Introduction
Cervical cancer is a gynecological disease that
continues to threaten the health of women globally. By integrating
viral DNA into human chromosomal DNA and activating proto-oncogenes
(or deactivating tumor suppressor genes), the Human papilloma virus
(HPV) is one of the primary causes of cervical cancer (80–90%)
(1). Currently, >100 HPV variants
have been identified, though few are carcinogenic (2). HPV types 16 and 18, classified as
high-risk HPVs, are able to promote the progression of premalignant
lesions, which unless treated, ultimately results in the
development of cancer (3). Of all
cervical cancer cases, ~70% have been associated with HPV 16 and 18
(4); furthermore, high-risk HPVs
secrete early protein 6 that forms a complex with p53, resulting in
its ubiquitination and subsequent degradation (5).
p53 is a powerful transcription factor that prevents
the malignant transformation of cells, and as such is frequently
inactivated following viral infection (6,7). Various
stress signals, including oncogene activation, DNA damage and
hypoxia, improve the stability of wild-type p53. Alterations in p53
expression level are associated with the transcription of specific
p53-responsive gene, including p21, Bcl-2-associated X protein
(Bax), p53 upregulated modulator of apoptosis (PUMA) and growth
arrest and DNA damage-inducible 45, which are involved in
p53-induced cell cycle arrest, metabolism, DNA repair, apoptosis
and senescence (8,9). Previous investigations have revealed
that mutations in p53 are present in ~50% human cancers (10). Under normal conditions, p53 is
tightly regulated by the mouse double minute 2 homolog (Mdm2)
protein via a self-regulating feedback loop (11,12). p53
is able to increase the expression level of Mdm2, which conversely
inhibits p53 in three ways: i) Mdm2 binds to the transactivation
domain of p53 and subsequently inhibits transcriptional activity;
ii) Mdm2 blocks the nuclear export of p53; or iii) Mdm2 may
function as an E3 ubiquitin protein ligase, targeting p53 for
ubiquitination and degradation (13). Furthermore, the overexpression of the
Mdm2 gene may result in loss of p53 function in numerous types of
malignant tumor (14). Therefore,
interrupting the p53-Mdm2 interaction (or Mdm2 itself) with
small-molecule inhibitors may reactivate p53 and inhibit tumor
growth (15,16); the development of nutlin-3 provides
important proof-of-concept for the design of small-molecule
inhibitors of Mdm2 (17).
The present study introduced the organic
small-molecule compound 2-[2-hydroxyl-1-(4-methoxy phenyl)
ethyl]-3-(4-benzyloxy phenyl) isoindolin-1-one (CDS-1548), which
altered the activity of cervical cancer cell lines by promoting the
accumulation of p53 and inducing apoptosis. In the present study,
the method by which CDS-1548 activates p53 and inhibits tumor cell
viability (by eliciting apoptosis and cell cycle arrest) was
investigated.
Materials and methods
Cell culture and reagents
The human cervical cancer cell line HeLa was
purchased from the Chinese Academy of Sciences Cell Bank (Shanghai,
China) and maintained in Dulbecco's modified Eagle's medium (DMEM;
Corning, Inc.) with 10% fetal bovine serum (cat. no., 626216; Omega
Bio-Tek, Inc.), 2 mmol/l L-glutamine (Sigma-Aldrich; Merck KGaA),
100 IU/ml penicillin (Sigma, Shanghai) and 100 µg/ml streptomycin
(Sigma-Aldrich; Merck KGaA). The following antibodies were
purchased from Santa Cruz Biotechnology, Inc.: Anti-caspase 3 (cat.
no. sc-56053), anti-caspase 8 (cat. no. sc-81656), anti-caspase 9
(cat. no. sc-133109), anti-cleaved poly (ADP-ribose) polymerase 1
(PARP-1; cat. no. sc-56196), anti-p53 (cat. no. sc-47698), anti-p21
(cat. no. sc-71811), anti-Mdm2 (cat. no. sc-5304), anti-cytochrome
c (cat. no. sc-13560), anti-cyclin B1 (cat. no. sc-245),
anti-cyclin-dependent kinase 1/2 (CDK1/2; cat. no. sc-53219),
anti-checkpoint kinase 1 (CHK1; cat. no. sc-56288), anti-checkpoint
2 (CHK2; cat. no. sc-136251), anti-M-phase phosphatase 3 (CDC25C;
cat. no. sc-327), H2AX, H2A histone family, member X (H2AX; cat.
no. sc-54606), anti-p-ataxia telangiectasia and Rad3-related
protein (ATR; cat. no. sc-515173), anti-B-cell lymphoma 2 (Bcl-2;
cat. no. sc-7382), anti-Bcl-2 homologous antagonist killer (BAK;
cat. no. sc-517390), anti-BAX (cat. no. sc-7480), anti-PUMA (cat.
no. sc-374223) and anti-β-actin (cat. no. sc-8432).
Anti-phosphorylated (p)-CHK1 (cat. no. 12302) and anti-p-CHK2 (cat.
no. 2197) were obtained from Cell Signaling Technology. Inc., and
anti-γ-H2AX histone family (cat. no. ab26350) was purchased from
Abcam. M-MLV Reverse Transcriptase (cat. no. 28025013) was obtained
from Thermo Fisher Scientific, Inc. Propidium iodide (PI),
4′,6-diamidino-2-phenylindole (DAPI),
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), and all other chemical reagents were obtained from
Sigma-Aldrich (Merck KGaA). The fluorescein isothiocyanate (FITC)
Annexin V Apoptosis Detection Kit I was purchased from Bioteke
Corporation. Caspase 3, 8 and 9 activity assay kits were obtained
from BestBio. Cell Mitochondria Isolation kit (cat. no. c3601) was
purchased from Beyotime Institute of Biotechnology. CDS-1548 was
synthesized by the Center for Combinatorial Chemistry and Drug
Discovery of Jilin University (Changchun, China) according to a
previously reported method (18,19).
Cell viability assay
An MTT assay was conducted to evaluate the effects
of CDS-1548 on cell viability. Briefly, 5×103 cells/well
were seeded into 96-well plates and incubated in serum-free DMEM
for 24 h at 37°C. The cells were washed with phosphate-buffered
saline (PBS, pH 7.0) and treated at 37°C with various
concentrations of CDS-1548 (0.14, 0.37, 1.1, 3.3, 11, 33 and 100
µM, to a final volume of 100 µl/well) for 12, 24 and 48 h.
Following treatment, 20 µl MTT solution was added and the cells
were incubated for an additional 3 h at 37°C. The culture medium
was subsequently removed and 150 µl dimethyl sulfoxide (DMSO) was
added to dissolve the formazan crystals. The absorbance of was
assessed at 495 nm using a microplate reader and equated to the
number of viable cells.
Nuclear staining
To examine the condensation and fragmentation of
cellular nuclei, 5×104 cells/well were seeded in to
24-well plates and incubated for 24 h; the medium was then replaced
with equal quantities of DMEM or DMEM + 2 µM CDS-1548. Following a
further 24-h incubation at 37°C, the cells were washed with PBS and
stained with DAPI (2.5 µg/ml) for 5 min at room temperature. The
morphology of the nuclei was determined by fluorescence microscopy
(magnification, ×10).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using
TRIzol® reagent (Thermo Fisher Scientific, Inc.) and 1
µg total RNA was used as a template for reverse transcription using
M-MLV reverse transcriptase (Thermo Fisher Scientific, Inc.),
according to previous methods (20,21). The
synthesized cDNA was subsequently amplified by qPCR using SYBR™
Green PCR Master Mix (Thermo Fisher Scientific, Inc.) in 7500 Fast
Real-Time PCR System, according to the manufacturer's protocols.
The following thermocycling conditions: Denaturation (94°C for 30
sec), annealing (55°C for 30 sec) and extension (72°C for 1 min)
for 30 cycles. The primers used were as follows: β-actin forward,
5′-TCTGGCACCACACCTTCTACAATG-3′, and reverse,
5′-GGATAGCACAGCCTGGATAGCAA-3′; p53 forward,
5′-GGCTCTGACTGTACCACCATCCA-3′, and reverse,
5′-GGCACAAACACGCACCTCAAAG-3′; p21 forward,
5′-GGAAGACCATGTGGACCTGT-3′, and reverse,
5′-GGCGTTTGGAGTGGTAGAAA-3′; and PUMA forward,
5′-TAGAGAGAGCGACGTGAC-3′, and reverse, 5′-CGGTATCTACAGCAGCGCAT-3.
mRNA levels were quantified using the 2−ΔΔCq method
(20) and normalized to the internal
reference gene β-actin. The experiments were repeated at least
three times.
Cell cycle status and apoptosis
assay
Early apoptotic cells were characterized by the
translocation of phosphatidylserine to the outer surface of the
cell membrane (22). Annexin V/PI
staining was used to detect the number of apoptotic cells,
according to the manufacturer's protocol. Briefly, 4×105
cells/well were incubated overnight in 6-well plates, and treated
with 2, 5, or 10 µM CDS-1548 prior to a further 24, 48 or 72-h
incubation at 37°C. Following treatment, the cells were harvested
in 15 ml tubes by trypsinization, and the medium was removed by
centrifugation at 3,000 × g for 5 min at 4°C. For the apoptotic
assay, cells were washed twice in ice-cold PBS, aspirated and
resuspended in binding buffer with FITC-annexin V and PI at room
temperature for 15 min (in the dark). Subsequently, cells were
resuspended in binding buffer and flow cytometricaly analyzed using
the Beckman Flow Cytometry Analyzer (Beckman CytoFLEX; Beckman
Coulter, Inc.,). For cell cycle analysis, following the
aforementioned CDS-1548 treatment, cells were rinsed in PBS,
resuspended in 5 ml PI solution (25 µg/ml RNase A, 50 µg/ml PI) and
incubated at 37°C for 30 min in the dark. The cell cycle
distribution was determined using the Beckman Flow Cytometry
Analyzer and the data were statistically analyzed using SPSS 19.0
software (IBM Corp.).
Western blotting
Following treatment with CDS-1548, the cells were
lysed at 4°C using ice-cold lysis buffer [50 mmol/l Tris (pH 8.0),
150 mmol/l NaCl, 0.1% SDS, 1% NP40 and 0.5% sodium deoxycholate]
supplemented with protease/phosphatase inhibitors (1% Cocktail and
1 mmol/l phenylmethylsulfonyl fluoride). The lysates were
centrifuged at 10,000 × g for 5 min at 4°C, and the supernatant was
collected. Protein quantification was performed using a Bradford
assay kit and 40 µg protein/lane was separated by SDS-PAGE (10%
gel). The proteins were transferred to polyvinylidene fluoride
membranes (EMD Millipore) and blocked using TBS with 5% fat-free
milk and 0.1% tween-20 for 1 h at room temperature. The membranes
were washed twice in TBST and incubated with primary antibodies
(1:200) overnight at 4°C. The membranes were rinsed three times
with ice-cold PBS and then incubated with peroxidase-conjugated
secondary antibodies (1:2,000) for 2 h at room temperature. The
protein bands were visualized using the BeyoECL Star kit (Beyotime
Institute of Biotechnology).
Mitochondria and cytosol
extraction
Briefly, cells (8×106) were seeded in a
T75 flask and then incubated overnight at 37°C. After 24-h
treatment with CDS-1548 (2, 5 and 10 µM), cells were scraped from
the plate in PBS and harvested by centrifugation at 500 × g for 5
min at 4°C. The cell pellets were resuspended in extraction buffer
from the Cell Mitochondria Isolation kit [20 mmol/l HEPES (pH 7.5),
1.5 mmol/l MgCl2, 10 mmol/l KCl, 1 mmol/l EDTA, 1 mmol/l
EGTA, 1 mmol/l DTT, 0.1 mmol/l phenylmethylsulfonyl fluoride, and
250 mmol/l sucrose], and homogenized using a microhomogenizer. The
homogenates were subsequently centrifuged at 750 × g for 10 min at
4°C. To isolate mitochondrial extracts, the supernatants were
re-centrifuged at 10,000 × g for 15 min at 4°C, and the precipitate
(mitochondria protein extract) and the supernatant (cytoplasmic
fractions.) were retrieved.
Determination of caspase activity
Caspase-3, 8 and9 activity were determined using
caspase-3/8/9 activity kits according to the manufacturer's
protocols. Briefly, following treatment with CDS-1548, the cells
were scraped from the culture plates in PBS (0.01 M, pH 7.4) and
centrifuged at 10,000 × g at 4°C. The cell pellets were lysed in
100 µl lysis buffer and the resulting suspension was centrifuged at
10,000 × g for 10 min at 4°C. An equal amount of supernatant was
incubated with the corresponding substrates in reaction buffer
containing dithiothreitol, and the absorbance at 405 nm was
determined using a microplate reader.
Statistical analysis
All quantitative data are expressed as the mean ±
standard deviation of three independent experiments. Statistical
differences were evaluated using analysis of variance and the Least
Significant Difference post hoc test. SPSS 19.0 (SPSS, Inc., IL,
USA) was used for statistical analysis and P<0.05 was considered
to indicate a statistically significant difference.
Results
Inhibitory effects of CDS-1548 on HeLa
cells
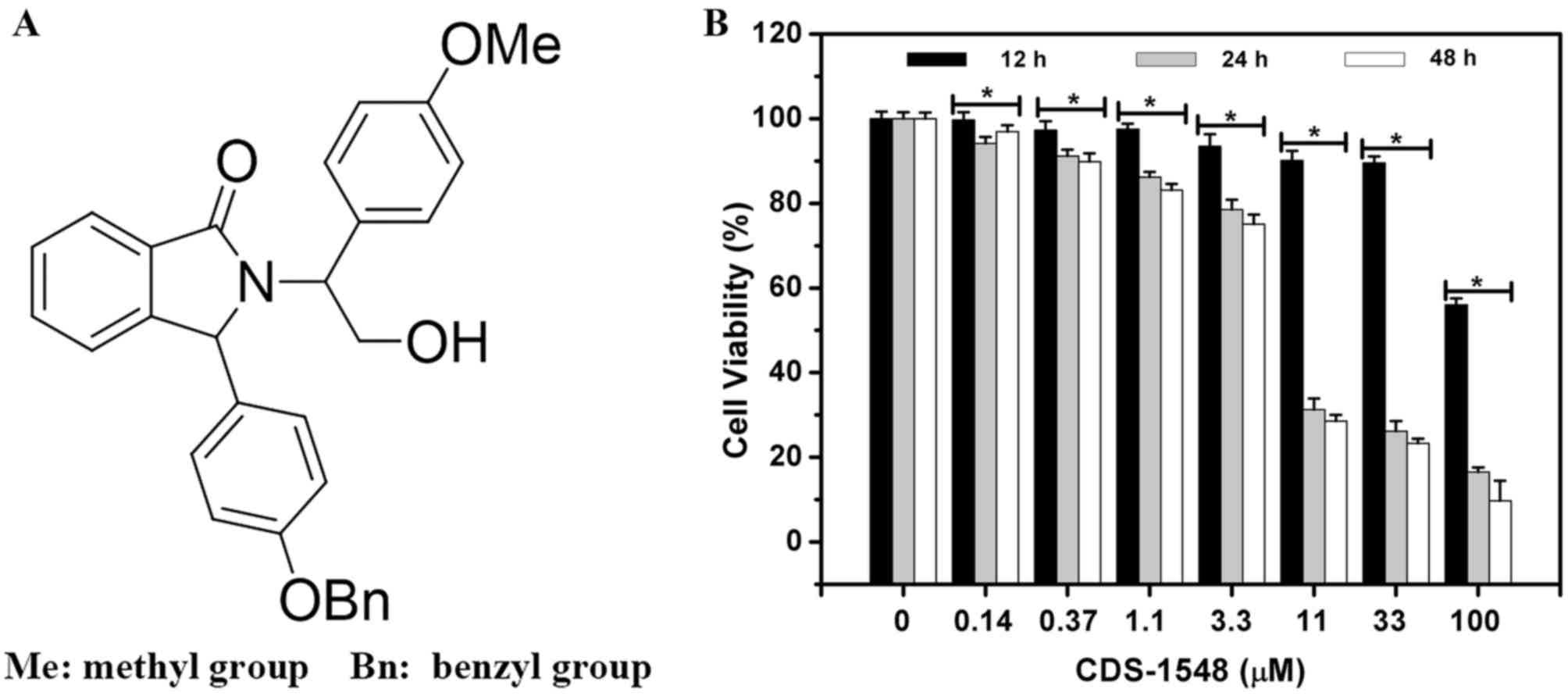
To assess the inhibitory effects of CDS-1548 on HeLa
cells (Fig. 1A), a
concentration-escalation experiment was performed (Fig. 1B). CDS-1548 reduced cell viability in
a dose- and time-dependent manner. The MTT assay revealed that the
half maximal inhibitory concentration (IC50) of CDS-1548
in HeLa cells was markedly reduced following increasing treatment
durations. The IC50 at 12, 24 and 48 h were 100.0, 4.3
and 4.0 µM, respectively. Notably, at 11 µM, CDS-1548 significantly
enhanced cell cytotoxicity following 24- and 48-h of treatment,
suggesting that this was the optimum concentration in HeLa cells.
However, significant differences were not observed between the 24-
and 48-treatments due to the cycling time of the HeLa cell
line.
CDS-1548 induces G2/M
arrest in HeLa cells
To determine whether CDS-1548 caused cell-cycle
arrest, a flow cytometric cell cycle assay was performed using HeLa
cells following 24 h of treatment with 0, 2, 5, or 10 µM CDS-1548.
A significantly increased percentage of G2/M-phase cells
was observed from 25.115% (0 µM) to 38.65% (2 µM), 73.03% (5 µM) or
89.62% (10 µM CDS-1548; Fig. 2A). To
further elucidate the potential molecular mechanism underlying
CDS-1548-induced cell-cycle arrest, the expression levels of
proteins associated with G2/M phase progression were
determined. The results indicated that CDS-1548 downregulated the
expression level of cyclin B1 in a dose-dependent manner,
upregulated the expression levels of p-CHK1 and decreased the
levels of CDC25C compared with the control-treated cells. However,
CDS-1548 did not alter the expression levels of CHK2 or its
phosphorylation (Fig. 2B).
Alterations in the expression levels of p-ATR and the histone
γ-H2AX, DNA-damage markers associated with CHK1, were also
determined. CDS-1548 treatment increased the level of γ-H2AX
expression and ATR phosphorylation. These data suggested that
CDS-1548 induced cell cycle arrest at the G2/M-phase via
DNA-damage checkpoint pathways.
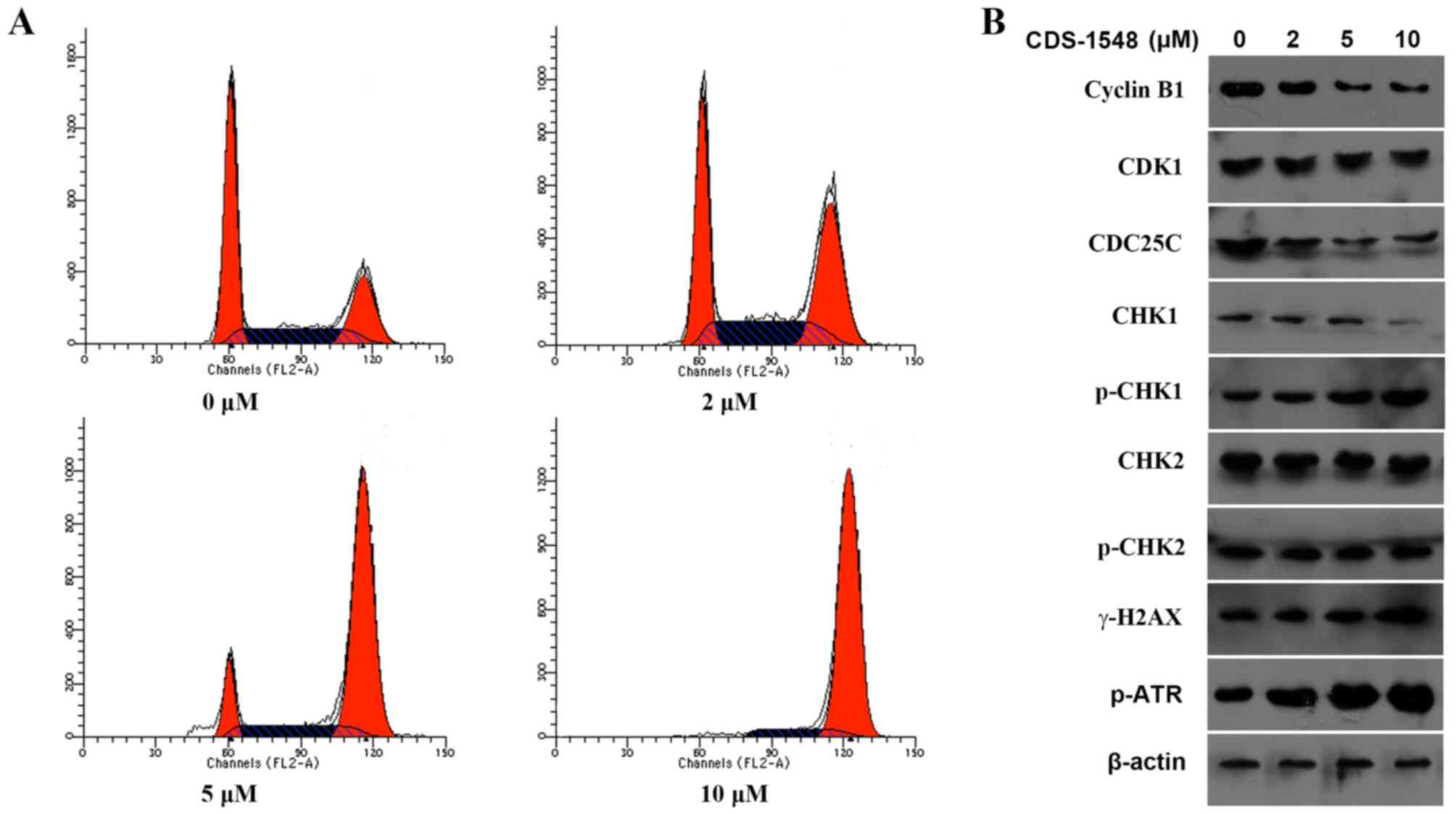 | Figure 2.CDS-1548 treatment induces cell cycle
arrest at the G2/M phase. (A) HeLa cells were treated
with 2, 5 or 10 µM CDS-1548 for 24 h, stained with propidium
iodide, and the DNA content analyzed by flow cytometry. N=3. (B)
HeLa cells were treated with 2, 5 or 10 µM CDS-1548 for 24 h. The
expression levels of proteins associated with the cell cycle were
detected by western blotting. CDS-1548, 2-[2-hydroxyl-1-(4-methoxy
phenyl) ethyl]-3-(4-benzyloxy phenyl) isoindolin-1-one; CKD1,
cyclin dependent kinase 1; CHK1, checkpoint kinase 1; CHK2,
checkpoint kinase 2; CDC25C, M-phase phosphatase 3; H2AX, H2A
histone family, member X; ATR, ataxia telangiectasia and
Rad3-related protein; p-, phosphorylated. |
CDS-1548 induces p53-dependent
apoptosis in human cervical cancer cells
The present study investigated whether the
inhibitory effect of CDS-1548 on HeLa cells also induced cell
death. As illustrated in Fig. 3A,
chromatin condensation and apoptotic bodies were observed in the
nuclei subsequent to treatment with CDS-1548. Furthermore, flow
cytometric analysis indicated that CDS-1548 induced the
accumulation of early apoptotic cells (annexin V+/PI-), suggesting
that apoptosis in HeLa cells was dose- and time-dependent (Fig. 3B). After 24-h treatment with CDS-1548
(2, 5 and 10 µM) the percentages of early apoptotic cells were
2.82, 9.21 and 14.7%, and the percentages of necrotic cells
(annexin V+/PI+) were 3.69, 2.95 and 4.16% respectively. Compared
with 24-h CDS-1548 treatment, the proportions of early apoptotic
cells in the 48-h treatment group was significantly increased, to
3.33, 35.51 and 51.32%; the percentages of necrotic cells were
0.89, 3.05 and 6.41% for 2, 5, and 10 µM CDS-1548, respectively.
These results indicated that the inhibitory effects observed in
response to CDS-1548 were primarily associated with the induction
of apoptosis, and not necrosis in HeLa cells.
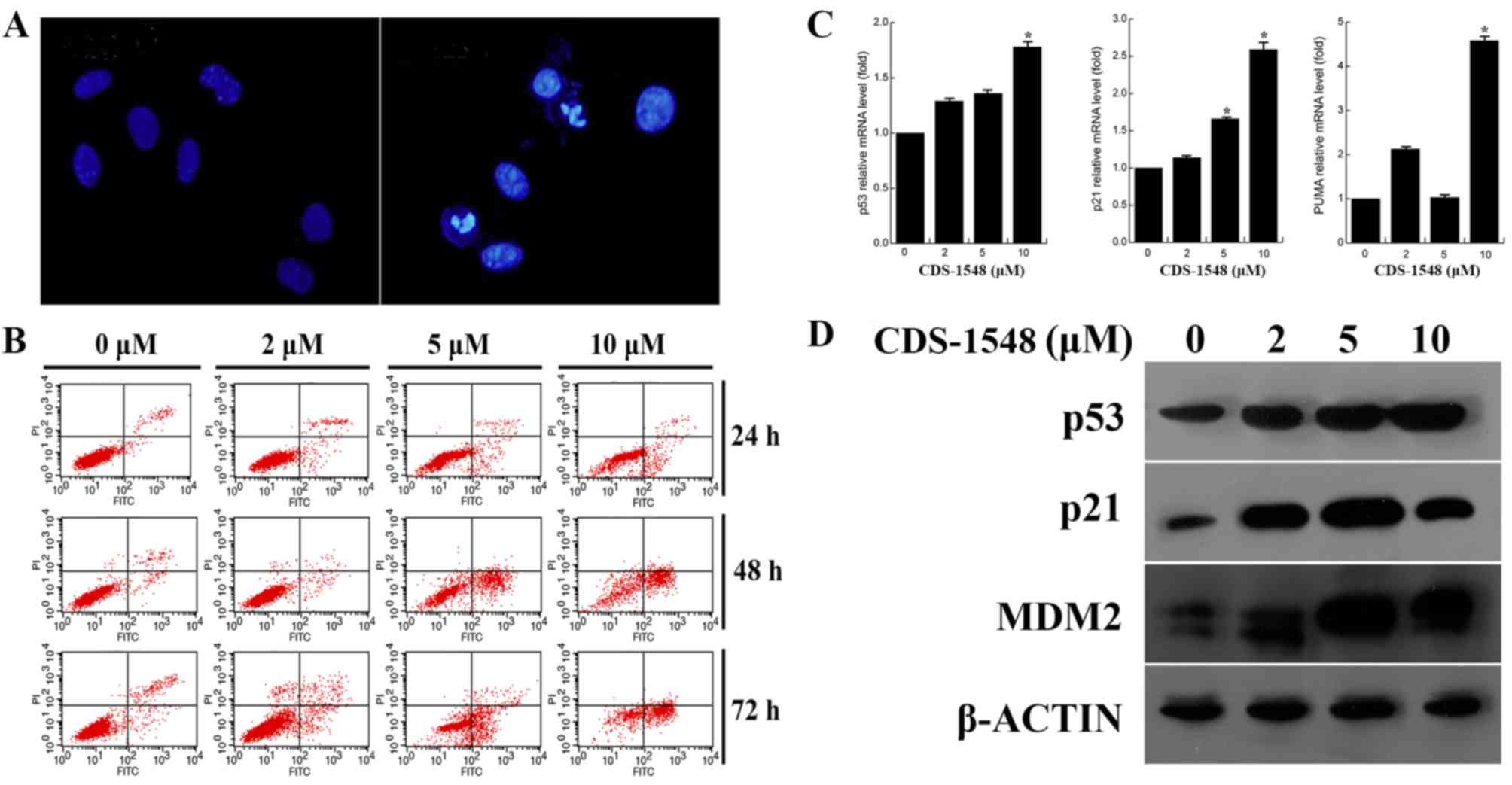 | Figure 3.CDS-1548 activates p53 and induces
apoptosis in HeLa cells. (A) Cells were treated with 5 µM CDS-1548
for 24 h, fixed and stained using DAPI. The morphology of the
nuclei was observed under a fluorescent microscope using a blue
filter. (B) HeLa cells were treated with 2, 5 or 10 µM CDS-1548 for
24, 48 or 72 h, and the apoptotic index was assessed using flow
cytometry. The administration of CDS-1548 markedly enhanced the
number of apoptotic cells, compared with the DMSO control. (C) HeLa
cells were treated with 2, 5 or 10 µM CDS-1548 for 12 h, and p53,
p21 and PUMA mRNA expression levels were determined using reverse
transcription-quantitative PCR. *P<0.05 vs. control. (D) HeLa
cells were treated with 2, 5 or 10 µM CDS-1548 for 24 h, and
protein expression levels of p53, p21 and Mdm2 was determined by
western blotting. CDS-1548, 2-[2-hydroxyl-1-(4-methoxy phenyl)
ethyl]-3-(4-benzyloxy phenyl) isoindolin-1-one; Mdm2, mouse double
minute 2 homolog; PUMA, p53 upregulated modulator of apoptosis. |
To confirm the effect of CDS-1548 on p53 activation,
the expression levels of p53, p-p53 and the downstream target genes
of p53 were determined. Fig. 3C and
D illustrate that the mRNA and protein expression levels of
p53, p21 and PUMA were increased in a dose-dependent manner in HeLa
cells, compared with control treated cells. Furthermore, the
upregulation of Mdm2 protein expression was observed. These results
demonstrated that p53 accumulated in HeLa cells following CDS-1548
treatment, resulting in an increase in the expression levels of
p21, Mdm2 and PUMA in a p53-associated manner.
CDS-1548 triggers apoptosis by
activating the mitochondria-mediated pathway
Previous investigations have demonstrated that
p53-mediated apoptotic cell death is associated with the intrinsic
mitochondrial pathway (23,24). The present study investigated the
expression of PARP and pro-caspase-3 (an effector caspase) to
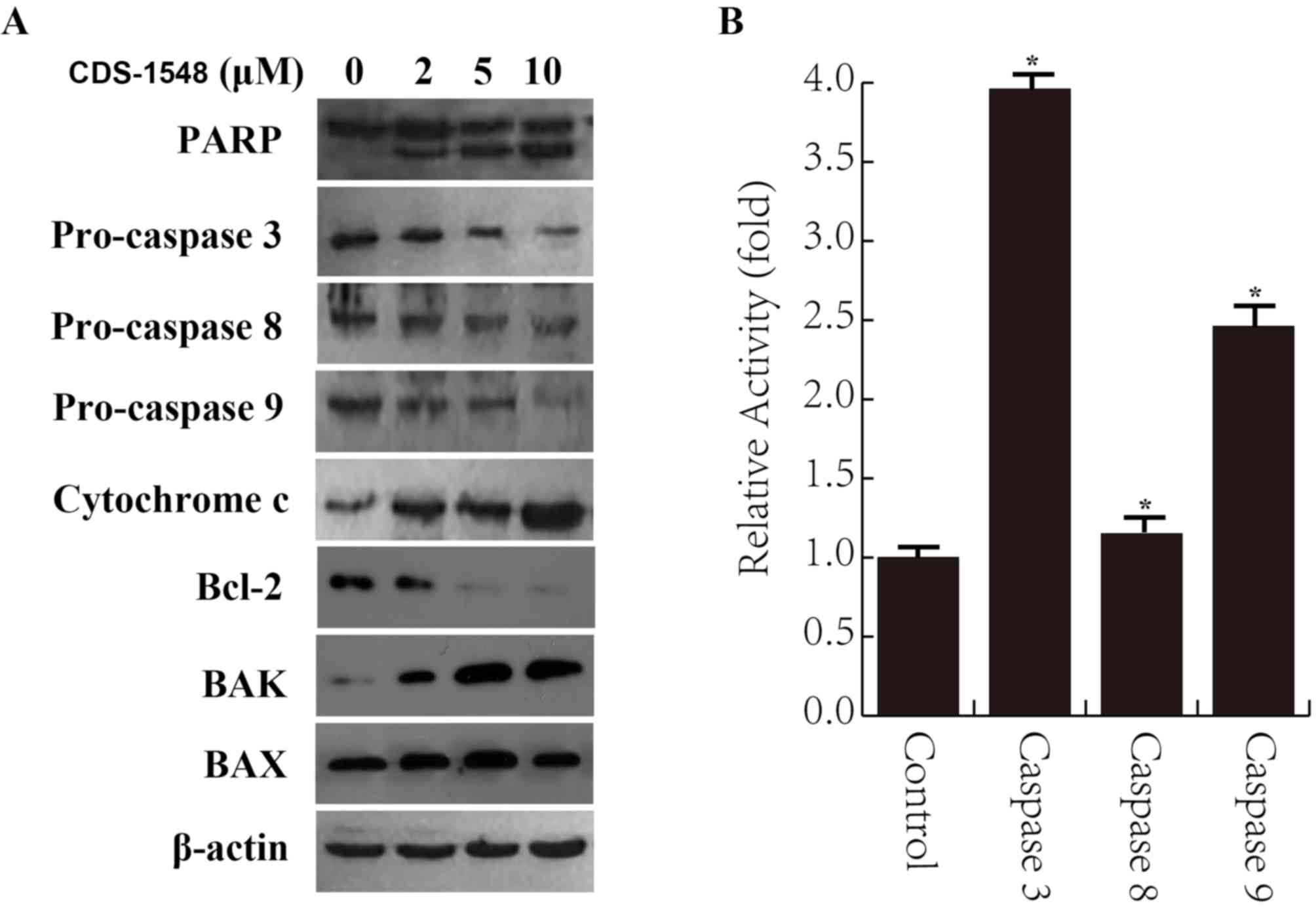
determine whether cell death was apoptotic in nature. As presented
in Fig. 4A, CDS-1548 resulted in the
cleavage of PARP and decreased the expression levels of pro-caspase
9 and 3 in a dose-dependent manner; the expression level of
pro-caspase 8 was not altered. These results suggested that
CDS-1548 induced apoptotic cell death. Furthermore, CDS-1548
treatment downregulated Bcl-2 expression levels and upregulated
those of BAK, but not BAX, resulting in the release of cytochrome c
into the cytosol of HeLa cells. The caspase activity assay further
indicated an increase in caspase 3 and 9 activity following
CDS-1548 treatment, while caspase 8 activity remained unaltered
(Fig. 4B). These results implied
that CDS-1548 treatment inhibited Bcl-2 expression, but promoted
BAK expression and cytochrome c release, inducing apoptosis via the
mitochondria-mediated signaling pathway.
Discussion
Cervical cancer is an important threat to female
health globally (25). Currently,
regular screening and vaccination are effective means of preventing
cervical cancer (26–29), though these methods are expensive and
may not be affordable in developing countries (30). Chemotherapeutic treatments, although
effective in a proportion of patients, are not completely curative
and are accompanied by severe side effects (31). As an alternative approach, small
molecule inhibitors that destroy cancerous cells with less toxic
effects on normal cells may be beneficial (32). In previous years, potent, selective
and efficacious small-molecule inhibitors have been successfully
developed, and a number of these compounds have been advanced into
clinical trials for the treatment of human cancers (33).
In the present study, the anticancer effect of
CDS-1548 on HeLa cells was evaluated. CDS-1548 is characterized by
two chiral centers, with 1H-isoindolin-1-one as a nuclear parent.
The results of the present study revealed that CDS-1548 treatment
induced the activation of p53 and elevated the mRNA and protein
expression levels of p53-targeted genes. Additionally, p21 may
suppress CDK1 and cyclin B1 expression by inhibiting either CDK
activity or impeding the formation of the CDK-cyclin B1 complex,
thereby resulting in cell cycle arrest at the G2/M phase
(34). Furthermore, the upregulated
expression level of PUMA, an important regulatory factor of
p53-mediated apoptosis (35), was
observed. It was hypothesized that CDS-1548-induced p53 activation
and elevated expression levels of p21 and PUMA served important
roles in the promotion of G2/M cell cycle arrest and
apoptosis. The inhibition of Bcl-2 expression, the upregulation of
BAK, the release of cytochrome c and the activation of caspase 3
and caspase 9 are considered to be markers of early-stage
apoptosis; however, the potential function of CDS-1548 as an
inhibitor of p53 requires further investigation by experimentation
with alternative cell lines and in vivo studies.
The proliferation of cancer cells depends on a
defect or dysfunction at the G1 checkpoint, and
subsequent entry into the S and G2 phases, where DNA
damage repair is initiated (36).
Therefore, cancer cells in the G2/M phase are sensitive
to the cytotoxic effects of chemotherapeutic drugs. More
importantly, the activation of p53 promotes apoptosis upon
G2/M phase arrest in response to DNA damage (37). The results of the present study
indicated that CDS-1548 induced cell cycle arrest at the
G2/M phase, which was accompanied by the downregulation
of cyclin B1, CDK1 and CDC25C. Additionally, the phosphorylation of
CHK1 and the upregulation of ATR expression (involved in the
DNA-damage response) were observed. Of note, there were no observed
changes in CHK2 protein expression level in HeLa cells.
Subsequently, it was confirmed that CDS-1548 treatment
significantly induced the phosphorylation of ATR and γ-H2AX in HeLa
cells, collectively suggesting that CDS-1548 induced p53-dependent
apoptosis by promoting G2/M phase arrest.
Further investigation is required to address the
potential molecular mechanisms involved in CDS-1548-mediated p53
activation. Specifically, the regulatory action of CDS-1548 on Mdm2
and p53 is an important feedback loop that requires further
clarification through in vitro and in vivo anti-tumor
activity assays.
In conclusion, the present study demonstrated that
the novel small-molecule inhibitor CDS-1548 possessed cytotoxicity
against cancer cells. Furthermore, CDS-1548 triggered apoptosis via
p53 accumulation and cell cycle arrest at the G2/M
phase, indicating that the use of small-molecule inhibitors that
target p53 may be a potential strategy for the treatment of
cervical cancer.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Special Foundation for Industry Innovation of Development and
Reform Commission of Jilin (grant no. 2018C049-4), the Foundation
of Education Department of Jilin Province (grant no. 2016133), the
Youth Foundation of Jilin Science and Technology Bureau (grant no.
20166026, 201750259) and the Major Programs of the Jilin Institute
of Chemical Technology (grant no. 20180101).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors contributions
YZ, XB and WS conceived and designed the
experiments. YZ, CH and YG performed the experiments. JR and MY
conducted the data analysis, and YZ produced the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Walboomers JM, Jacobs MV, Manos MM, Bosch
FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ and Muñoz N:
Human papillomavirus is a necessary cause of invasive cervical
cancer worldwide. J Pathol. 189:12–19. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bzhalava D, Eklund C and Dillner J:
International standardization and classification of human
papillomavirus types. Virology. 476:341–344. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kavanagh K, Pollock KG, Potts A, Love J,
Cuschieri K, Cubie H, Robertson C and Donaghy M: Introduction and
sustained high coverage of the HPV bivalent vaccine leads to a
reduction in prevalence of HPV 16/18 and closely related HPV types.
Br J Cancer. 110:2804–2811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schiffman M, Castle PE, Jeronimo J,
Rodriguez AC and Wacholder S: Human papillomavirus and cervical
cancer. Lancet. 370:890–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scheffner M: Ubiquitin, E6-AP, and their
role in p53 inactivation. Pharmacol Ther. 78:129–139. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oliner JD, Kinzler KW, Meltzer PS, George
DL and Vogelstein B: Amplification of a gene encoding a
p53-associated protein in human sarcomas. Nature. 358:80–83. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bates S and Vousden KH: Mechanisms of
p53-mediated apoptosis. Cell Mol Life Sci. 55:28–37. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Khoo KH, Verma CS and Lane DP: Drugging
the p53 pathway: Understanding the route to clinical efficacy. Nat
Rev Drug Discov. 13:217–236. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Soussi T and Wiman KG: Shaping genetic
alterations in human cancer: The p53 mutation paradigm. Cancer
Cell. 12:303–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oliner JD, Pietenpol JA, Thiagalingam S,
Gyuris J, Kinzler KW and Vogelstein B: Oncoprotein MDM2 conceals
the activation domain of tumour suppressor p53. Nature.
362:857–860. 1993. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ashkroft M and Vousden K: Regulation of
p53 stability. Oncogene. 18:7637–7643. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Michael D and Oren M: The p53-Mdm2 module
and the ubiquitin system. Semi Cancer Biol. 13:49–58. 2003.
View Article : Google Scholar
|
|
14
|
Freedman D, Wu L and Levine AJ: Functions
of the MDM2 oncoprotein. Cell Mol Life Sci. 5:96–107. 1999.
View Article : Google Scholar
|
|
15
|
Chène P, Fuchs J, Bohn J,
Garcı́a-Echeverrı́a C, Furet P and Fabbro D: A small synthetic
peptide, which inhibits the p53-hdm2 interaction, stimulates the
p53 pathway in tumour cell lines. J Mol Biol. 299:245–253. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang RW and Wang H: Antisense
oligonucleotide inhibitors of MDM2 oncogene expression. Methods Mol
Med. 85:205–222. 2003.PubMed/NCBI
|
|
17
|
Vassilev L, Vu BT, Graves B, Carvajal D,
Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et
al: In vivo activation of the p53 pathway by small-molecule
antagonists of MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu CM, Zheng LY, Pei YZ and Bai X:
Synthesis of novel 3-aryl isoindolinone derivatives. Chem Res Chin
Univ. 29:487–494. 2013. View Article : Google Scholar
|
|
19
|
Hu CM: Design, synthesis and anticancer
activity of novel 3-aryl isoindolinone derivatives: [D]. Changchun
Jilin Univ; 2012
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang M, Wu XC and Huang W: Raloxifene
upregulated mesangial cell MMP-2 activity via ER-β through
transcriptional regulation. Cell Biochem Biophys. 67:607–613. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li MO, Sarkisian MR, Mehal WZ, Rakic P and
Flavell RA: Phosphatidylserine receptor is required for clearance
of apoptotic cells. Science. 302:1560–1563. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin C, Knudson CM, Korsmeyer SJ and Van
Dyke T: Bax suppresses tumorigenesis and stimulates apoptosis in
vivo. Nature. 385:637–640. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miyashita T and Reed JC: Tumor suppressor
p53 is a direct transcriptional activator of the human bax gene.
Cell. 80:293–299. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dasari S, Wudayagiri R and Valluru L:
Cervical cancer: Biomarkers for diagnosis and treatment. Clin Chim
Acta. 445:7–11. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Canavan T and Doshi NR: Cervical cancer.
Am Fam Physician. 61:1369–1376. 2000.PubMed/NCBI
|
|
27
|
Luhn P, Walker J, Schiffman M, Zuna RE,
Dunn ST, Gold MA, Smith K, Mathews C, Allen RA, Zhang R, et al: The
role of co-factors in the progression from human papillomavirus
infection to cervical cancer. Gynecol Oncol. 128:265–270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arbyn M, Anttila A, Jordan J, Ronco G,
Schenck U, Segnan N, Wiener H, Herbert A and von Karsa L: European
guidelines for quality assurance in cervical cancer screening.
Second edition-summary document. Ann Oncol. 21:448–458. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Medeiros LR, Rosa DD, da Rosa MI, Bozzetti
MC and Zanini RR: Efficacy of human papillomavirus vaccines: A
systematic quantitative review. Int J Gynecol Cancer. 19:1166–1176.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cuzick J, Arbyn M, Sankaranarayanan R, Tsu
V, Ronco G, Mayrand MH, Dillner J and Meijer CJ: Overview of human
papillomavirus-based and other novel options for cervical cancer
screening in developed and developing countries. Vaccine. 26 (Suppl
10):k29–k41. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Segovia-Mendoza M, Jurado R, Mir R, Medina
LA, Prado-Garcia H and Garcia-Lopez P: Antihormonal agents as a
strategy to improve the effect of chemo-radiation in cervical
cancer: In vitro and in vivo study. BMC Cancer. 15:212015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cancer Genome Atlas Research Network:
Albert Einstein College of Medicine; Analytical Biological
Services; Barretos Cancer Hospital; Baylor College of Medicine;
Beckman Research Institute of City of Hope; Buck Institute for
Research on Aging; Canada's Michael Smith Genome Sciences Centre;
Harvard Medical School; Helen F. Graham Cancer Center &Research
Institute at Christiana Care Health Services, ; et al Integrated
genomic and molecular characterization of cervical cancer. Nature.
543:378–384. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao Y, Aguilar A, Bernard D and Wang SM:
Small-molecule inhibitors of the MDM2-p53 protein-protein
interaction (MDM2 Inhibitors) in clinical trials for cancer
treatment. J Med Chem. 58:1038–1052. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bucher N and Britten CD: G2 checkpoint
abrogation and checkpoint kinase-1 targeting in the treatment of
cancer. Br J Cancer. 98:523–528. 2008. View Article : Google Scholar : PubMed/NCBI
|