Introduction
Ketoconazole, a cytochrome P450 inhibitor with
antifungal effects, has been reported to exhibit anticancer
effects, including increased plasma levels of cytotoxic fenretinide
and enhanced intratumoral apoptosis in neuroblastoma (1). In addition, a combination of venetoclax
with ketoconazole has been reported to exhibit significant
therapeutic effects for patients with chronic lymphocytic leukemia
(2). Furthermore, ketoconazole is a
second-line hormonal agent in castration-resistant prostate cancer
as it reduces androgen biosynthesis (3).
Immunomodulatory protein cloned from Ganoderma
microsporum (GMI) contains 110 amino acids. Overall, ~83%
homology exists between fungal immunomodulatory proteins from
Ganoderma tsugae and GMI in the alignment of amino acid
sequences (4). It has been
demonstrated in A549 cells that GMI inhibits tumor necrosis factor
α-induced matrix metalloproteinase 9-mediated migration and
invasion (5). A number of signaling
pathways have been reported to be affected by GMI in the treatment
of various cancer types. In non-small-cell lung cancer, oral
administration of GMI induces activation of
Ca2+-dependent pathways, which is associated with a
decrease in cytosolic p53 (4). Chiu
et al (6) proposed that
induction of autophagy by GMI destroys multiple drug resistance
mechanisms via Akt/mammalian target of rapamycin inhibition in the
treatment of lung cancer.
In our previous study, GMI was identified to enhance
cisplatin-induced apoptosis via the autophagy/caspase-7 pathway in
lung cancer. The effects of GMI with low-dose cisplatin indicate
that GMI can serve as an adjuvant of cisplatin in the treatment of
lung cancer (7). Recently, GMI has
been demonstrated to induce oral cancer stem cell-elicited tumor
regression via blockage of the interleukin-6/signal transducer and
activator of transcription 3 signaling pathway (8).
Adenosine monophosphate-activated protein kinase
(AMPK) is an energy sensor activated by metabolic stress to
maintain cellular energy homeostasis (9). AMPK is activated by the upstream kinase
liver kinase B1 and is negatively regulated by phosphorylation of
the heterodimeric AMPK (9). Studies
regarding AMPK activation and inhibition of migration or invasion
have produced controversial results. Inhibition of AMPK results in
increased migration of pancreatic cancer cells (10). C-X-C motif chemokine ligand 12
prevents pancreatic ductal adenocarcinoma metastasis via
phosphorylated (p)-AMPK activation (10). Ginkgolic acid, a phenolic acid
extracted from ginkgo fruit, inhibits migration and invasion by
inducing AMPK activation in colon cancer cells (11).
Monocyte chemoattractant protein-1 (MCP-1; also
termed CCL2) is a major chemokine that induces infiltration and
migration of macrophages and monocytes (12). Both MCP-1 and its receptor CCR2 have
been reported to be induced and involved in various types of tumor.
Macrophages and microglia produce MCP-1, which is critical for
recruiting both regulatory T cells and myeloid-derived suppressor
cells in the glioma microenvironment (13). Blockage of the MCP-1-CCR2 complex in
combination with radiotherapy improves radiotherapeutic efficacy in
pancreatic ductal adenocarcinoma (12).
To the best of our knowledge, there is limited
understanding regarding the effects of ketoconazole, alone and in
combination with GMI, on melanoma. The aim of the current study was
to investigate the inhibitory effects of GMI combined with
ketoconazole on melanoma survival and metastasis. Results of the
present study revealed that a combination of GMI and ketoconazole
can inhibit the proliferation and migration of melanoma and reduce
the level of secreted MCP-1.
Materials and methods
Cell line and chemicals
A375.S2 human melanoma cells and Hs68 fibroblast
were purchased from the Food Industry Research and Development
Institute (Hsinchu, Taiwan). A375.S2 cells were incubated in
minimum essential medium (MEM) (Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 2 mM L-glutamine, 0.1 mM non-essential
amino acids, 1.5 g/l sodium bicarbonate and 1.0 mM sodium pyruvate.
The medium also contained 10% heat-inactivated fetal bovine serum
(FBS) (Gibco; Thermo Fisher Scientific, Inc) and antibiotics,
including 100 U/ml penicillin and 100 µg/ml streptomycin. The cells
were cultured in an incubator with a humidified atmosphere of 5%
CO2 at 37°C.
MTT was purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). Dorsomorphin dihydrochloride (catalog no.
3093) and ketoconazole (catalog no. 1103) were obtained from Tocris
Bioscience (Bristol, UK).
Production of GMI protein
GMI, kindly provided by Mycomagic Biotechnology Co.,
Ltd. (Taipei, Taiwan), was expressed and generated from G.
microsporum. GMI was cloned and expressed as described
previously (4).
Cytotoxicity assay
A375.S2 cells grown to 80% confluency were washed
twice with PBS and trypsinized with 1 ml 0.25% trypsin-0.03% EDTA.
The cells were then seeded at a density of 5×103
cells/well into 96-well microtiter tissue culture plates. The
seeded plates were incubated for 24 h at 37°C in 5% CO2.
The original media was then aspirated and fresh media containing
varying concentrations of GMI (0, 0.3, 0.6 and 1.2 µM) and/or
ketoconazole (0, 10 and 20 µM) was added to the 96-well plates,
followed by incubation at 37°C for 24 or 48 h. A cytotoxicity assay
was then performed using MTT and dimethyl sulfoxide was used to
dissolve the purple formazan. The absorbance was recorded at a
wavelength of 570 nm using a microtiter plate reader.
Western blot analysis
A375.S2 cell lysates were extracted by RIPA buffer
(catalog no., 9806; Cell Signaling Technology, Inc.). The protein
concentrations were determined by Bio-Rad Protein Assay (catalog
no. 5000006; Bio-Rad Laboratories, Inc.). Total lysates (50 µg)
were loaded per lane and resolved by 10% SDS-PAGE, and transferred
onto polyvinylidene difluoride (PVDF) membranes using a transblot
system (Bio-Rad Laboratories, Inc.). The PVDF membranes were then
incubated with blocking buffer [0.01 M Tris-HCl buffer (pH 7.5),
0.1 M NaC, 0.1% Tween-20 and 3% BSA] for 1 h at 25°C and washed.
Subsequently, the membranes were incubated overnight at 4°C with
the following primary antibodies: Anti-phosphorylated (p)-adenosine
monophosphate-activated protein kinase (AMPK)α (catalog no. 2535;
dilution 1:500), anti-AMPKα (catalog no. 2603; dilution 1:1,000),
anti-p-AMPKβ1 (catalog no. 4181; dilution 1:1,000), anti-AMPKβ1/2
(catalog no. 4150; dilution 1:1,000), anti-p-acetyl-CoA carboxylase
(ACC; catalog no. 3661; dilution 1:500), anti-dihydrosphingosine
1-phosphate phosphatase LCB3 (LC3B; catalog no. 3868; dilution
1:2,000), anti-survivin (catalog no. 2808; dilution 1:1,000),
anti-β-actin (catalog no. 3700; dilution 1:5,000) and anti-ACC
(catalog no. 3676; dilution 1:1,000), all obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Primary antibodies
were diluted to the appropriate volume in blocking buffer. The
membranes were then incubated with a horseradish
peroxidase-conjugated anti-rabbit secondary antibody (catalog no.
7074; dilution 1:5,000) for 1 h at room temperature. Immunoreactive
bands were visualized using an enhanced chemiluminescent system
(NEN Life Science Products, Inc., Boston, MA, USA). The band
density was analyzed by ImageJ version 1.34e software (National
Institutes of Health).
Assay for MCP-1 chemokines
A375.S2 cells (5×105 cells) were plated
onto a 6 cm dish (Nalge Nunc International, Penfield, NY, USA) with
or without 40 µM dorsomorphin dihydrochloride following treatment
with GMI (0 and 0.6 µM) and/or ketoconazole (0 and 20 µM) for 48 h
incubated at 37°C. The conditioned media were assayed for MCP-1
secretion by solid-phase enzyme-linked immunoabsorbent assay
(ELISA) using a MCP-1/CCL2 Human Uncoated ELISA kit (catalog no.
88-7399; eBioscience; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), according to the manufacturer's protocol.
Boyden chamber assay
Cell migration assays were performed using modified
Boyden chambers. MEM with 10% FBS was added to the lower chamber.
A375.S2 cells were pretreated with GMI (0, 0.6, and 1.2 µM) for 24
h at 37°C and collected, followed by resuspension at a density of
2×105 cells/well in MEM with 0.5% FBS in the upper
chamber. Following incubation for 24 h, cells on the membrane were
fixed with methanol and stained with 20% Giemsa solution (Merck
KGaA) for 2 h at 25°C. The stained cells were then counted under a
light microscope (magnification, ×100).
Apoptosis assay
The apoptosis assay was performed by Annexin V and
propidium iodide staining using the FITC Annexin V Apoptosis
Detection kit (catalog no. 556547; BD Biosciences, San Jose, CA,
USA). Following GMI (0, 0.6 and 1.2 µM) and ketoconazole (0, 10 and
20 µM) treatment for 24 h at 37°C, A375.S2 cells were trypsinized
and stained with Annexin V and propidium iodide solution, according
to the manufacturer's protocol. Following staining, the cells were
analyzed using a flow cytometer and CellQuest 5.1 software (BD
Biosciences).
Statistical analysis
Comparisons between two groups were performed by
one-way analysis of variance followed by Tukey's post hoc test
using Predictive Analytics 18 software (IBM Corporation). All data
are presented as the mean ± standard deviation. Each experiment was
performed in triplicate. P<0.05 was considered to indicate a
statistically significant difference.
Results
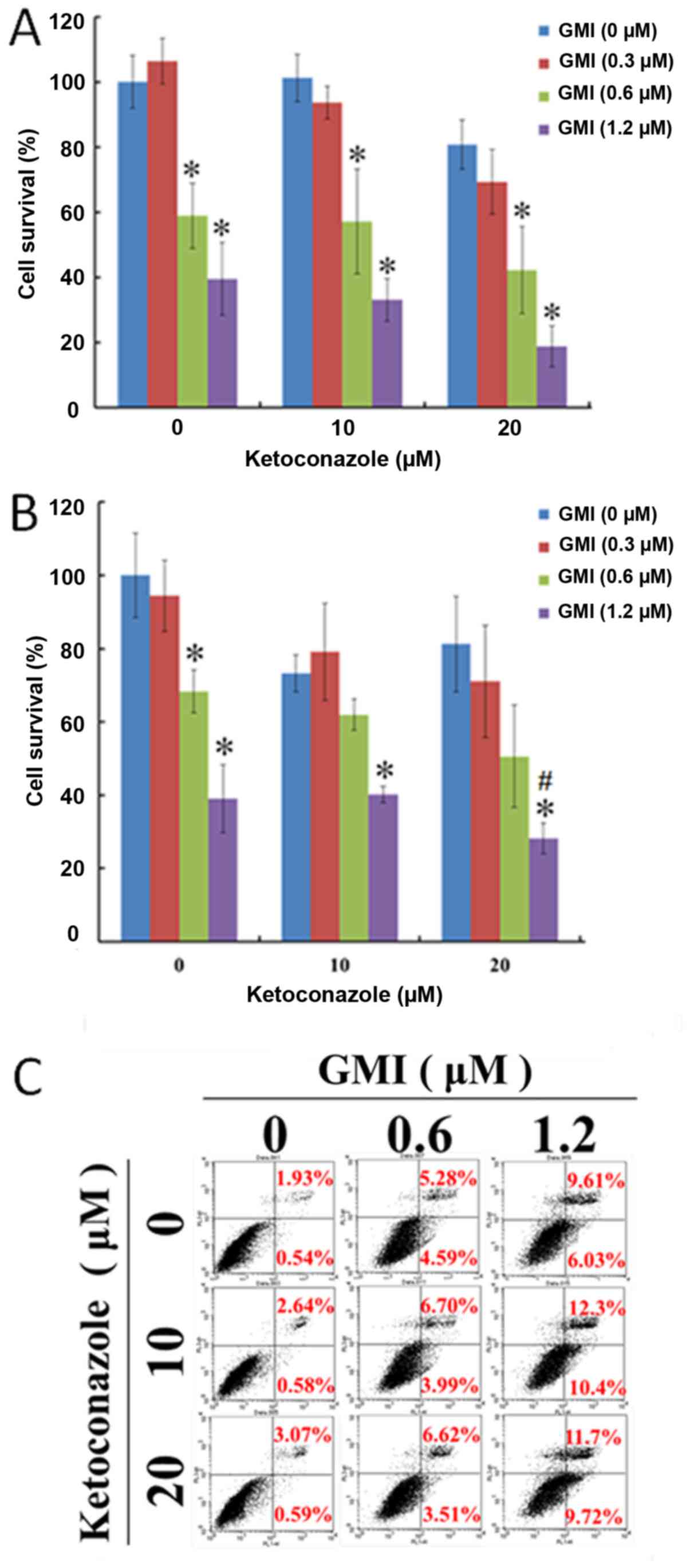
Ketoconazole enhances the cytotoxic
effect of GMI in melanoma cancer cells
The cell viability of A375.S2 cells treated with
varying concentrations of ketoconazole and GMI for 24 and 48 h was
analyzed by MTT assay. GMI was identified to mediate cytotoxicity
in A375.S2 cells. As presented in Fig.
1A and B, ketoconazole enhanced GMI-induced inhibition of cell
viability in a concentration-dependent manner in A375.S2 cells.
Using Annexin V/propidium iodide staining, it was revealed that GMI
induced apoptosis in a dose-dependent manner and ketoconazole
increased GMI-activated apoptosis in A375.S2 cells (Fig. 1C). In addition, it was identified
that treatment with 0.3 and 0.6 µM GMI did not decrease cell
viability in human skin fibroblast Hs68 cells (data not shown),
which suggests ketoconazole does not induce cytotoxicity in Hs68
cells.
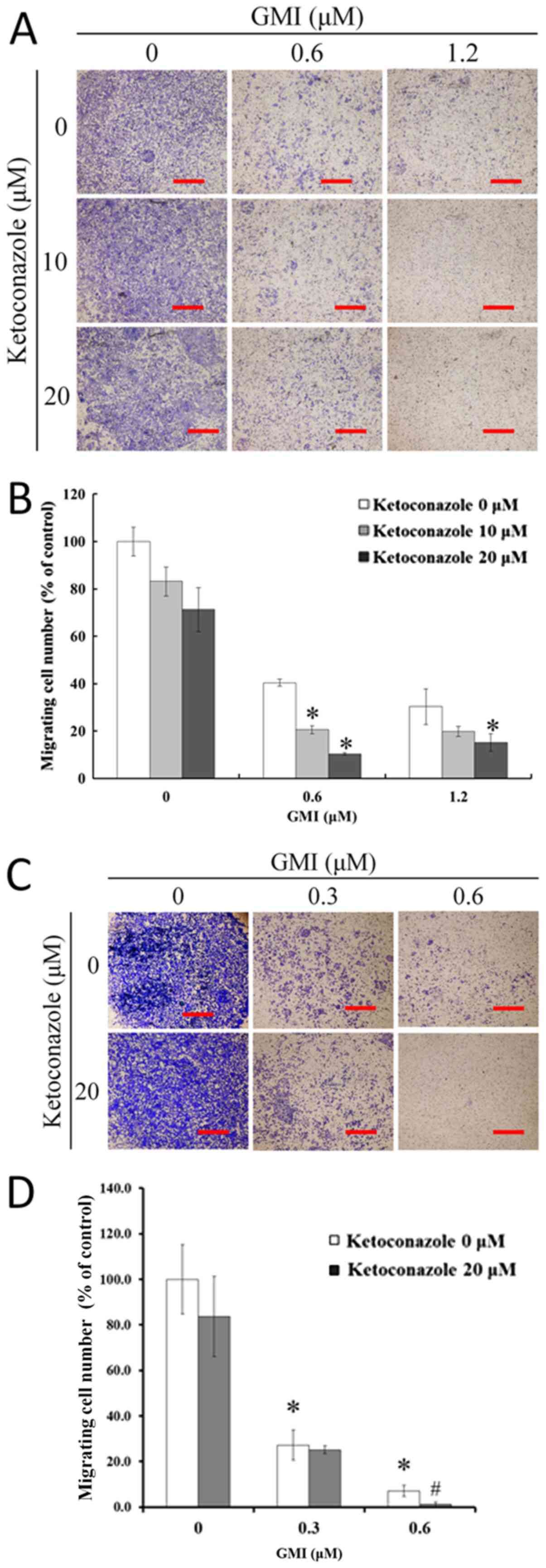
Ketoconazole enhances GMI-induced
inhibition of A375S2 cells migration
The effects of ketoconazole on GMI-inhibited cell
migration were then investigated using a modified Boyden chamber
assay to quantify the migratory potential of A375.S2 cells. The
results revealed that GMI induced a dose-dependent decrease in
migration (Fig. 2A). As presented in
Fig. 2B, following treatment with
0.6 µM GMI the proportion of migrated cells reduced to 39% and with
1.2 µM GMI with or without 20 µM ketoconazole the proportion of
migrated cells was <20%. Ketoconazole induced a dose-dependent
decrease in GMI-inhibited migration to <15%. Furthermore,
low-toxic concentrations (0.3 µM) of GMI and ketoconazole were used
for a migration assay. GMI (0.3 and 0.6 µM) significantly inhibited
the migratory ability of A375.S2 cells (Fig. 2C and D). This indicates that GMI
inhibits the migratory ability of A375.S2 cells in a
cytotoxic-independent manner.
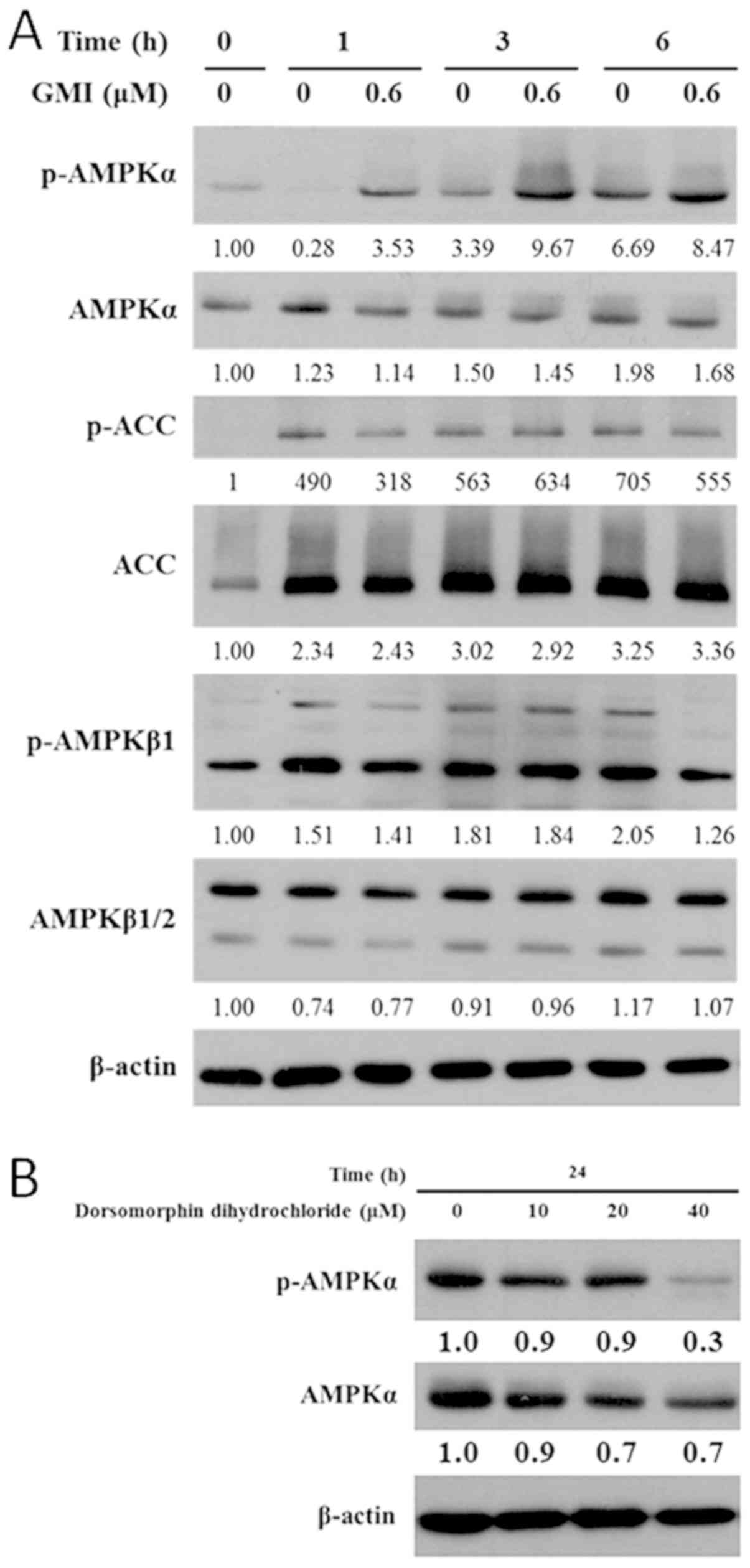
Ketoconazole reduces GMI-activated
p-AMPKα and autophagy but does not affect p-AMPKβ1 in A375.S2
cells
To investigate the effects of ketoconazole and GMI
on p-AMPK signaling and autophagy, the expression levels of
dihydrosphingosine 1-phosphate phosphatase LCB3 (LC3B), p-AMPKα,
p-AMPKβ1 and downstream p-ACC were measured by western blot
analysis. Activation of AMPK inhibits the metastatic potential of
tumor cells by reducing the activity of molecules associated with
the ERK signaling pathway, particularly berberine and
5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (14). To investigate which p-AMPK isoform is
regulated by GMI, the expression levels of p-AMPKα, p-AMPKβ1 and
p-ACC were determined. Treatment with 0.6 µM GMI for 6 h increased
p-AMPKα and p-ACC expression levels but did not affect the
expression level of p-AMPKβ1 (Fig.
3A). The AMPK inhibitor dorsomorphin dihydrochloride is a
cell-permeable compound that inhibits AMPK kinases, vascular
endothelial growth factor receptor 2 and activin-like kinase 2
(9). In the present study, treatment
with 40 µM dorsomorphin dihydrochloride for 24 h reduced the
expression level p-AMPKα (Fig.
3B).
As presented in Fig.
4A, treatment with GMI for 48 h increased the expression levels
of LC3B and p-AMPK in A375.S2 cells. Treatment with ketoconazole
and GMI for 48 h decreased the expression level of survivin but did
not affect the expression of LC3B, p-AMPK or p-ACC. In addition,
treatment with 40 µM dorsomorphin dihydrochloride inhibited
GMI-induced p-AMPK expression. The LC3B expression level increased
following treatment with dorsomorphin dihydrochloride. In addition,
dorsomorphin dihydrochloride inhibited the ketoconazole and
GMI-induced decrease in survivin expression level. Furthermore,
treatment with GMI and ketoconazole for 3 h increased the
expression level of activated p-AMPK (Fig. 4B), which suggests that treatment for
a short time period can increase AMPK activity.
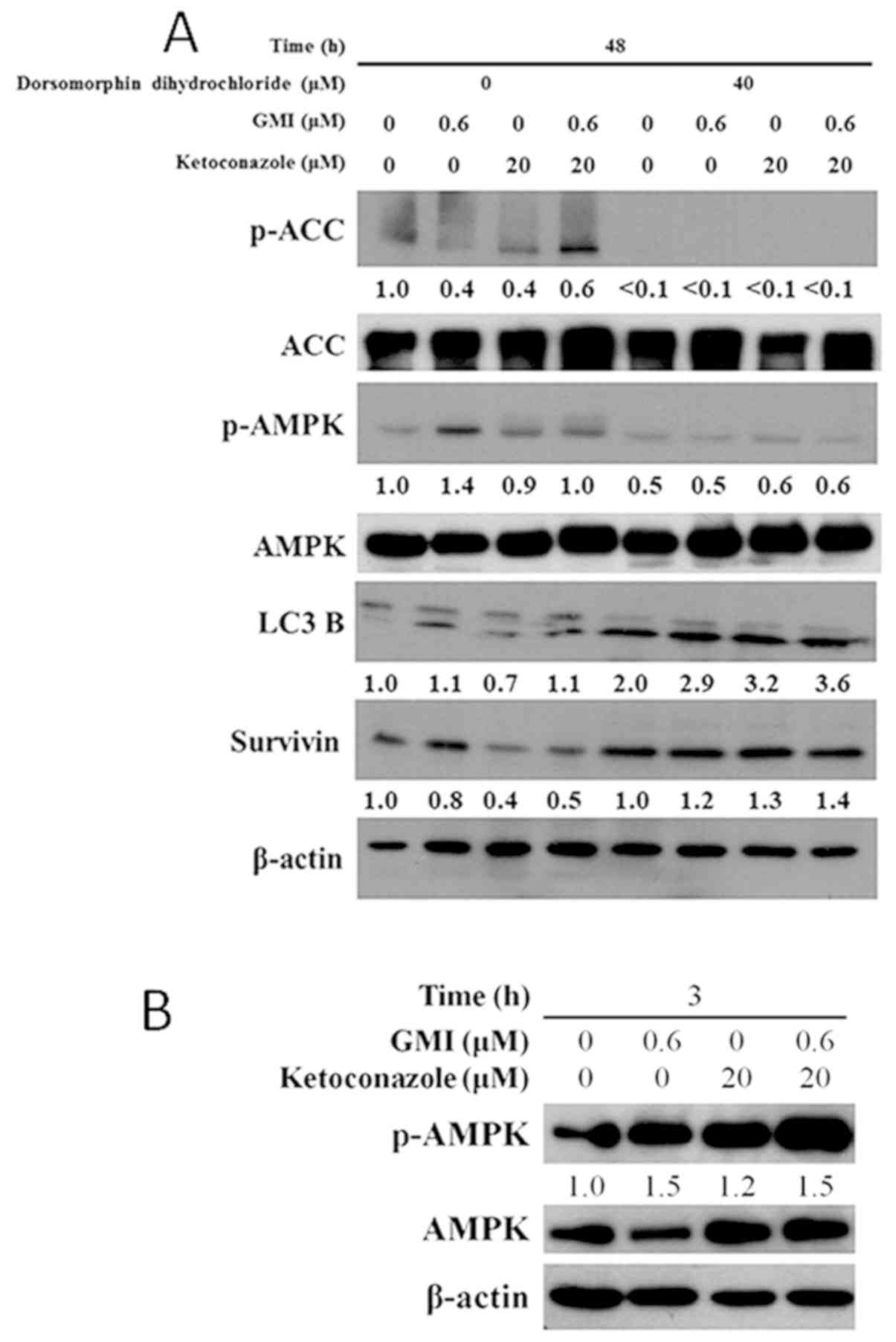 | Figure 4.Effects of GMI and dorsomorphin
dihydrochloride on the protein expression levels of proteins
associated with cell death and the AMPK signaling pathway. (A)
A375.S2 cells were pretreated with 40 mM dorsomorphin
dihydrochloride for 1 h and then treated with ketoconazole (0 or 20
µM) combined with GMI (0 or 0.6 µM) for 48 h. The expression levels
of p-AMPK, AMPK, p-ACC, ACC, LC3B (LC3B-I and LC3B-II) and survivin
were measured by western blot analysis. GMI and ketoconazole
increased the expression level of LC3B and reduced the expression
of survivin. (B) A375.S2 cells were treated with ketoconazole (0 or
20 µM) combined with GMI (0 or 0.6 µM) for 3 h. The expression
levels of p-AMPK, AMPK and β-actin were then measured by western
blot analysis. GMI, Ganoderma microsporum; AMPK, adenosine
monophosphate-activated protein kinase; p-, phosphorylated; ACC,
acetyl-CoA carboxylase; LC3B, dihydrosphingosine 1-phosphate
phosphatase LCB3. |
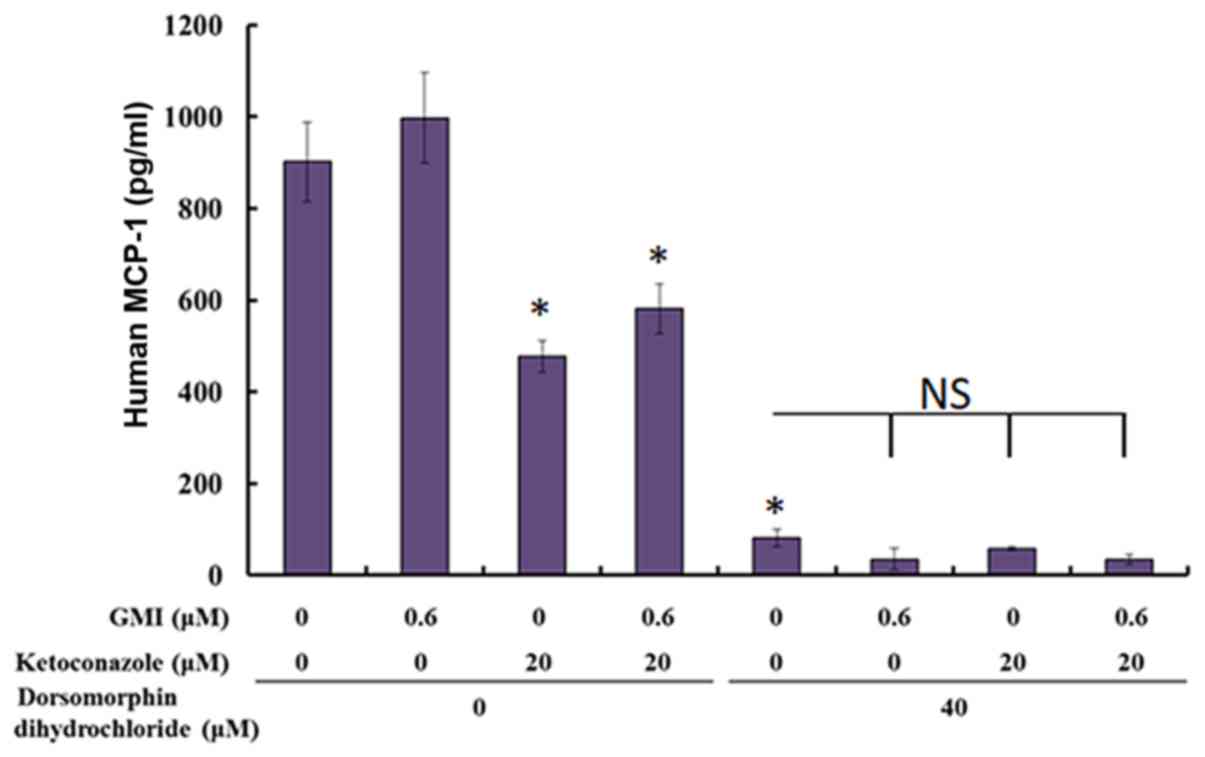
Ketoconazole and dorsomorphin
dihydrochloride reduce MCP-1 secretion in A375.S2 cells
In addition, MCP-1 is an important chemokine that
attracts macrophages to tumors (15). The present study used ELISA to
evaluate the secretion of MCP-1 following treatment with GMI and
ketoconazole. Ketoconazole was identified to significantly reduce
the level of MCP-1, whereas 0.6 µM GMI did not affect the level of
MCP-1. In addition, dorsomorphin dihydrochloride significantly
reduced the level of MCP-1 (Fig.
5).
Discussion
Ketoconazole, a cytochrome P450, family 3, subfamily
A (CYP3A) inhibitor, promotes androgen receptor (AR) nuclear
localization. Inhibition of CYP3A5 has been associated with
inhibitory effects in response to dihydrotestosterone treatment in
prostate cancer cells by decreasing nuclear AR localization and
reducing prostate-specific antigen levels (16). Therefore, co-treatment with CYP3A
inhibitor may offer a strategy for improving androgen deprivation
therapy (16). A combination of CYP
inhibitor and ERK inhibitor enhances anti-colon cancer cell
activity (17). Co-administration of
CYP3A inhibitor and ketoconazole with the proteasome inhibitor
bortezomib has been reported to increase the blood proteasome
inhibitory effect in patients with advanced solid tumors by 35%
(18). Ketoconazole also enhances
the effect of chemotherapeutic agents, including doxorubicin
(19), mitoxantrone (20) and docetaxel (3). Oral delivery of pacitaxel with a dual
CYP3A4 inhibitor has been demonstrated to effectively inhibit tumor
growth in B16 F10 melanoma tumor-bearing mice (21). The results of the present study
supported a therapeutic potential of a combination of ketoconazole
and GMI for melanoma treatment.
The AMPK-associated kinase NUAK family SNF1-like
kinase 2 exhibits oncogenic properties in melanoma cells during
cancer development and tumor progression (22). Metformin blocks melanoma invasion and
metastasis via AMPK activation (23). In the current study, GMI was revealed
to activate AMPK, which may serve a role in the inhibition of
migration. In addition, GMI appeared to have a small effect on the
expression of p-ACC. It has been reported that ACC activation
occurs via an AMPK-independent pathway (24). GMI triggers the AMPK-dependent and
the ACC-independent pathway.
The BRAF V600E mutation is present in ~50% of all
cases of melanoma and oral BRAF inhibitors, including PLX472,
inhibit tumors in patients with this mutation via inhibition of the
oncogenic mitogen-activated protein kinase signaling pathway
(25). Knight et al (25) revealed that treatment with PLX4720
decreases the expression level of MCP-1, which is associated with a
decrease in tumor growth. Combination therapy of PLX4720 and
anti-MCP-1 has been demonstrated to promote anti-melanoma activity
in mouse models (25). In the
present study, ketoconazole decreased the expression level of
MCP-1, which may lead to antitumor effects. Therefore, ketoconazole
may be applied for the treatment of melanoma cases with the BRAF
V600E mutation. In conclusion, the current study demonstrated that
a combination of ketoconazole with GMI exhibits therapeutic
potential against melanoma. Therefore, clinical trials of this
combination therapy are required in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by Chung Shan
Medical University Hospital, Taiwan (grant nos. CSH-2017-C-015 and
CSH-2014-C-023) and the Ministry of Science and Technology, Taiwan
(grant nos. MOST 104-2311-B-040-001, MOST 106-2314-B-040-017 and
MOST 107-2314-B-040-016-MY2).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YPH and JLK conceived and designed the study. CTL,
CTH, YCC and YDL performed experiments and analyzed the data. TYH,
PYL and YTK conducted data analysis and interpretation. YPH and JLK
wrote, edited and revised the manuscript. All authors discussed
results and collaboratively in drafting the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publications
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lopez-Barcons L, Maurer BJ, Kang MH and
Reynolds CP: P450 inhibitor ketoconazole increased the intratumor
drug levels and antitumor activity of fenretinide in human
neuroblastoma xenograft models. Int J Cancer. 141:405–413. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Agarwal SK, Salem AH, Danilov AV, Hu B,
Puvvada S, Gutierrez M, Chien D, Lewis LD and Wong SL: Effect of
ketoconazole, a strong CYP3A inhibitor, on the pharmacokinetics of
venetoclax, a BCL-2 inhibitor, in patients with non-Hodgkin
lymphoma. Br J Clin Pharmacol. 83:846–854. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Figg WD, Woo S, Zhu W, Chen X, Ajiboye AS,
Steinberg SM, Price DK, Wright JJ, Parnes HL, Arlen PM, et al: A
phase I clinical study of high dose ketoconazole plus weekly
docetaxel for metastatic castration resistant prostate cancer. J
Urol. 183:2219–2226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hsin IL, Ou CC, Wu TC, Jan MS, Wu MF, Chiu
LY, Lue KH and Ko JL: GMI, an immunomodulatory protein from
Ganoderma microsporum, induces autophagy in non-small cell
lung cancer cells. Autophagy. 7:873–882. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin CH, Hsiao YM, Ou CC, Lin YW, Chiu YL,
Lue KH, Chang JG and Ko JL: GMI, a Ganoderma immunomodulatory
protein, down-regulates tumor necrosis factor α-induced expression
of matrix metalloproteinase 9 via NF-κB pathway in human alveolar
epithelial A549 cells. J Agric Food Chem. 58:12014–12021. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chiu LY, Hu ME, Yang TY, Hsin IL, Ko JL,
Tsai KJ and Sheu GT: Immunomodulatory protein from Ganoderma
microsporum induces pro-death autophagy through Akt-mTOR-p70S6K
pathway inhibition in multidrug resistant lung cancer cells. PLoS
One. 10:e01257742015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hsin IL, Ou CC, Wu MF, Jan MS, Hsiao YM,
Lin CH and Ko JL: GMI, an immunomodulatory protein from
Ganoderma microsporum, potentiates cisplatin-induced
apoptosis via autophagy in lung cancer cells. Mol Pharm.
12:1534–1543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang TY, Yu CC, Hsieh PL, Liao YW, Yu CH
and Chou MY: GMI ablates cancer stemness and cisplatin resistance
in oral carcinomas stem cells through IL-6/Stat3 signaling
inhibition. Oncotarget. 8:70422–70430. 2017.PubMed/NCBI
|
|
9
|
Hardie DG: AMPK: Positive and negative
regulation, and its role in whole-body energy homeostasis. Curr
Opin Cell Biol. 33:1–7. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roy I, McAllister DM, Gorse E, Dixon K,
Piper CT, Zimmerman NP, Getschman AE, Tsai S, Engle DD, Evans DB,
et al: Pancreatic cancer cell migration and metastasis is regulated
by chemokine-biased agonism and bioenergetic signaling. Cancer Res.
75:3529–3542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Qiao L, Zheng J, Jin X, Wei G, Wang G, Sun
X and Li X: Ginkgolic acid inhibits the invasiveness of colon
cancer cells through AMPK activation. Oncol Lett. 14:5831–5838.
2017.PubMed/NCBI
|
|
12
|
Kalbasi A, Komar C, Tooker GM, Liu M, Lee
JW, Gladney WL, Ben-Josef E and Beatty GL: Tumor-derived CCL2
mediates resistance to radiotherapy in pancreatic ductal
adenocarcinoma. Clin Cancer Res. 23:137–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang AL, Miska J, Wainwright DA, Dey M,
Rivetta CV, Yu D, Kanojia D, Pituch KC, Qiao J, Pytel P, et al:
CCL2 produced by the glioma microenvironment is essential for the
recruitment of regulatory T cells and myeloid-derived suppressor
cells. Cancer Res. 76:5671–5682. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim HS, Kim MJ, Kim EJ, Yang Y, Lee MS and
Lim JS: Berberine-induced AMPK activation inhibits the metastatic
potential of melanoma cells via reduction of ERK activity and COX-2
protein expression. Biochem Pharmacol. 83:385–394. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gazzaniga S, Bravo AI, Guglielmotti A, van
Rooijen N, Maschi F, Vecchi A, Mantovani A, Mordoh J and Wainstok
R: Targeting tumor-associated macrophages and inhibition of MCP-1
reduce angiogenesis and tumor growth in a human melanoma xenograft.
J Invest Dermatol. 127:2031–2041. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mitra R and Goodman OB Jr: CYP3A5
regulates prostate cancer cell growth by facilitating nuclear
translocation of AR. Prostate. 75:527–538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lim SM, Hwang JW, Ahn JB, Bae SK, Park CH,
Kim KY, Rha SY, Chung HC, Roh JK and Shin SJ: Combination of CYP
inhibitor with MEK/ERK inhibitor enhances the inhibitory effect on
ERK in BRAF mutant colon cancer cells. Anticancer Res.
33:2499–2508. 2013.PubMed/NCBI
|
|
18
|
Venkatakrishnan K, Rader M, Ramanathan RK,
Ramalingam S, Chen E, Riordan W, Trepicchio W, Cooper M, Karol M,
von Moltke L, et al: Effect of the CYP3A inhibitor ketoconazole on
the pharmacokinetics and pharmacodynamics of bortezomib in patients
with advanced solid tumors: A prospective, multicenter, open-label,
randomized, two-way crossover drug-drug interaction study. Clin
Ther 31 Pt. 2:2444–2458. 2009. View Article : Google Scholar
|
|
19
|
Sella A, Kilbourn R, Amato R, Bui C,
Zukiwski AA, Ellerhorst J and Logothetis CJ: Phase II study of
ketoconazole combined with weekly doxorubicin in patients with
androgen-independent prostate cancer. J Clin Oncol. 12:683–688.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Eklund J, Kozloff M, Vlamakis J, Starr A,
Mariott M, Gallot L, Jovanovic B, Schilder L, Robin E, Pins M and
Bergan RC: Phase II study of mitoxantrone and ketoconazole for
hormone-refractory prostate cancer. Cancer. 106:2459–2465. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patel K, Patil A, Mehta M, Gota V and
Vavia P: Oral delivery of paclitaxel nanocrystal (PNC) with a dual
Pgp-CYP3A4 inhibitor: Preparation, characterization and antitumor
activity. Int J Pharm. 472:214–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Namiki T, Tanemura A, Valencia JC, Coelho
SG, Passeron T, Kawaguchi M, Vieira WD, Ishikawa M, Nishijima W,
Izumo T, et al: AMP kinase-related kinase NUAK2 affects tumor
growth, migration, and clinical outcome of human melanoma. Proc
Natl Acad Sci USA. 108:6597–6602. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cerezo M, Tichet M, Abbe P, Ohanna M,
Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P,
Bertolotto C, et al: Metformin blocks melanoma invasion and
metastasis development in AMPK/p53-dependent manner. Mol Cancer
Ther. 12:1605–1615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fediuc S, Gaidhu MP and Ceddia RB:
Regulation of AMP-activated protein kinase and acetyl-CoA
carboxylase phosphorylation by palmitate in skeletal muscle cells.
J Lipid Res. 47:412–420. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Knight DA, Ngiow SF, Li M, Parmenter T,
Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al: Host
immunity contributes to the anti-melanoma activity of BRAF
inhibitors. J Clin Invest. 123:1371–1381. 2013. View Article : Google Scholar : PubMed/NCBI
|