on melanoma
and cervical cancer cell lines were investigated. A375, WM115 and
HeLa cells were treated with increasing concentration of compounds
5–7 (0 to 100 µM) for 48 h prior to assessing cell viability with
MTT assay. The results demonstrated that all compounds inhibited
cell proliferation of the three cell lines with different
IC50 ranging from 9.7 to 44.6 µM (Fig. 2A). Notably, compound 6 was the most
potent compound to induce melanoma and cervical cancer cell
toxicity. In addition, compound 6 was more toxic to melanoma cells
than cervical cancer cells. The effect of compound 6 on cell cycle
profile was then explored. To do so, melanoma and cervical cancer
cells were treated with 10 and 35.0 µM compound 6, respectively,
for 48 h prior to analyzing cell cycle. The results in Fig. 2B and C demonstrated that compound 6
induced a significant G2/M cell cycle arrest in all cell
lines, which was mainly at the expense of G1 phase cell
populations. In addition, A375 cell treatment with compound 6
caused an increase in the cell population in the G2/M
phase from 2.58±3.1% (control) to 24.59±2.4%. Similarly, WM115 cell
treatment with compound 6 increased cell population in the
G2/M from 12.49±2.6% (control) to 23.93±4.2%. In
cervical cancer cells, compound 6 treatment increased the
G2/M phase cell population from 13.23±3.5% (control) to
24.17±5.6%. Notably, whereas no significant effects of compound 6
were observed on the S phase of melanoma cells, HeLa cells treated
with compound 6 exhibited a significant increase in the S phase
cell population which raised from 9.24±2.3% (control) to
23.37±4.8%. These results suggested that compounds (5–7),
particularly compound 6, may inhibit cancer cells proliferation and
induce G2/M cell cycle arrest in cancer cells.
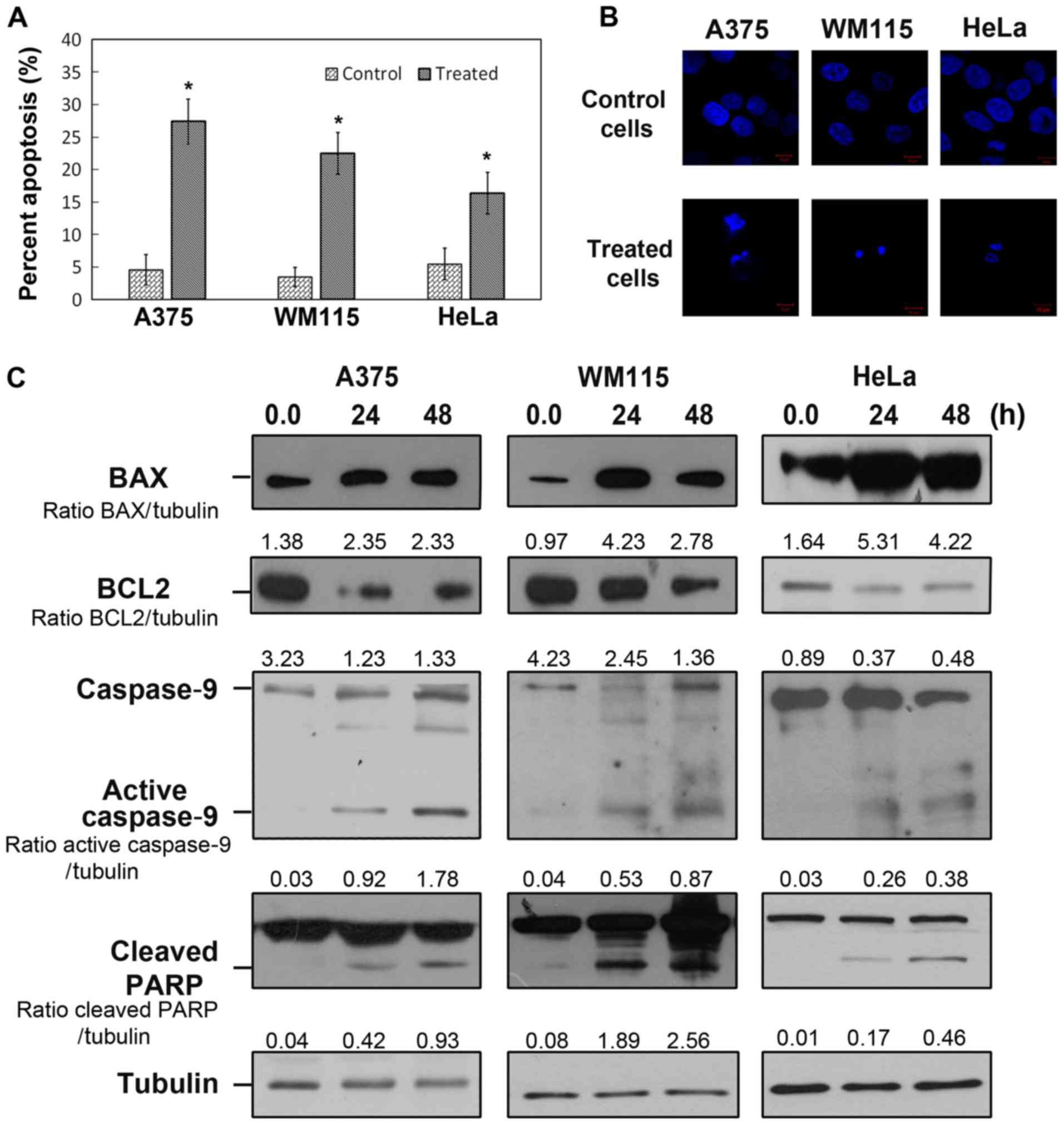
Compound 6 induces intrinsic
apoptosis
To determine whether compound 6 induced apoptotic
cell death, melanoma and cervical cancer cells were treated with 10
and 35.0 µM compound 6, respectively, for 48 h and stained with PI
and Annexin V-FITC. The results from flow cytometry demonstrated
that compound 6 significantly increased apoptosis in all cancer
cell lines (Fig. 3A). In addition,
the levels of apoptotic melanoma cells were higher than the levels
of apoptotic HeLa cells following compound 6 treatment. A375 cells
treated with 10 µM of compound 6 for 48 h increased significantly
from 4.58±2.34% (control) to 27.38±3.4%. Similar results were
obtained for WM115 cells that exhibited an increase in apoptosis
from 3.48±1.49% (control) to 22.49±3.23% following 48-h treatment
with 10 µM compound 6. However, the level of apoptosis induced by
35 µM compound 6 was only 16.38±3.23% in HeLa cells. In addition,
following nuclear staining and fluorescence microscopy analysis,
cancer cells treated with compound 6 exhibited fragmented
chromatin, which was characteristic of apoptotic cells (Fig. 3B). To further confirm that compound 6
induced apoptosis at a molecular level, and to investigate the
mechanisms underlying compound 6-induced cell death, western
blotting of key apoptotic proteins, including cleaved PARP, BAX,
BCL2 and caspase-9 were performed (Fig.
3C). The results demonstrated that PARP cleavage was increased
in all cancer cell lines following 24 and 48 h of treatment with
compound 6. Furthermore, the markers of intrinsic apoptosis,
including BAX cleaved caspase-9, were increased in the three cancer
cell lines following 48 h of treatment with compound 6. In
addition, the level of the anti-apoptotic protein BCL2 decreased in
all treated cells. These results indicated that compound 6 induced
intrinsic apoptotic pathways in melanoma and cervical cancer cell
lines.
Compound 6 inhibits AKT/mTOR
pathway
Previous studies have reported the ability of
different imidazo[1,2-a]pyridines to inhibit AKT/mTOR pathway in
cancer cells (12,13). These studies demonstrated that these
compounds bind to the ATP-binding site of PI3K with a high
affinity, which induces the PI3K/AKT/mTOR pathway inhibition. To
investigate whether compound 6 exerts its effect through the same
mechanism, western blotting of key proteins from this pathway,
including p-AKT (using the specific antibody for phosphorylated Ser
473 of Akt1), AKT and p-mTOR were performed. The results
demonstrated that both p-AKT and p-mTOR levels were reduced in all
cancer cells following 48-h treatment with compound 6 (Fig. 4A). These results indicated that
compound 6 inhibited AKT, which may regulate other important
proteins, including as p53.
 | Figure 4.Compound 6 inhibits AKT/mTOR pathway
and induces p53 dependent apoptosis. (A) Melanoma cells and HeLa
cells were treated with vehicle or compound 6 at 10 and 35 µM,
respectively, for 48 h. p-AKT, AKT, p-mTOR, p53, p21, cyclin B and
tubulin proteins were analyzed by western blotting. (B) A375 cells
were transfected with sip53 and treated with compound 6 (10 µM) for
48 h. Protein extracts were harvested and p53, p21 and PARP were
analyzed by western blotting. (C) Annexin V/propidium iodide double
staining exhibited a percentage of cell death induced by compound 6
after sip53 transfection lower than in the un-transfected group
(B). *P<0.05. AKT, protein kinase B; c, control; mTOR,
mechanistic target of rapamycin; p, phospho; si, small
interfering. |
p53 partially mediates compound 6
cytotoxicity
A previous study revealed that AKT facilitates p53
degradation by increasing MDM2 proto-oncogene expression (19). The effect of compound 6 on p53
response was therefore investigated. To do so, western blotting of
p53 and its downstream target p21was performed. As presented in
Fig. 4A, p53 and p21 protein levels
were increased in all cancer cells following treatment with
compound 6. In addition, treatment with compound 6 reduced cyclin B
level in all cancer cells which supports the G2/M cell
cycle arrest detected as shown in Fig.
2B and C. The potential roles of p53-mediated signaling in
compound 6-induced cell death were investigated by inhibiting p53
expression via si-RNA transfection. A375 cells transfected with
sip53 presented attenuated p21 expressions in control and compound
6-treated cells (Fig. 4B). This was
further evidenced by the decreased level of cleaved PARP observed
following compound 6 treatment in A375 sip53 cells (Fig. 4B). These observations were reinforced
by Annexin V assay, which demonstrated that compound 6 treatment
induced a moderate increase in apoptosis (15.65±3.23%) in A375
sip53 cells, compared with un-transfected cells (34.23±3.45%)
(Fig. 4C). These observations
suggested that compound 6 may induce apoptosis partly through p53
regulation.
Discussion
The PI3K-AKT signaling pathway is one of the most
investigated cascades in cancer cells. Increasing evidence has
confirmed the crucial role of the PI3K-AKT pathway in cancer
initiation and progression (18).
The phosphatase and tensin homolog (PTEN) tumor suppressor is the
most common regulator of this pathway, and tumors exhibiting PTEN
loss usually have a high p-AKT level (20). In vitro and in vivo
investigations have demonstrated that tumors with mutant
proto-oncogene B-Raf (BRAF) also present high p-AKT levels, which
may contribute to the development of BRAF-inhibitor resistance
(19,20). Similarly, PI3K-AKT pathway is
commonly dysregulated in cervical cancer, which indicates that it
may be a potential therapeutic target in the treatment of melanoma
and cervical cancer (21). Numerous
PI3K inhibitors have been developed and recently evaluated in
clinical trials. These comprise PI3K specific inhibitors and dual
targets inhibitors, including PI3K-mTOR and AKT inhibitors
(11,22). Subsequently, some
imidazo[1,2-a]pyridine compounds were developed and tested for
their PI3K-AKT pathway inhibiting ability (23–25). Our
group has therefore designed and synthesized novel
imidazo[1,2-a]pyridines (compounds 5–7) (15). The current study, aimed to
investigate the exact anticancer effect of these compounds in
melanoma and cervical cancer cells. Notably, compound 6 exhibited
the most potent cytotoxic effect in both types of cancer cell. The
anticancer effect of compound 6 and its mechanism of action in
melanoma and cervical cancer cell lines were therefore deeper
examined. The results from this study suggested that compound 6 may
be considered as a promising novel chemotherapeutic drug for
melanoma and cervical cancers treatment.
Compound 6 exhibited potent cytotoxicity in A375 and
WM115 cell lines with low IC50 values (<12 µM), which
was of crucial importance, considering that metastatic melanoma
cells seem to be resistant to chemotherapy. For example, the dose
of dacarbazine, which is the common current treatment of metastatic
melanoma, necessary to inhibit melanoma cell proliferation is ~25
µM (18). However, some imidazo[1,2-
a]pyridines presented similar or more potent cytotoxicity against
melanoma cancer cells (23,24). A novel series of
imidazo[1,2-a]pyridine compounds inhibit A375 human melanoma cell
line proliferation with a low IC50 <1 µM (23). A recent study that screened the
cytotoxic effects of novel pyrido-imidazodiazepinones reported that
seven of these compounds significantly inhibit melanoma cell growth
at 1 µM (24).
In the present study, cancer cell treatment with
compound 6 induced a decrease in p-AKT level. This effect was
observed after 24 h in HeLa cells, and was even more important
following 48-h treatment with compound 6 in melanoma cells.
Notably, AKT inhibition was mirrored with the inhibition of its
downstream target mTOR. Previous studies have reported that AKT and
target m-TOR inhibitions increase p21 expression and activate
checkpoint kinase 2, which results in G2/M cell cycle
arrest (26–28). Furthermore, these studies showed that
the blockage of cell cycle arrest at the G2/M phase is
associated with a decrease in the cell cycle regulatory protein
cyclin B level. Similarly, the results from the present study
demonstrated that compound 6 induced a decrease in cyclin B1 level.
Notably, compound 6 increased p53 and p21 levels, which could
explain the G2/M cell cycle arrest observed.
The PI3K/AKT/mTOR pathway is involved in cell
survival, and its inhibition results in high p53 and BAX apoptotic
proteins levels (18). Active BAX
disrupts the mitochondrial membrane integrity and induces the
cytochrome c release from mitochondria into the cytosol.
Cytoplasmic cytochrome c and active caspase-9 are then involved in
the apoptosome formation and caspase-3 activation (29). However, cytochrome c release does not
happen when the anti-apoptotic protein BCL2 is present. The present
study demonstrated that compound 6 stimulated intrinsic apoptosis
by increasing BAX and active caspase-9 levels, and decreasing BCL2
level. The results suggested that the G2/M cell cycle
arrest and intrinsic apoptosis induced by compound 6 may be
mediated by AKT/mTOR inhibition. Notably, p53 silencing
significantly decreased the compound 6-induced apoptosis, which
suggested that p53 may serve an important role in compound
6-induced cell apoptosis. Numerous imidazopyridine derivatives are
currently used in the clinic for the treatment of other diseases,
including alpidem (anxiolytic) and zolpidem (hypnotic), which
exhibit low toxicity levels (30).
This could indicate that the compounds tested in the present study
may present minor adverse effects.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Qatar Charity
under the Ibhath project for research grants, which is funded by
the Cooperation Council for the Arab States of the Gulf through the
Islamic Development Bank.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SA designed the biological study, supervised
different steps and was the major contributor in the writing of the
manuscript. AMA synthesized and tested the chemical compounds. RYM
supervised and characterized the synthesized compounds. MG
contributed to the tissue culture experiments. HA contributed to
the tissues culture, western blotting experiments and cell cycle
analysis. AYA contributed to the western blotting and tissue
culture experiments. EAA contributed to the western blotting
experiments. YMA contributed to the apoptosis assay. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
R, Torre L and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen YJ and Del Priore G: The role of
cisplatin alternative regimens with radiotherapy in cervical
cancer. Gynecol Oncol Rep. 11:38–40. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schadendorf D, Fisher DE, Garbe C,
Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA,
McArthur G, Ribas A, et al: Melanoma. Nat Rev Dis Primers.
1:150032015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Patel PM, Suciu S, Mortier L, Kruit WH,
Robert C, Schadendorf D, Trefzer U, Punt CA, Dummer R, Davidson N,
et al: Extended schedule, escalated dose temozolomide versus
dacarbazine in stage IV melanoma: Final results of a randomised
phase III study (EORTC 18032). Eur J Cancer. 47:1476–1483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Long GV, Stroyakovskiy D, Gogas H,
Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A,
Grob JJ, et al: Combined BRAF and MEK Inhibition versus BRAF
inhibition alone in melanoma. N Engl J Med. 371:1877–1888. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sosman JA, Kim KB, Schuchter L, Gonzalez
R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ,
Flaherty KT, et al: Survival in BRAF V600-mutant advanced melanoma
treated with vemurafenib. N Engl J Med. 366:707–714. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim O, Jeong Y, Lee H, Hong SS and Hong S:
Design and synthesis of imidazopyridine analogues as inhibitors of
phosphoinositide 3-kinase signaling and angiogenesis. J Med Chem.
54:2455–2466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ducray R, Jones CD, Jung FH, Simpson I,
Curwen J and Pass M: Novel imidazo[1,2-a]pyridine base d inhibitors
of the IGF-1 receptor tyrosine kinase: Optimization of the aniline.
Bioorg Med Chem Lett. 21:4702–4704. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goel R, Luxami V and Paul K:
Imidazo[1,2-a]pyridines: Promising drug candidate for antitumor
therapy. Curr Top Med Chem. 16:3590–3616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hayakawa M, Kawaguchi K, Kaizawa H,
Koizumi T, Ohishi T, Yamano M, Okada M, Ohta M, Tsukamoto S,
Raynaud FI, et al: Synthesis and biological evaluation of
sulfonylhydrazone-substituted imidazo[1,2-a]pyridines as novel PI3
kinase p110alpha inhibitors. Bioorg Med Chem. 15:5837–5844. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee H, Kim SJ, Jung KH, Son MK, Yan HH,
Hong S and Hong SS: A novel imidazopyridine PI3K inhibitor with
anticancer activity in non-small cell lung cancer cells. Oncol Rep.
30:863–869. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Almeida GM, Rafique J, Saba S, Siminski T,
Mota NSRS, Filho DW, Braga AL, Pedrosa RC and Ourique F: Novel
selenylated imidazo[1,2-a]pyridines for breast cancer chemotherapy:
Inhibition of cell proliferation by Akt-mediated regulation, DNA
cleavage and apoptosis. Biochem Biophys Res Commun. 503:1291–1297.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng X, Bauer P, Baumeister T, Buckmelter
AJ, Caligiuri M, Clodfelter KH, Han B, Ho YC, Kley N, Lin J, et al:
Structure-based discovery of novel amide-containing nicotinamide
phosphoribosyltransferase (Nampt) inhibitors. J Med Chem.
56:6413–6433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cai D, Byth KF and Shapiro GI: AZ703, an
imidazo[1,2-a]pyridine inhibitor of cyclin-dependent kinases 1 and
2, induces E2F-1-dependent apoptosis enhanced by depletion of
cyclin-dependent kinase 9. Cancer Res. 66:435–444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morjan RY, Qeshta BS, Al-Shayyah HT,
Gardiner JM, Abu-Thaher BA and Awadallah AM: Reaction of
nitrilimines with 5-aminotetrazole and 2-aminopyrimidine. Int J Org
Chem. 4:201–207. 2014. View Article : Google Scholar
|
|
16
|
Aliwaini S, Swarts AJ, Blanckenberg A,
Mapolie S and Prince S: A novel binuclear palladacycle complex
inhibits melanoma growth in vitro and in vivo through apoptosis and
autophagy. Biochem Pharmacol. 86:1650–1663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Qatati A and Aliwaini S: Combined
pitavastatin and dacarbazine treatment activates apoptosis and
autophagy resulting in synergistic cytotoxicity in melanoma cells.
Oncol Lett. 14:7993–7999. 2017.PubMed/NCBI
|
|
18
|
Davies MA, Stemke-Hale K, Lin E, Tellez C,
Deng W, Gopal YN, Woodman SE, Calderone TC, Ju Z, Lazar AJ, et al:
Integrated molecular and clinical analysis of AKT activation in
metastatic melanoma. Clin Cancer Res. 15:7538–7546. 2010.
View Article : Google Scholar
|
|
19
|
Astle MV, Hannan KM, Ng PY, Lee RS, George
AJ, Hsu AK, Haupt Y, Hannan RD and Pearson RB: AKT induces
senescence in human cells via mTORC1 and p53 in the absence of DNA
damage: Implications for targeting mTOR during malignancy.
Oncogene. 31:1949–1962. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vasudevan KM, Barbie DA, Davies MA,
Rabinovsky R, McNear CJ, Kim JJ, Hennessy BT, Tseng H, Pochanard P,
Kim SY, et al: AKT-independent signaling downstream of oncogenic
PIK3CA mutations in human cancer. Cancer Cell. 16:21–32. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bahrami A, Hasanzadeh M, Hassanian SM,
ShahidSales S, Ghayour-Mobarhan M, Ferns GA and Avan A: The
potential value of the PI3K/Akt/mTOR signaling pathway for
assessing prognosis in cervical cancer and as a target for therapy.
J Cell Biochem. 118:4163–4169. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang L, Dan HC, Sun M, Liu Q, Sun X,
Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, et al:
Akt/Protein kinase B signaling inhibitor-2, a selective small
molecule inhibitor of Akt signaling with antitumor activity in
cancer cells overexpressing Akt. Cancer Res. 64:4394–4399. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garamvölgyi R, Dobos J, Sipos A, Boros S,
Illyés E, Baska F, Kékesi L, Szabadkai I, Szántai-Kis C, Kéri G and
Őrfi L: Design and synthesis of new imidazo[1,2-a]pyridine and
imidazo[1,2-a]pyrazine derivatives with antiproliferative activity
against melanoma cells. Eur J Med Chem. 108:623–643. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bellet V, Lichon L, Arama DP, Gallud A,
Lisowski V, Maillard LT, Garcia M, Martinez J and Masurier N:
Imidazopyridine-fused [1,3]-diazepinones part 2: Structure-activity
relationships and antiproliferative activity against melanoma
cells. Eur J Med Chem. 125:1225–1234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ingersoll MA, Lyons AS, Muniyan S, D'Cunha
N, Robinson T, Hoelting K, Dwyer JG, Bu XR, Batra SK and Lin MF:
Novel imidazopyridine derivatives possess anti-tumor effect on
human castration-resistant prostate cancer cells. PLoS One.
10:e01318112015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kuo PL, Hsu YL and Cho CY: Plumbagin
induces G2-M arrest and autophagy by inhibiting the AKT/mammalian
target of rapamycin pathway in breast cancer cells. Mol Cancer
Ther. 5:3209–3221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang MC, Chen YJ, Liou EJ, Tseng WY, Chan
CP, Lin HJ, Liao WC, Chang YC, Jeng PY and Jeng JH:
7-Ketocholesterol induces ATM/ATR, Chk1/Chk2, PI3K/Akt signaling,
cytotoxicity and IL-8 production in endothelial cells. Oncotarget.
7:74473–74483. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hirose Y, Katayama M, Mirzoeva OK, Berger
MS and Pieper RO: Akt activation suppresses Chk2-mediated,
methylating agent-induced G 2 arrest and protects from
temozolomide- induced mitotic catastrophe and cellular senescence.
Cancer Res. 65:4861–4869. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aliwaini S, Peres J, Kröger WL,
Blanckenberg A, de la Mare J, Edkins AL, Mapolie S and Prince S:
The palladacycle, AJ-5, exhibits anti-tumour and anti-cancer stem
cell activity in breast cancer cells. Cancer Lett. 357:206–218.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hanson SM, Morlock EV, Satyshur KA and
Czajkowski C: Structural requirements for eszopiclone and zolpidem
binding to the gamma-aminobutyric acid type-A (GABAA) receptor are
different. J Med Chem. 51:7243–7252. 2008. View Article : Google Scholar : PubMed/NCBI
|