Introduction
The tyrosine kinase inhibitors (TKI) imatinib (IMA)
and nilotinib (NIL) have significantly improved the treatment of
chronic myeloid leukemia (CML), Philadelphia-chromosome positive
acute lymphoblastic leukemia (Ph+ ALL), gastro-intestinal stromal
tumors (GIST), and other malignant diseases. The molecular target
of IMA is the oncogenic tyrosine kinase (TK) BCR-ABL1, which is
necessary and sufficient to develop CML. IMA was licensed for the
treatment of adults in the year 2001 (1–7) and
without age restriction in 2003 (8).
However, leukemic cells were quickly found to develop resistance or
intolerance to IMA therapy (9),
which led to the development of NIL (Tasigna®, Novartis,
Basel, Switzerland), a second generation TKI. NIL is an
aminopyrimidine-derivative based on IMA, and has an increased
potency and selectivity for the oncogenic BCR-ABL1 kinase, leading
to a 20- to 50-fold higher inhibitory activity in IMA-sensitive
cells and 3 to 7 times higher inhibitory activity in IMA-resistant
cells (10). NIL was approved for
the treatment of adult patients insensitive to IMA in 2008
(11).
IMA and NIL exhibit several off-target effects on
non-oncogenic TKs, such as platelet derived growth factor receptor
(PDGF-R) and colony stimulating factor 1 receptor (CSF1R, c-FMS),
which are involved in the bone remodeling cycle (12). IMA impairs bone metabolism in CML
patients under prolonged treatment (13–15) as
demonstrated by growth retardation in up to 73% of pediatric CML
patients (12). Furthermore, CML
patients undergoing IMA treatment, frequently experience
hypophosphatemia, which is associated with reduction in serum
25-hydroxyvitamin D3 (calcidiol) and
1.25-dihydroxyvitamin D3 (calcitriol) (16). Calcitriol regulates a wide variety of
genes associated with bone formation, as demonstrated by the role
of calcidiol in promoting mineralization and reducing osteoblast
proliferation and osteoclast differentiation. In vitro
studies have demonstrated that IMA and NIL interact with the
vitamin D metabolism pathway by competitively inhibiting CYP27B1,
the enzyme involved in hydroxylating calcidiol to its active form
calcitriol (17,18). However, the underlying
pathophysiological mechanisms remain poorly defined.
Bone formation is carried out by osteoblasts
producing bone matrix and mineral crystals whereas bone resorption
is carried out by osteoclasts resorbing bone matrix through
proteolytic enzymes and acidic dissolution of the minerals. One of
the key pathways regulating osteoclastogenesis is the receptor
activator of nuclear factor κB ligand (RANKL) pathway. Its receptor
RANK is expressed on osteoclast precursors. Upon binding of RANKL,
osteoclast differentiation is initiated through the activation of
specific downstream signaling pathways.
Osteoprotegerin (OPG) functions as a decoy receptor
for RANKL and prevents binding of RANKL to its receptor RANK,
consequently serving as a negative regulator of osteoclastogenesis.
Thus, the RANKL/OPG ratio is an essential determinant of bone mass
and skeletal integrity (19).
Calcitriol and other hormones such as parathyroid hormone (PTH)
control the expression of RANKL. As TKIs are known to interfere
with vitamin D metabolism and suppress longitudinal growth in
children, we investigated whether TKIs exert direct effects on
osteoblasts and the RANKL cascade in vitro.
Materials and methods
Cell culture
Human osteosarcoma cells (SaOS-2; Leibniz Institute
DSMZ-German Collection of Microorganisms and Cell Cultures,
Braunschweig, Germany) were seeded at a density of 1×105
cells/cm2 and grown in a humidified 5% CO2
atmosphere at 37°C. For proliferation McCoy's 5A medium without
phenol red (BioConcept, Allschwil, Switzerland) supplemented with
15% fetal bovine calf serum and 1% Penicillin and Streptomycin was
used. Ten days after seeding, osteogenic differentiation was
induced by growing the cells in Alpha Minimal Essential Medium
supplemented with 15% fetal bovine calf serum, 10 mM
β-glycerophosphate, 2 mM L-glutamine, 300 µM L-ascorbic acid
2-phospate (all from Sigma Aldrich, Germany) and 1% Penicillin and
Streptomycin.
For hemacolor and immunofluorescence staining cells
were grown in 12-well-plates on cover slips (ø 13 mm), while for
RNA analyses, cells were grown in culture flasks for up to 25 days.
Medium was changed 3 times a week. IMA or NIL (Novartis, Basel,
Switzerland) was added to the cell culture medium at a
concentration of 1 µM, respectively. After 5, 10, 15 or 25 days of
incubation, mineralization capacity and the RANKL cascade were
assessed.
Cell staining and microscopy
Hemacolor staining (Biomed, Oberschleißheim,
Germany) was performed according to the manufacturer's
instructions. After staining, samples were mounted with Celltexx
(Engelbrecht Medizin und Labortechnik GmbH, Edermünde, Germany).
Digital images were obtained using an Axio Imager A1 (Zeiss, Jena,
Germany).
Immunofluorescence staining and
confocal microscopy
Cells were washed with PBS, fixed with 4%
paraformaldehyde (Roth, Karlsruhe, Germany), and stored in PBS at
4°C. All washing steps were performed twice for 10 min in PBS-T
[PBS supplemented with 0.005% Tween (Serva, Heidelberg, Germany)].
Cells were stained with the following primary antibodies: RANKL
mouse IgG2B, RANK mouse IgG2A and OPG mouse IgG1 (R&D,
Minneapolis, USA). The respective secondary antibodies Alexa
Flour® 546 Goat anti-mouse IgG2b, Alexa
Flour® 488 Goat anti-mouse IgG2A and Alexa
Fluor® 647 Goat ant-mouse IgG1 (Thermo Fisher
Scientific, Waltham, USA) were applied for detection. The primary
antibodies [dissolved in PBS supplemented with 0.001% Tween and 1%
BSA (Sigma-Aldrich, Steinheim, Germany)] were applied at 8 µg/ml
over night at 4°C. After washing the secondary antibody (diluted
1:200) was applied for 1 h. After another washing step, DAPI
(Sigma-Aldrich, Steinheim, Germany) staining was prepared at 1
µg/ml in PBS and applied for 5 min in the dark at room temperature
followed by a 1 min PBS wash. Finally, the samples were mounted
with Fluorescence Mounting Medium (DAKO, Jena, Germany).
Fluorescence microscopy was performed using a
DeltaVision microscope system composed of softWoRx 5.5.1 software
(Applied Precision, United Kingdom), Olympus IX71 microscope
(Olympus, Tokyo, Japan), Cool SnapHQ camera (Photometrics, Tucson,
Arizona) and Plan Apo N 60×1.4 numerical oil objective (Nikon,
Tokyo, Japan). Cell boundaries were observed by thresholding the
bright-field image and overlaying it with the fluorescence image. Z
stacks with 10 focal planes were collected at 7 regions of interest
for each sample and each channel (DAPI, FITC, TRITC, CY5). Analysis
of images and measuring of fluorescence intensity were performed
manually using Fuji software (FujiFilm, Tokyo, Japan) (20). Z projection was done using average
intensity projection.
RNA isolation and quantification
RNA was extracted using the RNeasy® Plus
Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's
instructions. RNA was dissolved in 20 µl RNase-free water
(Invitrogen, Carlsbad, USA) and concentrations were measured by an
Infinite® 200 Pro plate reader (Tecan, Männedorf,
Switzerland). Samples with a 260/280 ratio of ≥2 were classified as
pure RNA and used for further investigations.
Reverse transcription of 0.5 µg template RNA with
random hexamer primers was carried out using iScript cDNA Synthesis
kit (Bio-Rad Lab, Munich, Germany) according to the manufacturer's
instructions. Reverse transcription-quantitative PCR was performed
in 20 µl reaction volumes containing 0.5 ng/µl of diluted cDNA, 10
µl of iQ SYBR Green Supermix (Bio-Rad Lab, Munic, Germany), 0.6 µl
of 10 pmol/µl forward primer, and 0.6 µl of 10 pmol/µl reverse
primer. A Stratagene MX300P cycler (SABiosciences, Qiagen, Hilden,
Germany) was used with the following profile: 95°C for 3 min
followed by 40 cycles of 95°C for 15 sec, 60°C for 30 sec, and 30
sec at 72°C with one final cycle of 55°C for 1 min and 30 sec at
95°C. The results were calculated using the ΔΔCq method and
presented as fold increase relative to GAPDH expression.
Primers were purchased from MWG Biotech AG
(Ebersberg, Germany). The sequences were: RANKL, forward:
5′-GCCAGTGGGAGATGTTAG-3′, reverse: 5′-TTAGCTGCAAGTTTTCCC-3′
(21); RANK, forward:
5′-AGGGAAAGCACTCACAGCTAAT-3′, reverse: 5′-ACATGCTCCCTGCTGACC-3′
(22); OPG, forward:
5′-GCTAACCTCACCTTCGAG-3′, reverse: 5′-TGATTGGACCTGGTTACC-3′
(21); ALP, forward:
5′-CAACCCTGGGGAGGAGAC-3′, reverse: 5′-GCATTGGTGTTGTACGTCTTG-3′;
OSX, forward: 5′-CAAAGAAGCCGTACTCTGTGG-3′, reverse:
5′-TGAAAGGAGCCCATTAGTGC-3′; OCN, forward:
5′-TGAGAGCCCTCACACTCCTC-3′, reverse: 5′-ACCTTTGCTGGACTCTGCAC-3′;
Wnt1, forward: 5′-CGCTGGAACTGTCCCACT-3′, reverse:
5′-AACGCCGTTTCTCGACAG-3′; Wnt10b, forward:
5′-TTCTCTCGGGATTTCTTGGA-3′, reverse: 5′-TCCGCTTCAGGTTTTCAGTT-3′;
GAPDH, forward: 5′-ACAGTCCATGCCATCACTGCC-3′, reverse:
5′-GCCTGCTTCACCACCTTCTTG-3′ (23).
Statistical analysis
Statistical analyses at defined time points of
incubation were performed using one-way analysis of variance and
multiple comparisons were conducted using a post-hoc Bonferroni
adjustment to evaluate the effects of IMA or NILtreated samples
compared to untreated controls (GraphPad Prism v.6.0 software;
GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as
means ± standard deviation (SD). In all cases, P<0.05 was
considered to indicate a statistically significant difference.
Mineralized matrix and immunofluorescence images
quantified using ImageJ v.1.49 software (National Institute of
Health, Bethesda, USA).
Results
TKIs inhibit osteoblast
mineralization
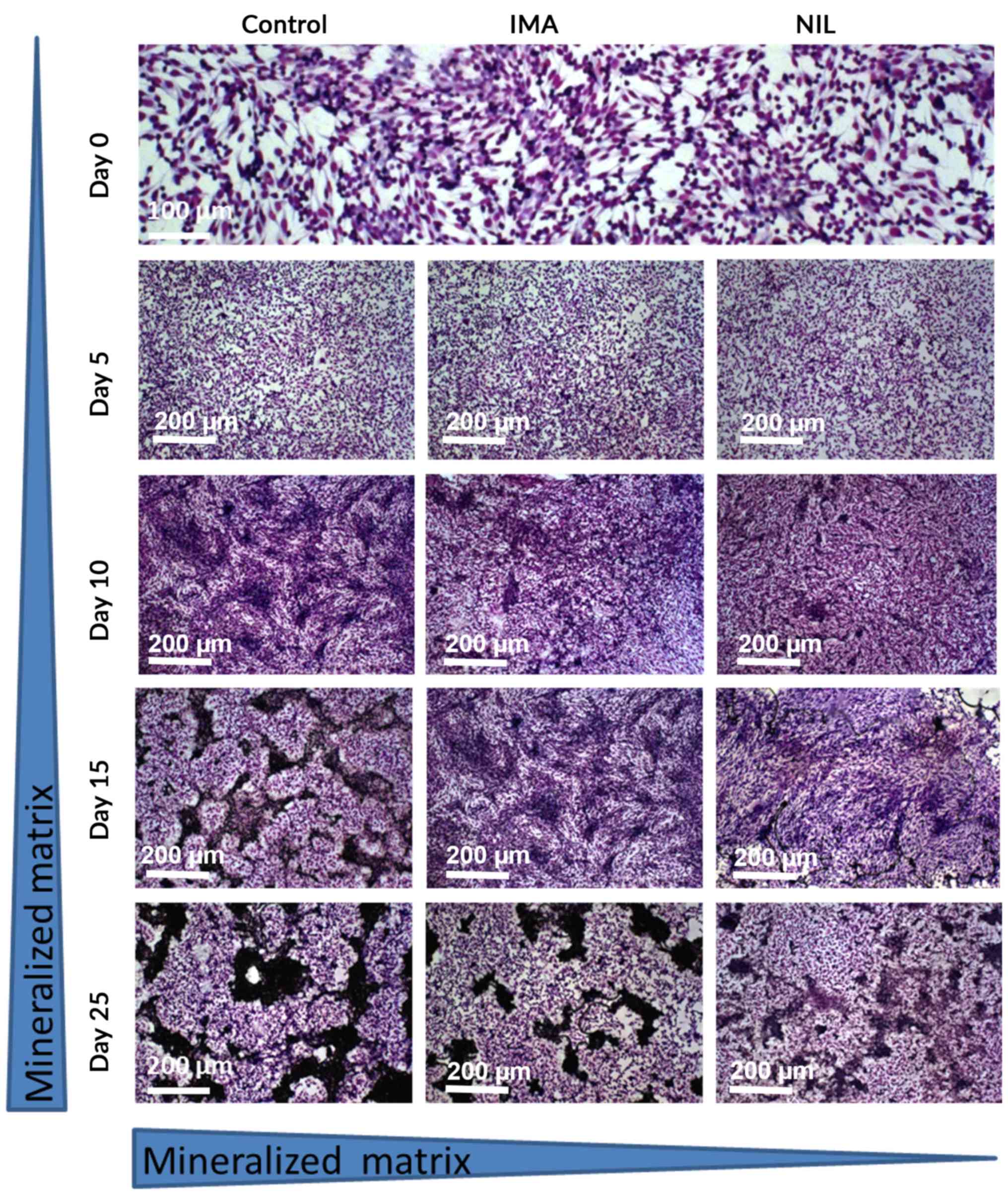
SaOS-2 cells were cultured for up to 25 days in the
presence of TKI [clinically effective concentration: 1 µM (24,25)] and
matrix mineralization was assessed at 5 day intervals. At day 25,
cultures treated with IMA exhibited 40% (a.u. 0.477±0.045; Fig. 1) less mineralization than untreated
controls (a.u. 0.788±0.053; Fig. 1).
This inhibitory effect was more pronounced with NIL, which reduced
bone matrix by 90% (a.u. 0.081±0.01; Fig. 1).
TKIs suppress the expression of
osteogenic markers and Wnt molecules in SaOS-2 cells
To study the mechanism underlying the reduced
mineralization following TKI treatment, we investigated specific
gene markers of osteoblastogenesis in SaOS-2 cells. After 25 days
of IMA or NIL treatment, osterix (OSX) expression was significantly
reduced by 3.2- and 4.9-fold, respectively compared to the control
(Fig. 2). IMA and NIL also reduced
the gene expression of ALP by 2.3- and 3.4-fold, respectively,
although only the latter was significant (Fig. 2). We also investigated the effects of
TKIs on Wnt1 and Wnt10b expression, two members of the Wnt
signaling pathway, which play a key role in maintaining bone
homeostasis (26). NIL had a more
pronounced effect on Wnt expression than IMA. In the presence of
IMA, Wnt1 and Wnt10b mRNA levels were reduced by 1.8- and 0.6-fold,
respectively, although these differences were not significant. By
comparison, NIL reduced Wnt1 and Wnt10b expression by
3.6-(P<0.05) and 1.2-fold, respectively.
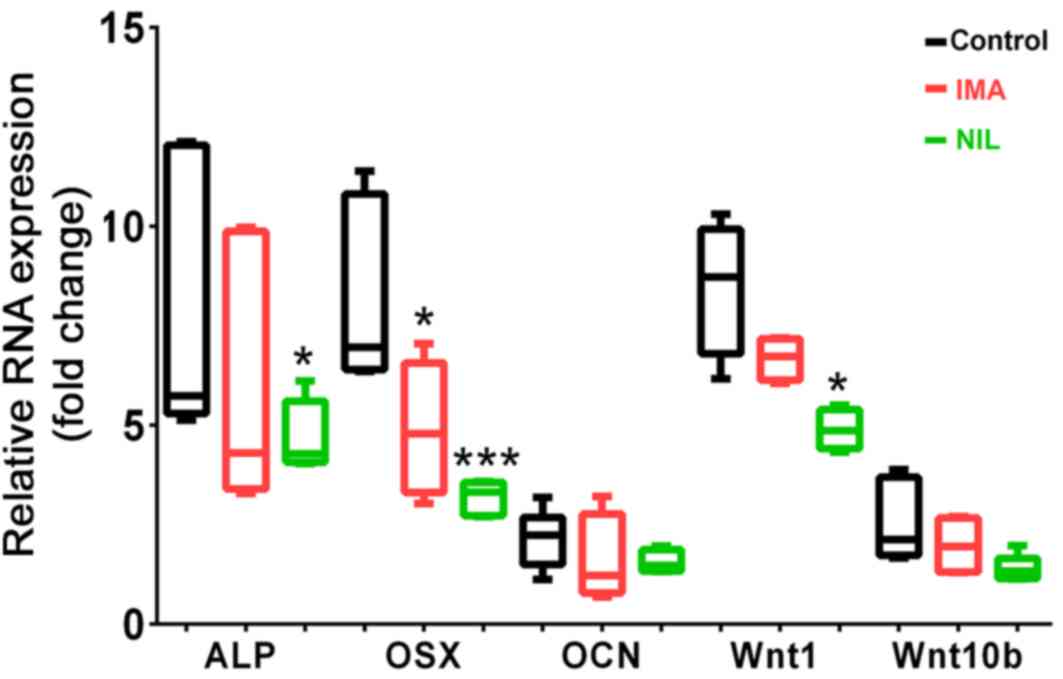 | Figure 2.Gene expression levels of osteogenic
markers in SaOS-2 cells treated for 25 days with IMA (1 µM, red
bars) and NIL (1 µM, green bars) in comparison with untreated
control (black bars). Reverse transcription-quantitative PCR
results of ALP, OSX, OCN, Wnt1 and Wnt10b were normalized to GAPDH
(n=4-6). *P<0.05 and ***P<0.001, vs. control. IMA, imatinib;
NIL, nilotinib; ALP, alkaline phosphatase; OSX, osterix; OCN,
osteocalcin. |
TKIs increase RANKL/OPG ratio in
osteoblasts
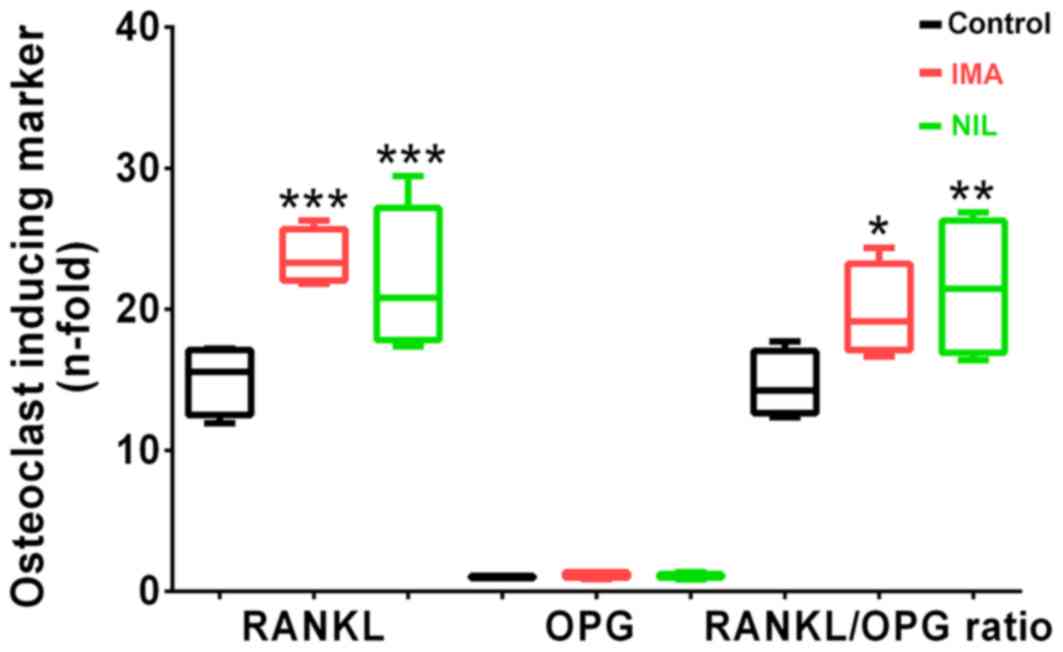
Finally, we investigated the expression levels of
RANKL and OPG, which are involved in the coupling of bone formation
and resorption. After 5 days of TKI treatment, we found no
differences in RANKL or OPG gene expression levels. However, after
25 days, IMA and NIL significantly increased RANKL expression by
160 and 150%, respectively, whereas OPG expression remained
unchanged compared to controls. This consequently resulted in an
increased RANKL/OPG ratio in osteoblasts treated with IMA (+160%)
and NIL (+150%) (Fig. 3). These
findings were verified by immunofluorescence staining of SaOS-2
cells reflected by medium fluorescence intensity (MFI) data,
showing an increased fluorescence signal for RANKL under IMA
(control: MFI/cell 0.71±0,06; IMA: MFI/cell 1.16±0.17) and NIL
treatment (NIL: MFI/cell 2.48±0.15) as well as a decreased
fluorescence signal for OPG under IMA (control: MFI/cell 75.6±6.48;
IMA: MFI/cell 35.4±4.82) and Nil (NIL: MFI/cell 37.4±7.16)
treatment (Fig. 4).
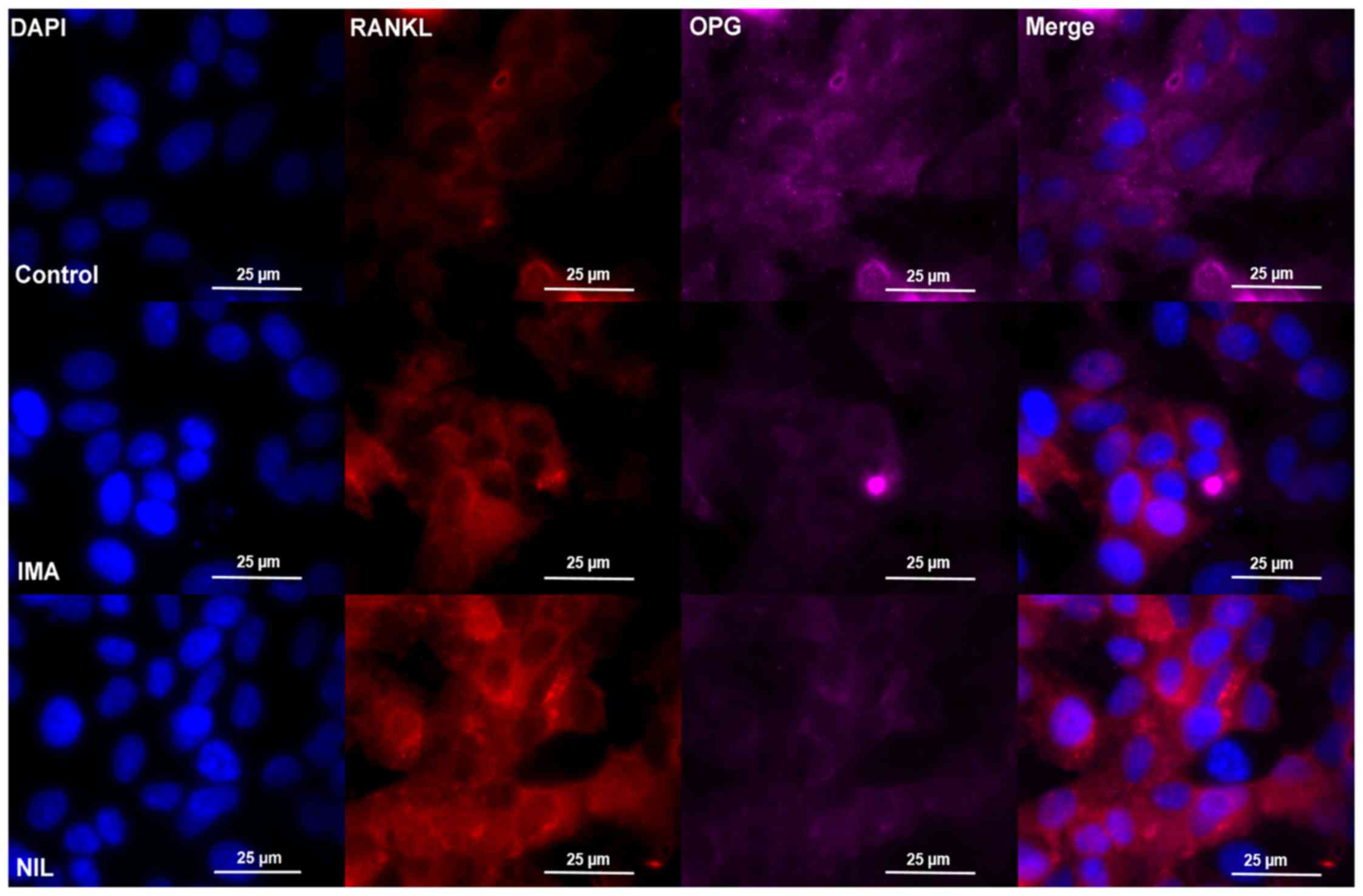 | Figure 4.Immunofluorescence staining of SaOS-2
cells after incubation without TKI (control, upper row), IMA (1 µM,
middle row) or NIL (1 µM, lower row) for 25 days, respectively.
Cells were stained with RANKL (red), OPG (purple), and DAPI (blue).
One representative image is shown per treatment. Each experiment
was performed three times. IMA, imatinib; NIL, nilotinib; RANKL,
receptor activator of nuclear factor κB ligand; OPG,
osteoprotegerin. |
Discussion
IMA and NIL were designed to inhibit BCR-ABL1, and
emerged as powerful tools in the management of CML. However, the
ATP binding domain in BCR-ABL1 that is targeted by IMA and NIL is
structurally similar to other TKs, and consequently results in
off-target effects. In CML patients receiving long-term IMA
treatment, this has been reported to result in impaired skeletal
growth. This is problematic because patients with CML need to
remain on TKI therapy for longer durations to effectively eliminate
all leukemic cell clones (27).
Thus, for long-term safety, it is important to understand the
effects of TKIs on bone remodeling.
In the current study, we report that TKI treatment
decreased mineralization in osteoblastic cells, which was
accompanied by decreased ALP and OSX expression. The effects on
mineralization became apparent after 15 days of osteogenic
induction in the presence of TKIs, but the maximal inhibitory
effect was observed at day 25. Importantly, NIL inhibited
osteoblastogenesis more potently than IMA. Our findings are
consistent with a prior report which showed that 5 weeks of IMA
treatment was associated with a significant reduction in bone
mineral density in juvenile growing rats (24). We also found that both TKIs increased
RANKL expression and decreased OPG levels, thereby resulting in an
increased RANKL/OPG ratio. This increase in osteoclastogenic
potential represents another mechanism by which TKIs may interfere
with bone remodeling processes, which is consistent with prior
reports by Tibullo et al (28,29). In
support of these findings at the clinical level, several groups
have reported adverse effects of TKIs. A concentration of 1 µM for
both drugs were defined as clinically effective due to the level
plasma concentration of IMA and NIL in human patients (25).
Within a few months of starting IMA treatment, adult
CML patients displayed alternations in mineral metabolism (13), reduced bone formation and bone mass
(30), and reduced OCN level
compared to healthy controls (13).
However, the effects of TKIs on bone remain
unresolved because there are several studies that have reported
contradictory findings. In vitro, TKIs were reported to
increase bone mineralization (28,29), OPG
levels (31), and the expression of
osteoblast specific genes (15,28,32). In
adult and pediatric patients, reduced as well as elevated
osteocalcin levels has been described (13,16,33,34)
during TKI treatment. Considering these differences, it is
pertinent to evaluate the design and context of these studies.
There is substantial heterogeneity in choice of in vitro
model system. Differences in cell lines (e.g., human vs. murine;
primary cell vs. cell line; malignant vs. non-malignant) and assays
may contribute to these discrepancies. Furthermore, the
interpretation of clinical studies is complicated by inherent
differences between adult and pediatric patients in which bone
turnover varies substantially. In adult CML patients, TKI appears
to promote bone formation, while in pediatric CML patients, TKI
treatment decreases bone formation through growth retardation
(12,35,36).
We also found that the pro-osteogenic Wnt signaling
pathway were down-regulated, specifically Wnt1. Wnt signaling is a
key regulator of osteoblast function and bone homeostasis. Prior
studies have shown that IMA reduces β-catenin expression, the main
transcription factor for canonical Wnt signaling (37). Moreover, co-treatment of IMA with Wnt
inhibitors potentiated the anti-leukemic effects of IMA (38). Thus, while suppression of Wnt
signaling has beneficial effects on cancer progression, bone health
may be compromised in the long-term.
Taken together, our study demonstrated that TKIs IMA
and NIL negatively regulate osteoblast function in vitro.
Moreover, TKI treatment was associated with an elevated RANKL/OPG
ratio thereby, providing a pro-osteoclastogenic environment.
Considering the previously described impact of TKIs on vitamin D
metabolism (17,18), which may further impair bone
metabolism, patients on long-term TKI treatment should have their
bone healthy regularly monitored.
Acknowledgements
The authors of the present study would like to thank
Dr. Ute Hempel (Institute of Physiological Chemistry, Medical
Faculty Carl Gustav Carus, Technische Universität Dresden, Dresden,
Germany) for supplying SaOS-2 cells as well as the helpful
suggestions and handling advice. The authors would also like to
thank Novartis International AG (Basel, Switzerland) for supplying
TKIs and for their financial support.
Funding
The present study was supported by Novartis
International AG (grant no. HTAS-079).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors contributions
AB, LMK, MS, SB and MR conceived and designed the
study. LMK managed the data and the overall study, verified the
results and prepared the visual aspects of the work. JTT and LMK
analyzed the data. AB and LMK acquired the funding. LMK, AW and SIK
performed the experiments. JTT, MS, SIK and MR provided the
reagents, materials, computing resources and analytical tools. AB,
MS, MR and SB supervised the study. LMK and JTT wrote the
manuscript. JTT, LMK, MS and MR critically analyzed the manuscript
for important intellectual content. All authors have read and
approved the final version.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The two TKIs used in the present study, IMA and NIL,
were obtained from Novartis International AG (Basel, Switzerland)
who also financially supported the study.
References
|
1
|
Champagne MA, Capdeville R, Krailo M, Qu
W, Peng B, Rosamilia M, Therrien M, Zoellner U, Blaney SM and
Bernstein M; Children's Oncology Group phase 1 study, : Imatinib
mesylate (STI571) for treatment of children with Philadelphia
chromosome-positive leukemia: Results from a children's oncology
group phase 1 study. Blood. 104:2655–2660. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Druker BJ, Tamura S, Buchdunger E, Ohno S,
Segal GM, Fanning S, Zimmermann J and Lydon NB: Effects of a
selective inhibitor of the Abl tyrosine kinase on the growth of
Bcr-Abl positive cells. Nat Med. 2:561–566. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Druker BJ, Talpaz M, Resta DJ, Peng B,
Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R,
Ohno-Jones S and Sawyers CL: Efficacy and safety of a specific
inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid
leukemia. N Engl J Med. 344:1031–1037. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Grigg A and Hughes T: Role of allogeneic
stem cell transplantation for adult chronic myeloid leukemia in the
imatinib era. Biol Blood Marrow Transplant. 12:795–807. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Millot F, Guilhot J, Nelken B, Leblanc T,
De Bont ES, Bekassy AN, Gadner H, Sufliarska S, Stary J,
Gschaidmeier H, et al: Imatinib mesylate is effective in children
with chronic myelogenous leukemia in late chronic and advanced
phase and in relapse after stem cell transplantation. Leukemia.
20:187–192. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Roy L, Guilhot J, Krahnke T,
Guerci-Bresler A, Druker BJ, Larson RA, O'Brien S, So C, Massimini
G and Guilhot F: Survival advantage from imatinib compared with the
combination interferon-alpha plus cytarabine in chronic-phase
chronic myelogenous leukemia: Historical comparison between two
phase 3 trials. Blood. 108:1478–1484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iijima R, Byrne RA, Dibra A, Ndrepepa G,
Spaulding C, Laarman GJ, Menichelli M, Valgimigli M, Di Lorenzo E,
Kaiser C, et al: Drug-eluting stents versus bare-metal stents in
diabetic patients with ST-segment elevation acute myocardial
infarction: A pooled analysis of individual patient data from seven
randomized trials. Rev Esp Cardiol. 62:354–364. 2009.(In English,
Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hobernicht SL, Schweiger B, Zeitler P,
Wang M and Hunger SP: Acquired growth hormone deficiency in a girl
with chronic myelogenous leukemia treated with tyrosine kinase
inhibitor therapy. Pediatr Blood Cancer. 56:671–673. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deguchi Y, Kimura S, Ashihara E, Niwa T,
Hodohara K, Fujiyama Y and Maekawa T: Comparison of imatinib,
dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines.
Leuk Res. 32:980–983. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kantarjian H, Giles F, Wunderle L, Bhalla
K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W,
et al: Nilotinib in imatinib-resistant CML and Philadelphia
chromosome-positive ALL. N Engl J Med. 354:2542–2551. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jabbour E, El AS, Cortes J and Kantarjian
H: Nilotinib: A novel Bcr-Abl tyrosine kinase inhibitor for the
treatment of leukemias. Expert Opin Investig Drugs. 17:1127–1136.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shima H, Tokuyama M, Tanizawa A, Tono C,
Hamamoto K, Muramatsu H, Watanabe A, Hotta N, Ito M, Kurosawa H, et
al: Distinct impact of imatinib on growth at prepubertal and
pubertal ages of children with chronic myeloid leukemia. J Pediatr.
159:676–681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Berman E, Nicolaides M, Maki RG, Fleisher
M, Chanel S, Scheu K, Wilson BA, Heller G and Sauter NP: Altered
bone and mineral metabolism in patients receiving imatinib
mesylate. N Engl J Med. 354:2006–2113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fierro F, Illmer T, Jing D, Schleyer E,
Ehninger G, Boxberger S and Bornhäuser M: Inhibition of
platelet-derived growth factor receptorbeta by imatinib mesylate
suppresses proliferation and alters differentiation of human
mesenchymal stem cells in vitro. Cell Prolif. 40:355–366. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fitter S, Dewar AL, Kostakis P, To LB,
Hughes TP, Roberts MM, Lynch K, Vernon-Roberts B and Zannettino AC:
Long-term imatinib therapy promotes bone formation in CML patients.
Blood. 111:2538–2547. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jaeger BA, Tauer JT, Ulmer A, Kuhlisch E,
Roth HJ and Suttorp M: Changes in bone metabolic parameters in
children with chronic myeloid leukemia on imatinib treatment. Med
Sci Monit. 18:CR721–CR728. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kroschwald L, Suttorp M, Tauer JT,
Zimmermann N, Gunther C and Bauer A: Offtarget effect of imatinib
and nilotinib on human vitamin D3 metabolism. Mol Med Rep.
17:1382–1388. 2018.PubMed/NCBI
|
|
18
|
Mehlig LM, Garve C, Tauer JT, Suttorp M
and Bauer A: Inhibitory effects of imatinib on vitamin D(3)
synthesis in human keratinocytes. Mol Med Rep. 11:3143–3147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Boyce BF and Xing L: Biology of RANK,
RANKL, and osteoprotegerin. Arthritis Res Ther. 9 (Suppl 1):S12007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mori K, Le GB, Berreur M, Riet A, Moreau
A, Blanchard F, Chevalier C, Guisle-Marsollier I, Léger J, Guicheux
J, et al: Human osteosarcoma cells express functional receptor
activator of nuclear factor-kappa B. J Pathol. 211:555–562. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiong MY, Liu LQ, Liu SQ, Liu ZH and Gao
HF: Effects of osteoprotegerin, RANK and RANKL on bone destruction
and collapse in avascular necrosis femoral head. Am J Transl Res.
8:3133–3140. 2016.PubMed/NCBI
|
|
23
|
Stephens AS, Stephens SR and Morrison NA:
Internal control genes for quantitative RT-PCR expression analysis
in mouse osteoblasts, osteoclasts and macrophages. BMC Res Notes.
4:4102011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tauer JT, Hofbauer LC, Jung R, Gerdes S,
Glauche I, Erben RG and Suttorp M: Impact of long-term exposure to
the tyrosine kinase inhibitor imatinib on the skeleton of growing
rats. PLoS One. 10:e01311922015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gandia P, Arellano C, Lafont T, Huguet F,
Malard L and Chatelut E: Should therapeutic drug monitoring of the
unbound fraction of imatinib and its main active metabolite
N-desmethyl-imatinib be developed? Cancer Chemother Pharmacol.
71:531–536. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Baron R and Kneissel M: WNT signaling in
bone homeostasis and disease: From human mutations to treatments.
Nat Med. 19:179–192. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suttorp M, Schulze P, Glauche I, Gohring
G, von Neuhoff N, Metzler M, Sedlacek P, de Bont ESJM, Balduzzi A,
Lausen B, et al: Front-line imatinib treatment in children and
adolescents with chronic myeloid leukemia: Results from a phase III
trial. Leukemia. 32:1657–1669. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tibullo D, Giallongo C, La CP, Berretta S,
Stagno F, Chiarenza A, Conticello C, Palumbo GA and Di Raimondo F:
Effects of imatinib mesylate in osteoblastogenesis. Exp Hematol.
37:461–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tibullo D, Barbagallo I, Giallongo C, La
CP, Branca A, Conticello C, Stagno F, Chiarenza A, Palumbo GA and
Di Raimondo F: Effects of second-generation tyrosine kinase
inhibitors towards osteogenic differentiation of human mesenchymal
cells of healthy donors. Hematol Oncol. 30:27–33. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
O'Sullivan S, Naot D, Callon KE, Watson M,
Gamble GD, Ladefoged M, Karsdal MA, Browett P, Cornish J and Grey
A: Imatinib mesylate does not increase bone volume in vivo. Calcif
Tissue Int. 88:16–22. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Sullivan S, Tay ML, Lin JM, Bava U,
Callon K, Cornish J, Naot D and Grey A: Tyrosine kinase inhibitors
regulate OPG through inhibition of PDGFRβ. PLoS One.
11:e01647272016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
O'Sullivan S, Naot D, Callon K, Porteous
F, Horne A, Wattie D, Watson M, Cornish J, Browett P and Grey A:
Imatinib promotes osteoblast differentiation by inhibiting PDGFR
signaling and inhibits osteoclastogenesis by both direct and
stromal cell-dependent mechanisms. J Bone Miner Res. 22:1679–1689.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
O'Sullivan S, Horne A, Wattie D, Porteous
F, Callon K, Gamble G, Ebeling P, Browett P and Grey A: Decreased
bone turnover despite persistent secondary hyperparathyroidism
during prolonged treatment with imatinib. J Clin Endocrinol Metab.
94:1131–1136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vandyke K, Fitter S, Dewar AL, Hughes TP
and Zannettino AC: Dysregulation of bone remodeling by imatinib
mesylate. Blood. 115:766–774. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Narayanan KR, Bansal D, Walia R, Sachdeva
N, Bhansali A, Varma N and Marwaha RK: Growth failure in children
with chronic myeloid leukemia receiving imatinib is due to
disruption of GH/IGF-1 axis. Pediatr Blood Cancer. 60:1148–1153.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Millot F, Guilhot J, Baruchel A, Petit A,
Leblanc T, Bertrand Y, Mazingue F, Lutz P, Vérité C, Berthou C, et
al: Growth deceleration in children treated with imatinib for
chronic myeloid leukaemia. Eur J Cancer. 50:3206–3211. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhou L, An N, Haydon RC, Zhou Q, Cheng H,
Peng Y, Jiang W, Luu HH, Vanichakarn P, Szatkowski JP, et al:
Tyrosine kinase inhibitor STI-571/Gleevec down-regulates the
beta-catenin signaling activity. Cancer Lett. 193:161–170. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suknuntha K, Thita T, Togarrati PP,
Ratanachamnong P, Wongtrakoongate P, Srihirun S, Slukvin I and
Hongeng S: Wnt signaling inhibitor FH535 selectively inhibits cell
proliferation and potentiates imatinib-induced apoptosis in myeloid
leukemia cell lines. Int J Hematol. 105:196–205. 2017. View Article : Google Scholar : PubMed/NCBI
|