Introduction
Cancer is a disease that poses a threat to human
health worldwide, and it has become a major public health concern
in China (1,2). Typically, cancer can be caused by the
accumulation of molecular alterations in genes that control cell
survival, growth, proliferation and differentiation within the
nascent tumor (3). Currently, the
molecular profile of cancer can be effectively assessed using gene
sequencing technologies and advanced analytical approaches
(4). Advances in technology provide
unprecedented speed and resolution to find causative mutations in
the patient's genome that underlie cancer development and
progression, ranging from point mutations to chromosomal
translocations (4,5). In particular, there are somatic
alterations that are unique to tumor cell genomes and specific
inherited or ‘germline’ genomic alterations that are known to
confer increased susceptibility to cancer development (4,5).
Concurrent cancers are rare, and can increase the economic burden
on a family and society. However, the molecular mechanisms
underlying the development and progression of concurrent cancers
remain unclear.
Next generation sequencing (NGS), also known as
high-throughput parallel sequencing technology, provides an
unbiased way to examine the molecular pathogenesis of diseases and
expands the impact of genomic analyses in biomedical research
(6,7). Furthermore, NGS has been widely used in
scientific research, and for the clinical diagnosis and treatment
of cancer (8–11). The exome represents only ~2% of the
human genome, but contains ~85% of known disease-associated
variants, making whole exome sequencing (WES) a significant
alternative to whole genome sequencing (WGS) (12,13).
Compared with WGS, WES has significant advantages, including
reduced costs, faster data analysis and easier data management
(13). Therefore, investigating the
molecular alterations in concurrent cancers using WES technology is
a more cost-effective approach.
In the present study, WES technology was used to
identify causative variants in a Chinese family in which two
members were diagnosed with concurrent cancer. Following filtering
of the raw data and exhaustive annotations, it was identified that
NADH:ubiquinone oxidoreductase core subunit S7 (NDUFS7) was
a candidate gene with somatic mutations (g.1391151G>A and
g.1393289G>C), and a subset of 16 genes were candidate genes
with germline alterations in patients with concurrent cancer. The
results of the present study may improve the current understanding
of the molecular mechanism of concurrent cancer and provide a basis
for further studies.
Materials and methods
Patients and samples
In the present study, patients with concurrent
cancer were identified using the following criteria: i) Each tumor
had confirmed evidence of malignancy; ii) each tumor was distinct;
and iii) the probability that one tumor had metastasized from the
other was excluded based on pathological and immunohistochemical
analysis (14). The present study
examined peripheral blood samples and tumor tissues from two
patients with concurrent cancer in one family. Tumor tissues
(>200 mg) were obtained during resection, placed in
cryopreservation tubes and immediately placed in liquid nitrogen or
−80°C freezer. A peripheral blood sample (5 ml) from an unaffected
family member was also collected (Table
I). All patients were recruited at the Second People's Hospital
of Yichang (Hubei, China) between December 2014 and January 2017.
The experiments were performed with the understanding of each
patient, and all patients signed a written informed consent. The
present study was performed in accordance with The Code of Ethics
of the World Medical Association of The Declaration of Helsinki
(15). The present study was
approved by the Medical Ethics Committee of Yichang Second People's
Hospital, Three Gorges University.
 | Table I.Sample information. |
Table I.
Sample information.
| Family member | Age, years | Sex | Sample ID | Sample type |
|---|
| II-1 | 68 | Female | BT15061701HNDQ | Breast tumor
tissue |
|
|
|
| BT15061702HNDQ | Rectal tumor
tissue |
|
|
|
| BT15082203HNDE | Peripheral
blood |
| II-5 | 61 | Male | BT15061703HNDQ | Liver tumor
tissue |
|
|
|
| BT15061704HNDQ | Lymphoma
tissue |
|
|
|
| BT15082201HNDE | Peripheral
blood |
| II-3 | 63 | Male | BT15082202HNDE | Peripheral
blood |
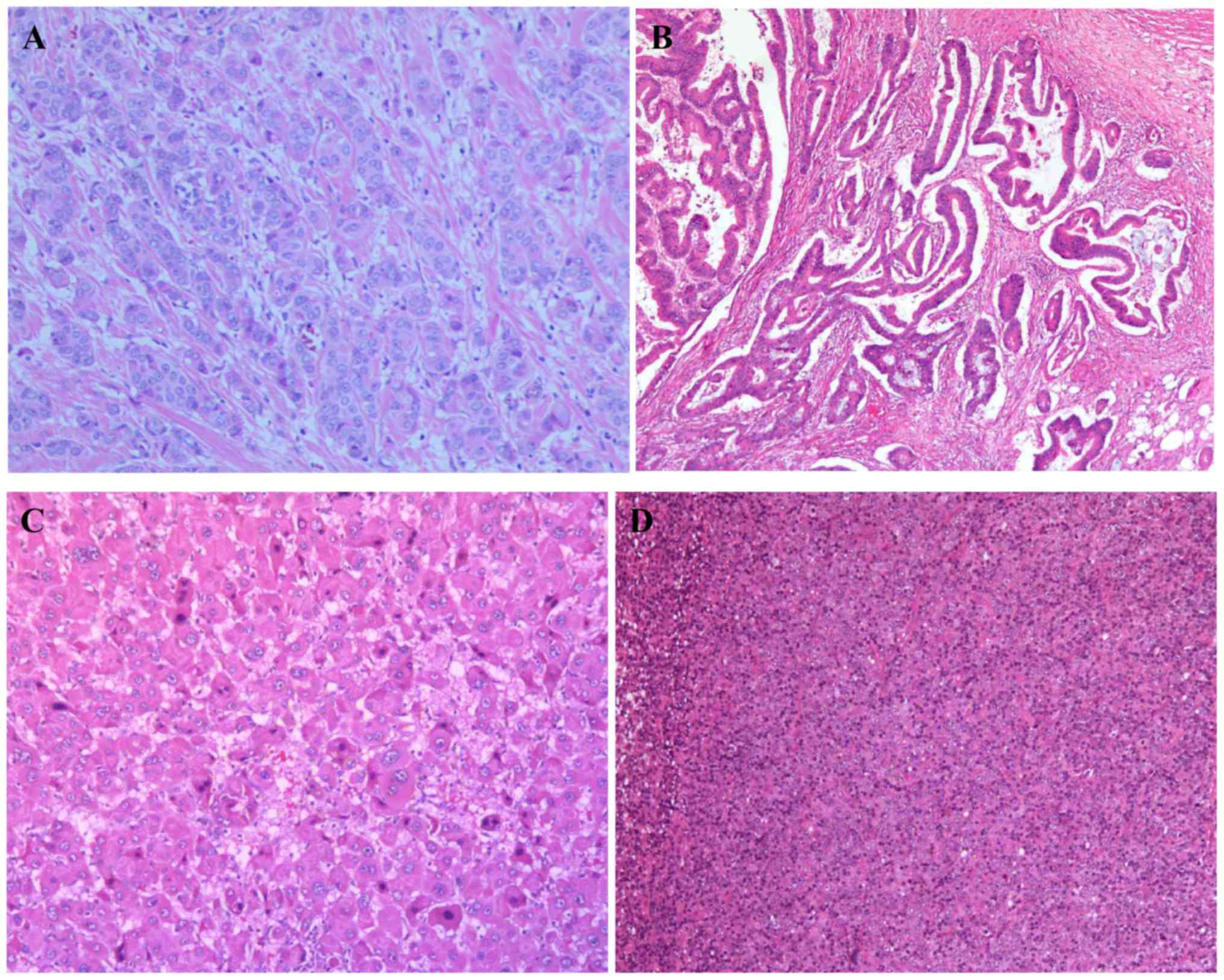
Hematoxylin and eosin (H&E)
staining of tumor tissues
Each tissue sample was fixed in 10% formaldehyde
overnight at 4°C prior to embedding (FFPE) in paraffin. The FFPE
tissue blocks were cut in 4 µm sections on a microtome at room
temperature and then fixed onto standard glass histological slides.
Each section was dewaxed in xylene, rehydrated through graded
ethanol and underwent H&E staining at room temperature. Each
section was incubated with hematoxylin for 5–10 min followed by
eosin for 1 min. A coverslip was then added to each slide with
Pertex mounting medium. The stained slides were scanned on an
Olympus light microscope at magnifications of ×100 or ×200. All
specimens were anonymized prior to receipt. All H&E stained
sections were examined by two pathologists.
DNA extraction, library preparation
and sequencing
DNA was extracted from peripheral blood and tumor
tissue samples using a QIAamp DNA Mini kit (Qiagen GmbH) according
to the manufacturer's protocol. To construct a library, we
performed exome capture using an Agilent SureSelect Human All
ExonV5 kit (Agilent Technologies, Inc.) following the
manufacturer's protocol. A total of 0.5 µg DNA per sample was used
as input material for the DNA library preparations. Genomic DNA
samples were fragmented by sonication to a size of ~350 bp (duty
factor 10%, peak incident power 175, cycles per burst 200,
treatment time 180 sec, bath temperature 4–8°C). Next, DNA
fragments were end polished, A-tailed and ligated with the
full-length adapter for sequencing, followed by further polymerase
chain reaction (PCR) amplification using KAPA HiFi HotStart
ReadyMix (Roche Diagnostics). The primers were based on the P5 and
P7 flow cell sequences, and were suitable for the amplification of
libraries prepared with full-length adapters. The primer sequences
were as follows: P5: 5′-AATGATACGGCGACCACCGAGATC-3′; P7:
5′-CAAGCAGAAGACGGCATACGA-3′. Thermocycling conditions: Initial
denaturation at 98°C for 45 sec, denaturation at 98°C for 15 sec,
annealing at 60°C for 30 sec, extension at 72°C for 30 sec, library
amplification with 3 cycles, a final extension at 72°C for 1 min
and hold at 4°C. Subsequently, PCR products were purified by the
AMPure XP system (Beckman Coulter, Inc.), libraries were analyzed
for size distribution by Agilent2100 Bioanalyzer (Agilent
Technologies, Inc.) and quantified by quantitative PCR (3 nM) using
KAPA Library Quantification kits (Roche Diagnostics) according to
the manufacturer's protocol. The primers were the same as for the
amplification procedure. The thermocycling conditions were as
follows: Initial denaturation at 95°C for 5 min, followed by 35
cycles of denaturation at 95°C for 30 sec and combined
annealing/extension at 60°C for 45 sec. A total of 6 pre-diluted
DNA standards and appropriately diluted NGS libraries were
amplified at the same time. The average Cq value for each DNA
standard was plotted against its known concentration to generate a
standard curve. The standard curve was used to convert the average
Cq values for diluted libraries to concentration, from which the
working concentration of each library was calculated. After the
quality test, the qualified library was sequenced as 100 bp
paired-end reads on an Ion Torrent platform (Applied Biosystems;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols.
Bioinformatics analysis
Firstly, clean data were obtained following
filtering of the low-quality reads, including reads with adapter
sequences, reads with proportion of N >10% and reads with
low-quality base numbers >5. The Burrows-Wheeler transform
methods were adopted to map these reads to the human reference
genome University of California, Santa Cruz (UCSC) hg19 (16,17).
Subsequently, the Picard and Genome Analysis Toolkit (GATK; version
3.2) methods were adopted for duplicate removal, local realignment
and base quality recalibration, as previously described (18,19).
Finally, the GATK Unified Genotyper software (version 3.0; Broad
Institute) was used for SNV annotation. The value of QualByDepth
(QD) describes the quality of variation per unit depth, and the
higher the QD, the higher the reliability of the variation in
general (20). QD >2.0 was set
for ‘good’ SNV (FILTER=Pass), which could distinguish well between
reliable and unreliable variations.
Variants were annotated using the ANNOVAR software
tool (21). Annotations for function
(exonic, intronic and untranslated region), reference genes, exonic
function (synonymous, non-synonymous, stop-gain, frameshift and
unknown), amino acid changes, 1,000 Genomes Project database
(22), single nucleotide
polymorphism database (dbsnp 138; ftp://ftp-trace.ncbi.nih.gov/snp/organisms/) and the
Cancer Gene Census (CGC) database (23) were performed. Varscan2
(VarScan.v2.3.9; http://varscan.sourceforge.net/) (24) was used to identify somatic mutations
in four paired tumor tissue and peripheral blood samples from the
two patients with concurrent cancer.
WES generated a large volume of data, and several
filtering criteria were applied to the dataset. Firstly, the
sequencing quality score of a given base, Q, was defined by the
following equation: Q=−10log10(e) where e was the estimated
probability of the base call being wrong. A quality score of 20
represented an error rate of 1 in 100, with a corresponding call
accuracy of 99%. Variants with a low-quality score (<20) were
removed. Secondly, 1,000 Genomes Pilot Project database stores data
from normal people. Minor allele frequency (MAF) is the percentage
of alleles that are relatively rare in a population. Every
variation at every location has a MAF value. MAF=1% is generally
used as the boundary line for judging a correlation with disease,
but the value is not absolute. Importantly, the threshold has to be
analyzed in combination with the incidence of diseases. Concurrent
cancers are rare; therefore, the variants with a reported MAF of
>0.005 were filtered out. Then, synonymous changes were removed,
and only the protein-altering variants were analyzed.
Results
Description of the pedigree
The patients with concurrent cancer were from Hubei,
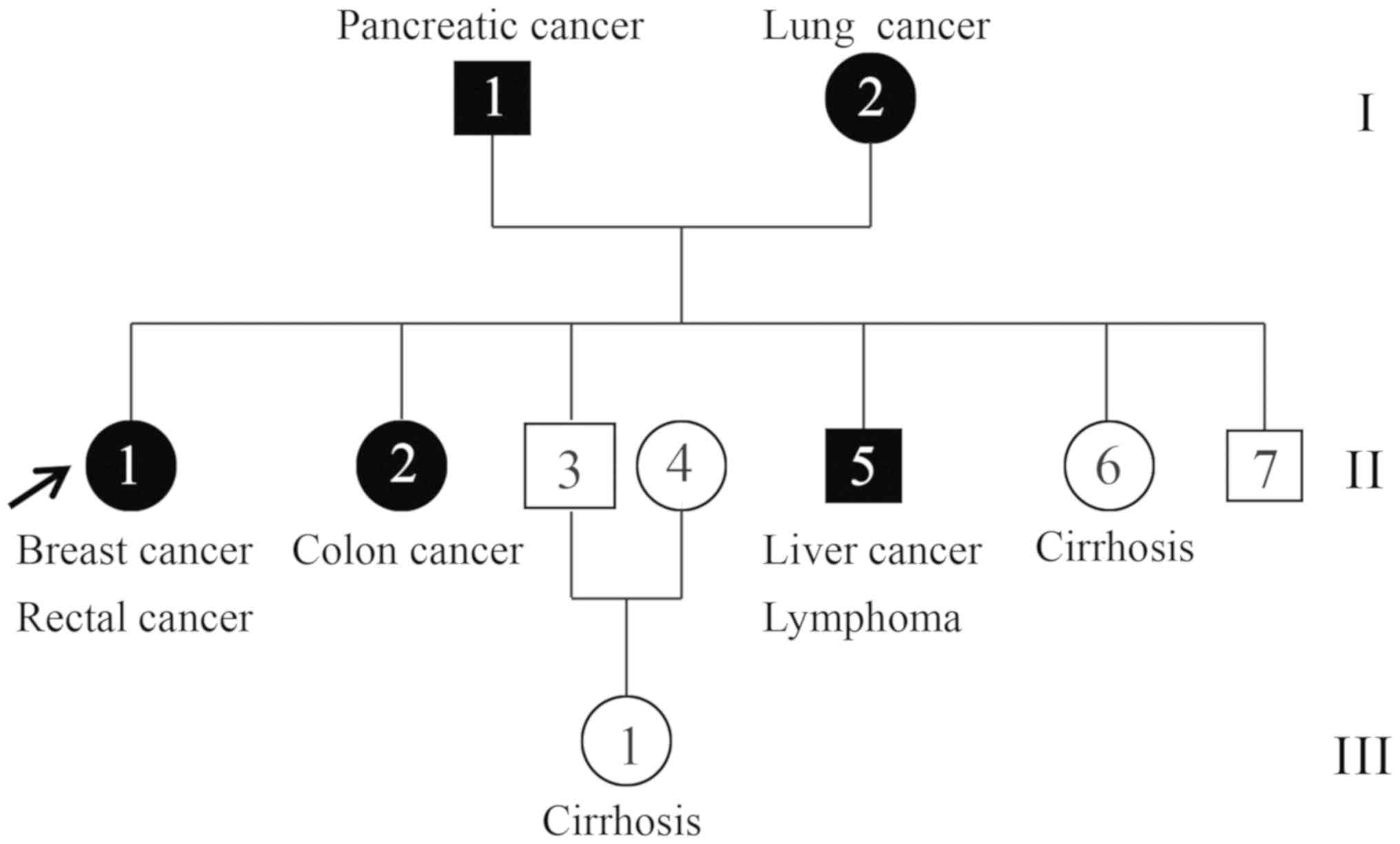
China. In total, two affected individuals (II-1 and II-5) and one
unaffected individual (II-3) were recruited (Fig. 1). The proband (II-1) was a
68-year-old female who presented breast cancer at pathological
tumor-node-metastasis (pTNM) stage pT2N2M0 and rectal cancer at
stage IIIA (pTNM stage, pT4N0M0) according to the National
Comprehensive Cancer Network guidelines and the pTNM staging system
(25–27). The younger brother (II-5) of the
proband was a 61-year-old patient with concurrent cancer who
suffered from liver cancer (pT3N0M0) and lymphoma at stage IIIA
(28,29). The two concurrent cancer cases in the
family were histologically confirmed at the hospital (30), and their tumor tissue and peripheral
blood samples were collected for WES (Fig. 2). Additionally, peripheral blood was
collected from one healthy family member (II-3), a 63-year-old
male, and its exome was also sequenced in the present study.
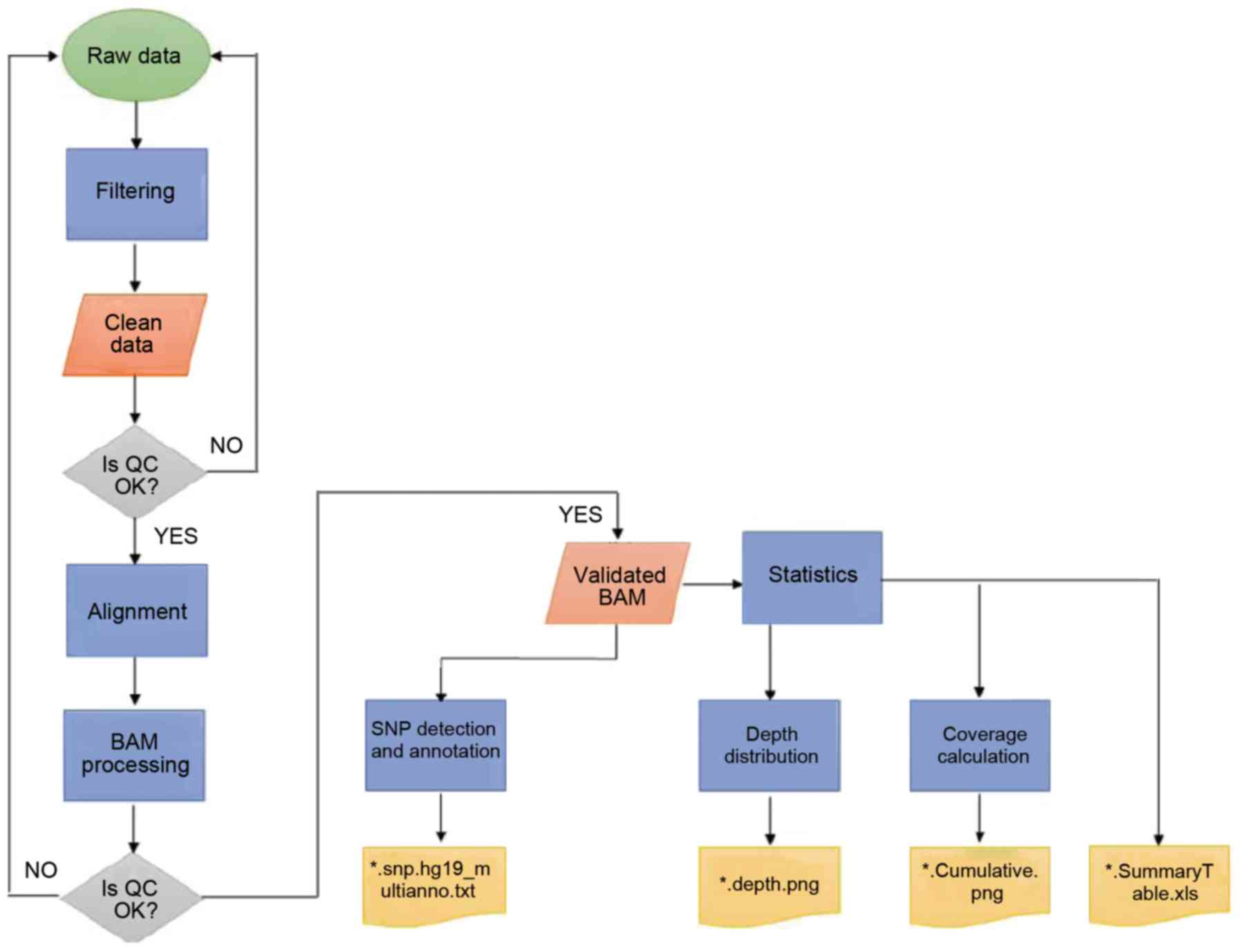
WES
To study the molecular pathogenesis of concurrent
cancer, the exomes of two affected individuals (II-1 and II-5) and
one unaffected individual (II-3) were sequenced. Large volumes of
raw data were generated and further analyzed (Fig. 3). Quality scores across all bases
were >20, which ensured the reliability of following analyses
(31). Clean data were achieved by
applying the aforementioned filtering processes. Following mapping
to the human reference genome UCSC hg19 (17), local realignment and base quality
recalibration, valid exome sequences with an average of 58X depth
for each targeted base and ≥82.08% of the exonic positions covered
>10X were obtained (Table II).
The ratio of transition/transversion in seven samples of one
unaffected (II-3) and two affected individuals (II-1 and II-5)
ranged between 2.2 and 2.4 (Table
III).
| Table II.Mean coverage and the percentage of
exonic positions with coverage >10X. |
Table II.
Mean coverage and the percentage of
exonic positions with coverage >10X.
| Sample | Mean coverage | Coverage >10X
(%) |
|---|
| BT15061701HNDQ | 55.79X | 82.08 |
| BT15061702HNDQ | 67.14X | 90.42 |
| BT15082203HNDE | 50.01X | 93.65 |
| BT15061703HNDQ | 57.28X | 87.11 |
| BT15061704HNDQ | 75.05X | 87.97 |
| BT15082201HNDE | 51.42X | 92.75 |
| BT15082202HNDE | 52.29X | 92.66 |
| Table III.Ratio of transition/transversion in
seven samples. |
Table III.
Ratio of transition/transversion in
seven samples.
| Sample | Transition | Tranversion | Ratio |
|---|
| BT15061701HNDQ | 266339 | 117292 | 2.270735 |
| BT15061702HNDQ | 175790 | 71318 | 2.464876 |
| BT15082203HNDE | 99653 | 43257 | 2.303743 |
| BT15061703HNDQ | 194071 | 80041 | 2.424645 |
| BT15061704HNDQ | 259392 | 111664 | 2.322969 |
| BT15082201HNDE | 68142 | 28732 | 2.371641 |
| BT15082202HNDE | 69564 | 29347 | 2.370396 |
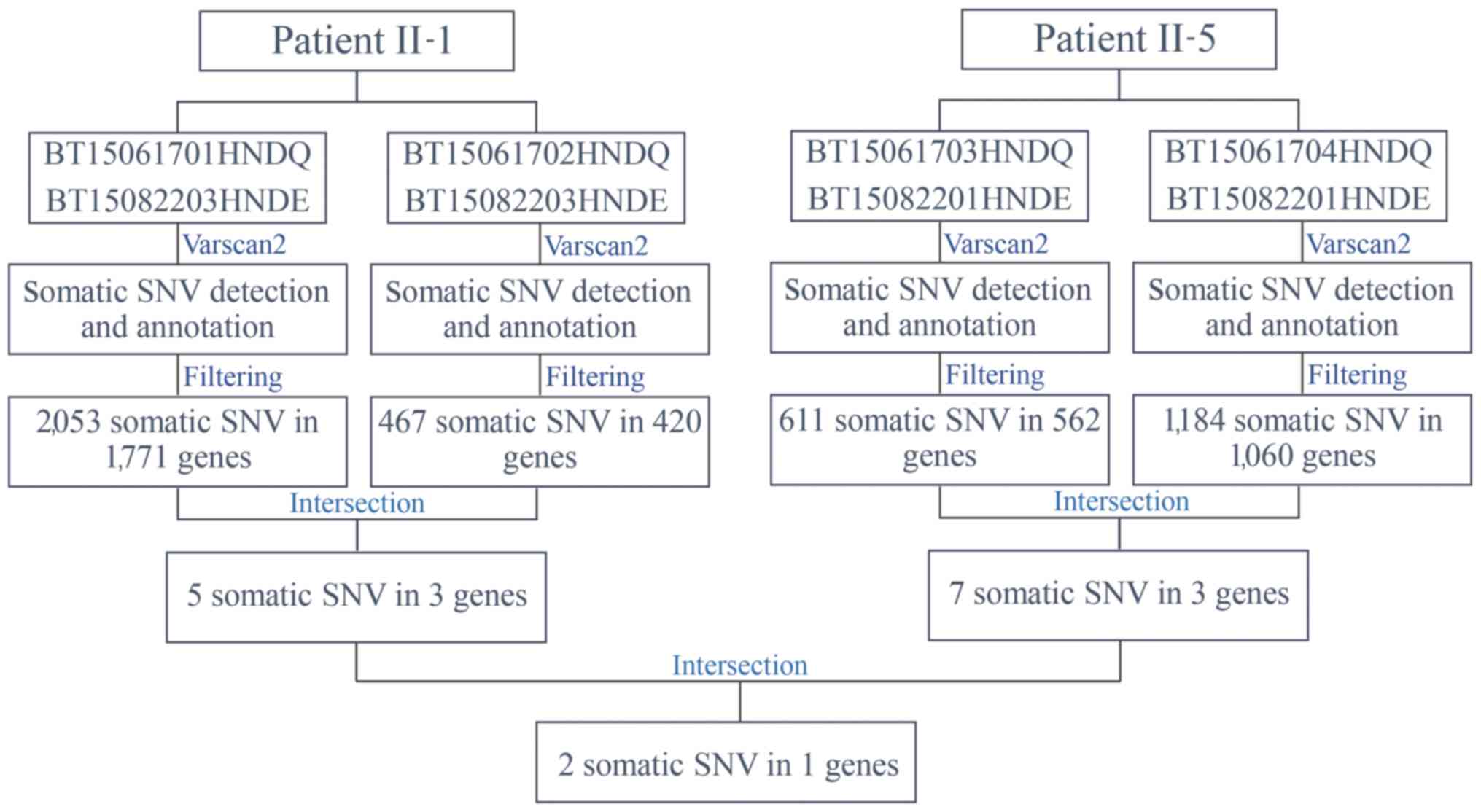
Somatic mutations
Varscan2 (32) was
used to identify somatic mutations in four paired tumor tissue and
peripheral blood samples from the two patients with concurrent
cancer: i) BT15061701HNDQ and BT15082203HNDE; ii) BT15061702HNDQ
and BT15082203HNDE; iii) BT15061703HNDQ and BT15082201HNDE; and iv)
BT15061704HNDQ and BT15082201HNDE (Fig.
4). Subsequent to detection and annotation of somatic SNV, the
present study focused on missense variants in the exonic regions or
splice sites. Furthermore, variants with a reported frequency
>0.005 in the 1,000 Genomes Pilot Project data were removed
(22). As a result, 2,053 somatic
mutations in 1,771 genes in breast tumor tissues, and 467 somatic
mutations in 420 genes in rectal tumor tissues, from patient II-1
were detected. Further analysis for intersection revealed that five
somatic mutations in three genes occurred simultaneously in the two
types of tumor tissues from patient II-1 (Table IV). Similarly, seven somatic
mutations in three genes occurred simultaneously in the two types
of tumor tissues from patient II-5 (Table V). Among these, NDUFS7 emerged
as a candidate gene with somatic mutations (g.1391151G>A and
g.1393289G>C) in two patients with concurrent cancer (Table VI). NDUFS7 (g.1391151G>A and
g.1393289G>C) is homozygous in breast cancer and liver cancer,
while it is heterozygous in rectal cancer and lymphoma.
| Table IV.Somatic mutations identified in the
two types of tumor tissues from patient II-1. |
Table IV.
Somatic mutations identified in the
two types of tumor tissues from patient II-1.
| Gene | Nucleotide
mutation | Mutation type | Amino acid
alteration |
|---|
| LDHAL6B |
g.59500155G>A | Missense
variant |
NM_033195:exon1:c.G1016A:p.S339N |
|
|
g.59500166A>G | Missense
variant |
NM_033195:exon1:c.A1027G:p.I343V |
| CDC27 |
g.45234707T>A | Missense
variant |
NM_001293091:exon5:c.A336T:p.L112F |
| NDUFS7 |
g.1391151G>A | Missense
variant |
NM_024407:exon6:c.G442A:p.V148I |
|
|
g.1393289G>C | Missense
variant |
NM_024407:exon7:c.G504C:p.R168S |
| Table V.Somatic mutations identified in the
two types of tumor tissues from patient II-5. |
Table V.
Somatic mutations identified in the
two types of tumor tissues from patient II-5.
| Gene | Nucleotide
mutation | Mutation type | Amino acid
alteration |
|---|
| NDUFS8 |
g.67803789A>G | Missense
variant |
NM_002496:exon6:c.A442G:p.T148A |
|
|
g.67803790C>T | Missense
variant |
NM_002496:exon6:c.C443T:p.T148I |
|
|
g.67803810T>A | Missense
variant |
NM_002496:exon6:c.T463A:p.F155I |
|
|
g.67803812C>G | Missense
variant |
NM_002496:exon6:c.C465G:p.F155L |
| SDHB |
g.17350532C>G | Missense
variant |
NM_003000:exon6:c.G578C:p.S193T |
| NDUFS7 |
g.1391151G>A | Missense
variant |
NM_024407:exon6:c.G442A:p.V148I |
|
|
g.1393289G>C | Missense
variant |
NM_024407:exon7:c.G504C:p.R168S |
| Table VI.NDUFS7 is a candidate gene
with somatic mutations in the two patients with concurrent
cancer. |
Table VI.
NDUFS7 is a candidate gene
with somatic mutations in the two patients with concurrent
cancer.
| Gene | Nucleotide
mutation | Mutation type | Amino acid
alteration |
|---|
| NDUFS7 |
g.1391151G>A | Missense
variant |
NM_024407:exon6:c.G442A:p.V148I |
|
|
g.1393289G>C | Missense
variant |
NM_024407:exon7:c.G504C:p.R168S |
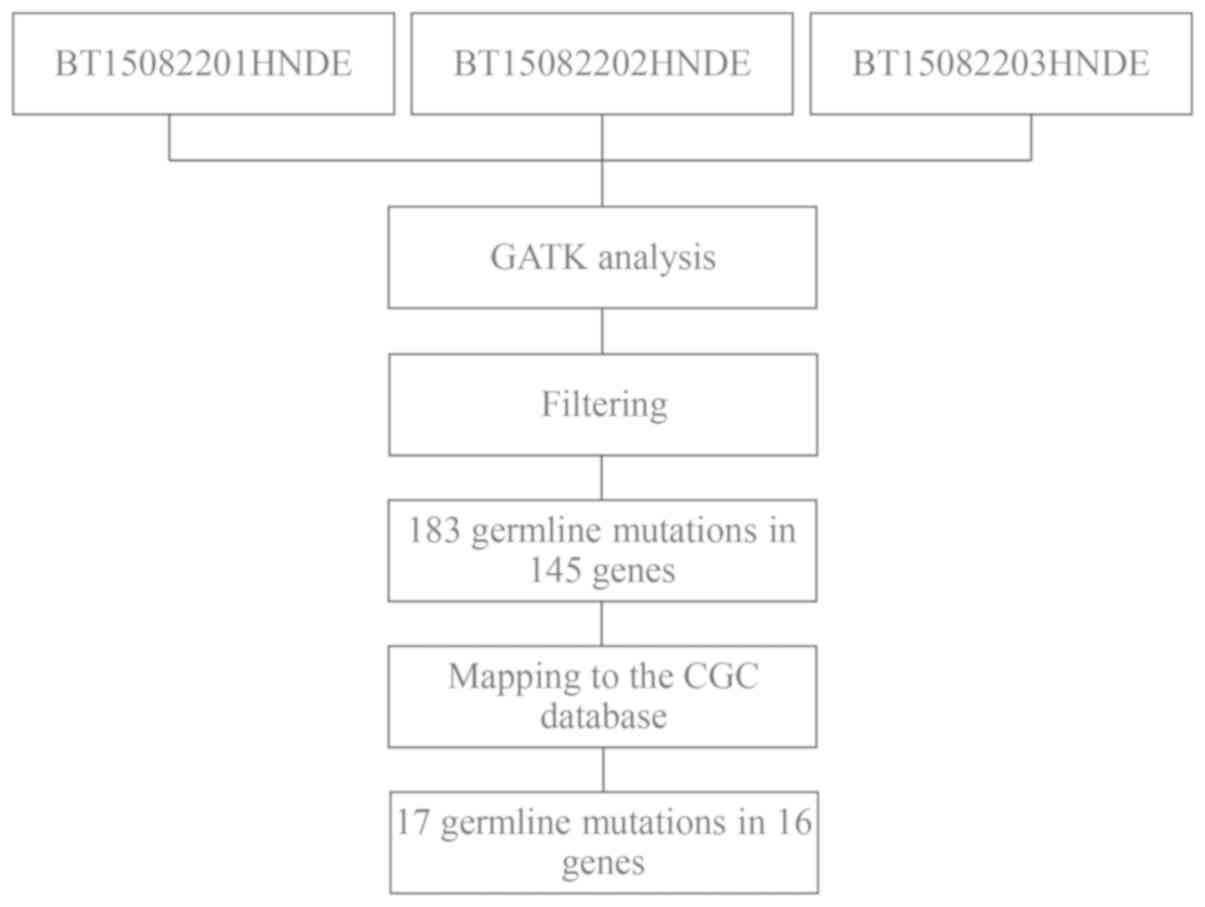
Germline mutations
To further investigate the molecular pathogenesis of
concurrent cancer, germline mutations were analyzed using GATK
methods (Fig. 5). Variants in
peripheral blood samples that were shared by the two affected
individuals (BT15082201HNDE and BT15082203HNDE), but were not
present in the unaffected individual (BT15082202HNDE) were
selected. Similarly, the present study focused only on missense
variants in the exonic region or splice site, and filtered out
variants with a reported frequency >0.005 in the 1,000 Genomes
Pilot Project data. As a result, 183 missense SNVs were detected in
145 candidate genes. Subsequently, these candidate SNVs were
further mapped to the CGC database to examine probable germline
mutations and genes (33). Finally,
17 SNVs in 16 genes emerged as candidate germline mutations in
patients with concurrent cancer (Table
VII).
| Table VII.Candidate germline mutations in the
two patients with concurrent cancer. |
Table VII.
Candidate germline mutations in the
two patients with concurrent cancer.
| Gene | Nucleotide
mutation | Mutation type | Amino acid
alteration |
|---|
| PGLYRP4 |
g.153314126C>T | Missense
variant |
NM_020393:exon6:c.G602A:p.R201Q |
| PEAR1 |
g.156876633C>A | Missense
variant |
NM_001080471:exon6:c.C605A:p.T202N |
| COL6A3 |
g.238243350C>T | Missense
variant |
NM_057166:exon38:c.G7327A:p.A2443T |
| KIF1A |
g.241659323C>T | Missense
variant |
NM_004321:exon43:c.G4586A:p.R1529Q |
| ZNF717 |
g.75786450G>A | Missense
variant |
NM_001128223:exon5:c.C2324T:p.T775M |
|
|
g.75788434G>T | Missense
variant |
NM_001128223:exon5:c.C340A:p.Q114K |
| ZNF141 | g.338156T>A | Missense
variant |
NM_003441:exon3:c.T163A:p.C55S |
| SSPO |
g.149523309C>A | Missense
variant |
NM_198455:exon101:c.C14392A:p.P4798T |
| EPPK1 |
g.144940706C>T | Missense
variant |
NM_031308:exon2:c.G6716A:p.R2239H |
| ZDHHC21 |
g.14619085C>T | Missense
variant |
NM_178566:exon10:c.G677A:p.R226Q |
| CNTRL |
g.123886324C>T | Missense
variant |
NM_007018:exon11:c.C1766T:p.T589M |
| ASCC1 |
g.73973043C>T | Missense
variant |
NM_001198798:exon2:c.G14A:p.R5H |
| OR8U1 |
g.56143823G>A | Missense
variant |
NM_001005204:exon1:c.G724A:p.G242S |
| PABPC3 |
g.25671688G>C | Missense
variant |
NM_030979:exon1:c.G1352C:p.G451A |
| NPIPB6 |
g.28354223G>A | Missense
variant |
NM_001282524:exon7:c.C983T:p.P328L |
| RAB36 |
g.23488846G>A | Missense
variant |
NM_004914:exon2:c.G241A:p.D81N |
| CCDC117 |
g.29169761T>G | Missense
variant |
NM_001284264:exon2:c.T234G:p.D78E |
The present study investigated the molecular
alterations in concurrent cancer. By sequencing the exomes of two
affected individuals and one unaffected individual in a Chinese
family with concurrent cancer, the present study identified
NDUFS7 as a candidate gene with somatic mutations
(g.1391151G>A and g.1393289G>C), and 17 SNVs in 16 genes as
candidate germline mutations. The present results provided insights
into the causative alterations of concurrent cancer at the
molecular level.
Discussion
It is an ongoing aim of cancer research to
understand the causative mutations underlying cancer development
and progression. Somatic mutations can occur in any non-germ cell
of the body following conception, whereas germline mutations are
inherited from the parents (4,5). During
the past decades, comprehensive efforts have been made by
scientists to improve the resolution and reduce the cost of
sequencing methods. The genomic landscapes of common forms of human
cancer have been identified (34–36).
However, the molecular mechanisms of concurrent cancers remain
unknown. Currently, there are no specific approaches to treat
concurrent cancer. Patients with concurrent cancer are treated just
like other common forms of human cancers.
Tumors evolve from benign to malignant lesions by
acquiring a series of mutations over time. Somatic mutations that
occur in tumor cell genomes serve a vital role in cancer
development, including the initiation of tumorigenesis. In common
solid tumors, including those derived from breast, colon, brain or
pancreas, an average of 33–66 genes may display subtle somatic
mutations that would be expected to alter the protein products
(5). Of these mutations, ~95% are
single-base substitutions, of which 90.7% result in missense
changes, 7.6% result in nonsense changes and 1.7% result in
alterations of splice sites or untranslated regions adjacent to the
start and stop codons (5). In the
present study, NDUFS7 emerged as a candidate gene with
somatic mutations in cases of concurrent cancer. The NDUFS7
gene encodes a protein that is a subunit of complex I in the
mitochondrial respiratory chain (37). Mutations in this gene cause Leigh
syndrome due to mitochondrial complex I deficiency (38,39).
Leigh syndrome is a severe neurological disorder that causes
bilaterally symmetrical necrotic lesions in subcortical brain
regions (38,39). To the best of our knowledge, the
present study is the first one that identified NDUFS7 as a
somatic mutation gene in concurrent cancer. Further studies are
required to verify how these somatic mutations in the NDUFS7
gene may cause functional alterations associated with the
development of cancer.
Germline mutations inherited from the parents can
increase susceptibility to cancer development (4,5). An
assessment of the components of the germline genome of patients may
improve the current understanding of the pathogenesis of various
types of cancer. The present study identified a total of 17
germline mutations in 16 candidate genes in peripheral blood
samples from patients with concurrent cancer, including the
peptidoglycan recognition protein 4, platelet endothelial
aggregation receptor 1 (PEAR1), collagen type VI α3 chain,
kinesin family member 1A, zinc finger protein 717 (ZNF717),
zinc finger protein 141 (ZNF141), SCO-spondin, epiplakin 1,
zinc finger DHHC-type containing 21 (ZDHHC21), centriolin,
activating signal cointegrator 1 complex subunit 1, olfactory
receptor family 8 subfamily U member 1, poly(A) binding protein
cytoplasmic 3, nuclear pore complex interacting protein family
member B6, RAB36 member RAS oncogene family and coiled-coil domain
containing 117 genes. Following mapping to the CGC database, it was
identified that mutations in these genes had been previously
reported to be involved in cancer progression. For example,
mutations in the ZDHHC21 gene have been reported in rectal
cancer but not in breast cancer, and mutations in the PEAR1,
ZNF717 and ZNF141 genes were detected in liver cancer
but not in lymphoma, as assessed by the Catalogue Of Somatic
Mutations In Cancer (40–42). Notably, to the best of our knowledge,
the present study is the first to suggest that mutations in these
genes may increase susceptibility to concurrent cancer.
In conclusion, the present study focused on a rare
case of a three-generation family in which two members had
developed concurrent cancer. Through WES and bioinformatics
analysis, probable somatic mutations were identified in the
NDUFS7 gene, and germline mutations were identified in 16
candidate genes. Although further studies are required to validate
these variants, to the best of our knowledge, the results of the
present study are the first to suggest a specific molecular profile
associated with concurrent cancer.
Acknowledgements
Not applicable.
Funding
This study was funded by grants from the Yichang
Science and Technology Fund of Hubei Province (grant no.
A16-301-27) and the Hubei Natural Science Foundation of China
(grant no. 2017CFB455cfb455).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XW and SW conceived the study and designed the
experiments. YY, HL and LJ performed experiments, analyzed the data
and wrote the manuscript. LZ, AL and FD collected and analyzed the
clinical data. XZ and MM performed the histological examination of
the two patients with concurrent cancer. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed involving human
participants were in accordance with the ethical standards of the
Medical Ethics Committee of Yichang Second People's Hospital, and
with the Declaration of Helsinki (15). Informed consent was obtained from all
patients included in the present study.
Patient consent for publication
Informed consent for the publication of the present
study was obtained from all participants.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen WQ, Zheng RS, Zeng HM, Zhang SW, Zhao
P and He J: Trend analysis and projection of cancer incidence in
China between 1989 and 2008. Zhonghua Zhong Liu Za Zhi. 34:517–524.
2012.(In Chinese). PubMed/NCBI
|
|
2
|
Zeng HM, Zheng RS, Zhang SW, Zhao P, He J
and Chen WQ: Trend analysis of cancer mortality in China between
1989 and 2008. Zhonghua Zhong Liu Za Zhi. 34:525–531. 2012.(In
Chinese). PubMed/NCBI
|
|
3
|
Siravegna G, Marsoni S, Siena S and
Bardelli A: Integrating liquid biopsies into the management of
cancer. Nat Rev Clin Oncol. 14:531–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mardis ER: Genome sequencing and cancer.
Curr Opinion Genetics Dev. 22:245–250. 2012. View Article : Google Scholar
|
|
5
|
Vogelstein B, Papadopoulos N, Velculescu
VE, Zhou S, Diaz LA Jr and Kinzler KW: Cancer genome landscapes.
Science. 339:1546–1558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mardis ER: New strategies and emerging
technologies for massively parallel sequencing: Applications in
medical research. Genome Med. 1:402009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mardis ER: A decade's perspective on DNA
sequencing technology. Nature. 470:198–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Esteban-Jurado C, Vila-Casadesus M, Garre
P, Lozano JJ, Pristoupilova A, Beltran S, Muñoz J, Ocaña T,
Balaguer F, López-Cerón M, et al: Whole-exome sequencing identifies
rare pathogenic variants in new predisposition genes for familial
colorectal cancer. Genet Med. 17:131–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gulbahce N, Magbanua MJM, Chin R, Agarwal
MR, Luo X, Liu J, Hayden DM, Mao Q, Ciotlos S, Li Z, et al:
Quantitative whole genome sequencing of circulating tumor cells
enables personalized combination therapy of metastatic cancer.
Cancer Res. 77:4530–4541. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sabour L, Sabour M and Ghorbian S:
Clinical applications of next-generation sequencing in cancer
diagnosis. Pathol Oncol Res. 23:225–234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sunami K, Takahashi H, Tsuchihara K,
Takeda M, Suzuki T, Naito Y, Sakai K, Dosaka-Akita H, Ishioka C,
Kodera Y, et al: Clinical practice guidance for next-generation
sequencing in cancer diagnosis and treatment (Edition 1.0). Cancer
Sci. 109:2980–2985. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Dijk EL, Auger H, Jaszczyszyn Y and
Thermes C: Ten years of next-generation sequencing technology.
Trends Genet. 30:418–426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rabbani B, Tekin M and Mahdieh N: The
promise of whole-exome sequencing in medical genetics. J Hum Genet.
59:5–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Swaroop VS, Winawer SJ, Kurtz RC and
Lipkin M: Multiple primary malignant tumors. Gastroenterology.
93:779–783. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gandevia B and Tovell A: Declaration of
Helsinki. Med J Aust. 2:320–321. 1964.PubMed/NCBI
|
|
16
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meyer LR, Zweig AS, Hinrichs AS, Karolchik
D, Kuhn RM, Wong M, Sloan CA, Rosenbloom KR, Roe G, Rhead B, et al:
The UCSC Genome Browser database: Extensions and updates 2013.
Nucleic Acids Res. 41:D64–D69. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The Genome Analysis Toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: the Genome Analysis Toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–33. 2013.
|
|
21
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
1000 Genomes Project Consortium, ;
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA,
Hurles ME and McVean GA: A map of human genome variation from
population-scale sequencing. Nature. 467:1061–1073. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sondka Z, Bamford S, Cole CG, Ward SA,
Dunham I and Forbes SA: The COSMIC Cancer Gene Census: Describing
genetic dysfunction across all human cancers. Nat Rev Cancer.
18:696–705. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koboldt DC, Chen K, Wylie T, Larson DE,
McLellan MD, Mardis ER, Weinstock GM, Wilson RK and Ding L:
VarScan: Variant detection in massively parallel sequencing of
individual and pooled samples. Bioinformatics. 25:2283–2285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Amin MB, Edge SB and Greene FL: AJCC
Cancer Staging Manual. 8th. Springer; New York, NY: 2017,
View Article : Google Scholar
|
|
26
|
Gradishar WJ, Anderson BO, Balassanian R,
Blair SL, Burstein HJ, Cyr A, Elias AD, Farrar WB, Forero A,
Giordano SH, et al: NCCN Guidelines Insights: Breast cancer,
Version 1.2017. J Natl Compr Canc Netw. 15:433–451. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Benson AB III, Venook AP, Bekaii-Saab T,
Chan E, Chen YJ, Cooper HS, Engstrom PF, Enzinger PC, Fenton MJ,
Fuchs CS, et al: Rectal cancer, Version 2.2015. J Natl Compr Canc
Netw. 13:719–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Benson AB III, D'Angelica MI, Abbott DE,
Abrams TA, Alberts SR, Saenz DA, Are C, Brown DB, Chang DT, Covey
AM, et al: NCCN Guidelines Insights: Hepatobiliary cancers, Version
1.2017. J Natl Compr Canc Netw. 15:563–573. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wierda WG, Zelenetz AD, Gordon LI,
Abramson JS, Advani RH, Andreadis CB, Bartlett N, Byrd JC, Caimi P,
Fayad LE, et al: NCCN Guidelines Insights: Chronic Lymphocytic
Leukemia/Small Lymphocytic Lymphoma, Version 1.2017. J Natl Compr
Canc Netw. 15:293–311. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feldman AT and Wolfe D: Tissue processing
and hematoxylin and eosin staining. Methods Mol Biol. 1180:31–43.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
1000 Genomes Project Consortium, ;
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker
RE, Kang HM, Marth GT and McVean GA: An integrated map of genetic
variation from 1,092 human genomes. Nature. 491:56–65. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cibulskis K, Lawrence MS, Carter SL,
Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES
and Getz G: Sensitive detection of somatic point mutations in
impure and heterogeneous cancer samples. Nat Biotechnol.
31:213–219. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Forbes SA, Bindal N, Bamford S, Cole C,
Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, et al:
COSMIC: Mining complete cancer genomes in the Catalogue of Somatic
Mutations in Cancer. Nucleic Acids Res. 39:D945–D950. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cancer Genome Atlas Network: Comprehensive
molecular portraits of human breast tumours. Nature. 490:61–70.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM,
Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al: Genomic portrait
of resectable hepatocellular carcinomas: Implications of RB1 and
FGF19 aberrations for patient stratification. Hepatology.
60:1972–1982. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reddy A, Zhang J, Davis NS, Moffitt AB,
Love CL, Waldrop A, Leppa S, Pasanen A, Meriranta L,
Karjalainen-Lindsberg M L, et al: Genetic and functional drivers of
diffuse large B cell lymphoma. Cell. 171:481–494.e15. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Triepels RH, van den Heuvel LP, Loeffen
JL, Buskens CA, Smeets RJ, Rubio Gozalbo ME, Budde SM, Mariman EC,
Wijburg FA, Barth PG, et al: Leigh syndrome associated with a
mutation in the NDUFS7 (PSST) nuclear encoded subunit of complex I.
Ann Neurol. 45:787–790. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lebon S, Minai L, Chretien D, Corcos J,
Serre V, Kadhom N, Steffann J, Pauchard JY, Munnich A, Bonnefont JP
and Rötig A: A novel mutation of the NDUFS7 gene leads to
activation of a cryptic exon and impaired assembly of mitochondrial
complex I in a patient with Leigh syndrome. Mol Genet Metab.
92:104–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lebon S, Rodriguez D, Bridoux D, Zerrad A,
Rotig A, Munnich A, Legrand A and Slama A: A novel mutation in the
human complex I NDUFS7 subunit associated with Leigh syndrome. Mol
Genet Metab. 90:379–382. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen H, Sun X, Ge W, Qian Y, Bai R and
Zheng S: A seven-gene signature predicts overall survival of
patients with colorectal cancer. Oncotarget. 8:95054–95065.
2016.PubMed/NCBI
|
|
41
|
Duan M, Hao J, Cui S, Worthley DL, Zhang
S, Wang Z, Shi J, Liu L, Wang X, Ke A, et al: Diverse modes of
clonal evolution in HBV-related hepatocellular carcinoma revealed
by single-cell genome sequencing. Cell Res. 28:359–373. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Varrault A, Ciani E, Apiou F, Bilanges B,
Hoffmann A, Pantaloni C, Bockaert J, Spengler D and Journot L: hZAC
encodes a zinc finger protein with antiproliferative properties and
maps to a chromosomal region frequently lost in cancer. Proc Natl
Acad Sci USA. 95:8835–8840. 1998. View Article : Google Scholar : PubMed/NCBI
|