Introduction
Myeloid leukemia is a common hematological
malignancy that is subdivided into acute myelogenous leukemia (AML)
and chronic myelogenous leukemia (CML). CML is a myeloproliferative
disorder characterized by the Philadelphia chromosome, a fusion of
chromosomes 9 and 22, that gives rise to the Bcr/Abl oncogene
(1,2). Bcr/Abl kinase inhibitors, such as
imatinib, yield an outstanding clinical response in the chronic
phase of CML. However, certain patients that progress to the blast
phase with a decreased responsiveness develop resistance to Bcr/Abl
kinase inhibitors (3). Doxorubicin,
one of the most widely used antitumor drugs inleukemia treatment,
exerts its proapoptotic effects through disturbing DNA function and
inducing DNA damage. To date, the DA regimen (mainly containing
doxorubicin and cytatabine) has been the standard treatment for
AML, but is not effective for patients in the blast phase. New
effective approaches must be explored to enhance the therapeutic
efficacy against leukemia.
It has been established that the activation of the
mammalian target of rapamycin (mTOR) pathway is closely associated
with cell proliferation and differentiation in several types of
human cancer, such as breast, lung, and pancreatic cancer (4–7). In
addition, the mTOR pathway has been found to be activated in
leukemia cells, especially in cell lines expressing Bcr/Abl
(8–11). Previous studies demonstrated that the
mTOR pathway was activated in myeloid leukemia, including CML, and
mTOR inhibitor (rapamycin, sapanisertib) could arrest cells at the
G0/G1 phase and increase apoptosis in
leukemia cells (12–14). mTOR is a serine/threonine kinase
which plays important roles in the
phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Protein
Kinase B (AKT)/mTOR pathway. It was reported thatPI3K, induced by
Bcr-Abl through a direct association with its regulatory subunit,
could activate mTOR upon phosphorylation of AKT (15). A recent study showed that a
dualPI3K/mTOR inhibitor effectively inhibited cell proliferation
and induced apoptosis by downregulating the PI3K/mTOR pathway
(16). Further evidence that
rapamycin combined with celecoxib could enhance the antitumor
effects of single treatment on CML cellswas provided (17). Therefore, targeting the mTOR pathway
may increase cell sensitivity to chemotherapeutic drugs and serve
as a new therapeutic approach for leukemia. However, the precise
mechanisms through which rapamycin may have synergistic effects
with chemotherapeutic drugs in leukemia remained unclear.
The activation of mTOR (phosphorylated-mTOR) could
phosphorylatetwo major downstream targets, ribosomal protein S6
kinase (p70S6K) and eukaryotic initiation factor 4E-binding protein
1 (4E-BP1). Upon its phosphorylation, p70S6K induces protein
synthesis and cell survival, and increases the translation of
5′-terminal oligopyrimidine tract mRNAs (18–20).
This is important for mitogen-induced cell proliferation and
chemotherapeutic drug resistance in cancer cells (21,22). The
authors' previous study showed that p70S6K and 4E-BP1 were
overexpressed and phosphorylated in CML bone marrow and K562 cells
(12). It was therefore speculated
that rapamycin could enhance the antitumor effects of chemotherapy
on leukemia cells through downregulating mTOR signaling.
In the present study, cell growth and apoptosis in
K562 cells was investigated following rapamycin and doxorubicin
treatment. In addition, the effects of p70S6K-targeting siRNA and
doxorubicin on K562 cells were closely investigated. The present
study tried to confirm whether rapamycin enhanced the antitumor
effects of doxorubicin by downregulating the mTOR/p70S6K pathway in
this preliminary study. Determining the underlying mechanism may
help define this new therapeutic target for clinical application in
hematologic malignancies.
Materials and methods
Chemicals and reagents
The K562 cell line was purchased from the Beijing
Institute for Cancer Research. The primary antibodies used in this
study include: Rabbit anti-human mTOR (1:1,000; cat. no. 2983),
phosphorylated (phospho/p)-mTOR (1:1,000; cat. no. 5536), p70S6K
(1:800, cat. no. 2708) and phospho-p70S6K (1:500; cat. no. 9234)
were purchased from Cell Signaling Technology, Inc.; Rabbit
anti-human β-actin (1:10,000; cat. no. AC026) monoclonal antibody
was bought from ABclonalBiotech Co., Ltd.; rabbit anti-human Bax
(1:500; cat. no. sc-23959) and Bcl-xL (1:500; cat. no. sc-8392),
and mouse anti-human Bcl-2 (1:500; cat. no. sc-509) antibodies were
bought from Santa Cruz Biotechnology, Inc.; and rabbit anti-human
cyclin dependent kinase (CDK)4 (1:5,000; cat. no. ab108357), CDK6
(1:20,000; cat. no. ab124821), cyclin B1 (1:10,000; cat. no.
ab32053) and cyclin D1 (1:200; cat. no. ab16663) were purchased
from Abcam. Goat anti-Mouse IgG (H&L), horseradish peroxidase
(HRP) conjugated secondary antibody (1:10,000; cat. no: 074-1806;
S0002) and Goat anti-Rabbit IgG (H&L), HRP conjugated secondary
antibody (1:5,000; cat. no: 074-1506; S0001) were purchased from
Affinity Biosciences.
Cell culture and treatment
K562 cells were cultured in RPMI-1640 (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with penicillin (100 U/ml),
streptomycin (100 U/ml) and 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.). Doxorubicin and rapamycin were diluted in
solvent dimethyl sulfoxide (DMSO; Merck KGaA). K562 cells were
treated with various concentrations of rapamycin and/or
doxorubicin.
Cell counting kit-8 (CCK-8) assay
Doxorubicin was dissolved in DMSO at a concentration
of 1 mM as the primary stock solution and stored at 4°C. It was
reported that the distribution volume of doxorubicin varies
markedly in different patients or for different cell lines
(23–26). Therefore, to investigate its effects
on cell proliferation, K562 cells were cultured with various
concentrations of doxorubicin (0.03125, 0.0625, 0.125, 0.25, 0.5,
1, 2 and 4 µM) for 24 h at 37°C. After cells were incubated with
the CCK-8 reagent (DojindoMolecular Technologies, Inc.) for 4 h,
the cytotoxic effect of doxorubicinon K562 cells was measured on a
spectrophotometer microplate reader (BioTek Instruments, Inc.) at a
wavelength of 450 nm. Cell viability was assayed and compared to
the control group. The sensitivity of cells to doxorubicin was
measured by IC50. The concentration of rapamycin (10, 20
and 40 nM) was added to K562 cells according to the authors'
previous studies (12,14). The interaction between rapamycin and
doxorubicin was assessed using the combination index (CI) (27,28). The
CI was used to identify antagonistic (CI>1), additive (CI=1) or
synergistic (CI<1) interactions.
Flow cytometry (FCM)
K562 cells from different groups treated with
rapamycin (20 nM), doxorubicin (0.5 µM) and a combination of both,
were collected and washed twice with phosphate-buffered saline
(PBS) and harvested in the buffer (PBS-0.05% trypsin). K562 cells
were incubated with 5 µl Annexin V and 10 µl propidium iodide (PI)
in the dark for 5 min, and then resuspended in 1X binding buffer.
Briefly, cell apoptosis was analyzed using an Annexin V/PI
apoptosis detection kit [Hangzhou Multi Sciences (Lianke) Biotech
Co., Ltd.] according to the manufacturer's protocol.
siRNA transfection
K562 cells (1.0×105/ml) were seeded in
6-well plates and transfected with 50 nM siRNA duplexes using
Lipofectamine™ 2000 reagent according to the manufacturer's
protocol (Thermo Fisher Scientific, Inc.). A total of 48 h
following transfection of p70S6K siRNA (5′-GACAAAAUCCUCAAAUGUA-3′)
and negative control siRNA (5′-GGCTACGTCCAGGAGCGCA-3′; GE
Healthcare Dharmacon, Inc.), the inhibition efficiency at mRNA and
protein levels was estimated using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis.
Western blot analysis
Total cell proteins were extracted from K562 cells
treated with rapamycin and/or doxorubicin using lysis buffer (1%
Triton X-100, 150 mM NaCl, 2 mM EDTA, 50 mM Tris-HCl) supplemented
with phosphatase inhibitor cocktail. After protein concentration
was measured by Gen5 1.0 software (BioTek Instruments, Inc.) on a
spectrophotometer microplate reader (BioTek Instruments, Inc.),
20–40 µg protein was run on a 10–15% sodium dodecyl sulfate
polyacrylamide gel electrophoresis gel and transferred to a
polyvinylidinedifluoride membrane. Following blocking non-specific
binding sites with 5% nonfat dry milk in tris-buffered saline with
0.05% Tween at 37°C for 60 min, the membranes were incubated with
primary antibodies against mTOR, p-mTOR, p70S6K, p-p70S6K, Bcl-2,
Bax, Bcl-xL, CDK4, CDK6, Cyclin B1 and CyclinD1at 4°C overnight.
Next, the membranes were probed with horseradish
peroxidase-conjugated secondary antibody (1:5,000) for 60 min at
37°C. Subsequently, the immunoreactive membranes were developed
using Pierce™ ECL Western Blotting Substrate (cat. no. 32109;
Thermo Fisher Scientific, Inc. cat. no.32109) on Luminescent Image
Analyzer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). Band
density was quantified and normalized to β-actinby Image J software
(version 2; National Institutes of Health).
RT-qPCR
Total RNA was extracted using TRIzol reagent (Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
To obtain cDNA, RT was done with the following conditions: 25°C for
5 min, 42°C for 60 min and 70°C for 15 min. After cDNA was obtained
by RT (Promega Corporation), PCR amplification was performed with
forward and reverse primers using the SYBR PrimeScript RT-PCR kit
(Takara Bio, Inc.) in an Mx3005p instrument (Agilent Technologies
GmbH). Amplification was performed with the following thermocycling
conditions: 10 min at 95°C, followed by 40 cycles of 95°C for 15
sec and 60°C for 1 min. The primers used in the present study are
listed in Table I. Cq values of the
test genes were normalized to the average Cq values of β-actin.
Fold-change was calculated using the 2−ΔΔCq method
(29).
 | Table I.Primers for reverse
transcription-quantitative PCR amplification. |
Table I.
Primers for reverse
transcription-quantitative PCR amplification.
| Primers | Sequence | Product length
(bp) |
|---|
| CDK4-sense |
5′-AGTTCGTGAGGTGGCTTTAC-3′ |
|
| CDK4-antisense |
5′-GCCTTGTCCAGATATGTCCTTAG-3′ | 162 |
| CDK6-sense |
5′-CTGCCTTGTTGGCAAAGTATC-3′ |
|
| CDK6-antisense |
5′-CCAGGTAGAAGGACTGCATTAG-3′ | 229 |
| Cyclin
D1-sense |
5′-CCACTCCTACGATACGCTACTA-3′ |
|
| Cyclin
D1-antisense |
5′-GGACTGAAAGTGCTTGGAAATG-3′ | 219 |
| Cyclin B-sense |
5′-GGTGTCACTGCCATGTTTATTG-3′ |
|
| Cyclin
B-antisense |
5′-CGAAGGAAGTGCAAAGGTAGA-3′ | 179 |
| p70S6K-sense |
5′-CAAGGTGAGGGAGATAGGGATA-3′ |
|
|
p70S6K-antisense |
5′-AAGGAAGGTAGACAGCAGAAAC-3′ | 134 |
Statistical analysis
All experiments were performed at least three times.
Values were presented as the mean ± standard deviation. Statistical
differences were performed by one-way analysis of variance with
least significant difference Duncan's post-hoc test using SPSS 21.0
for Windows (IBM, Corps.). P<0.05 was considered to indicate a
statistically significant difference between groups.
Results
Rapamycin and doxorubicin decreases
the viability of K562 cells
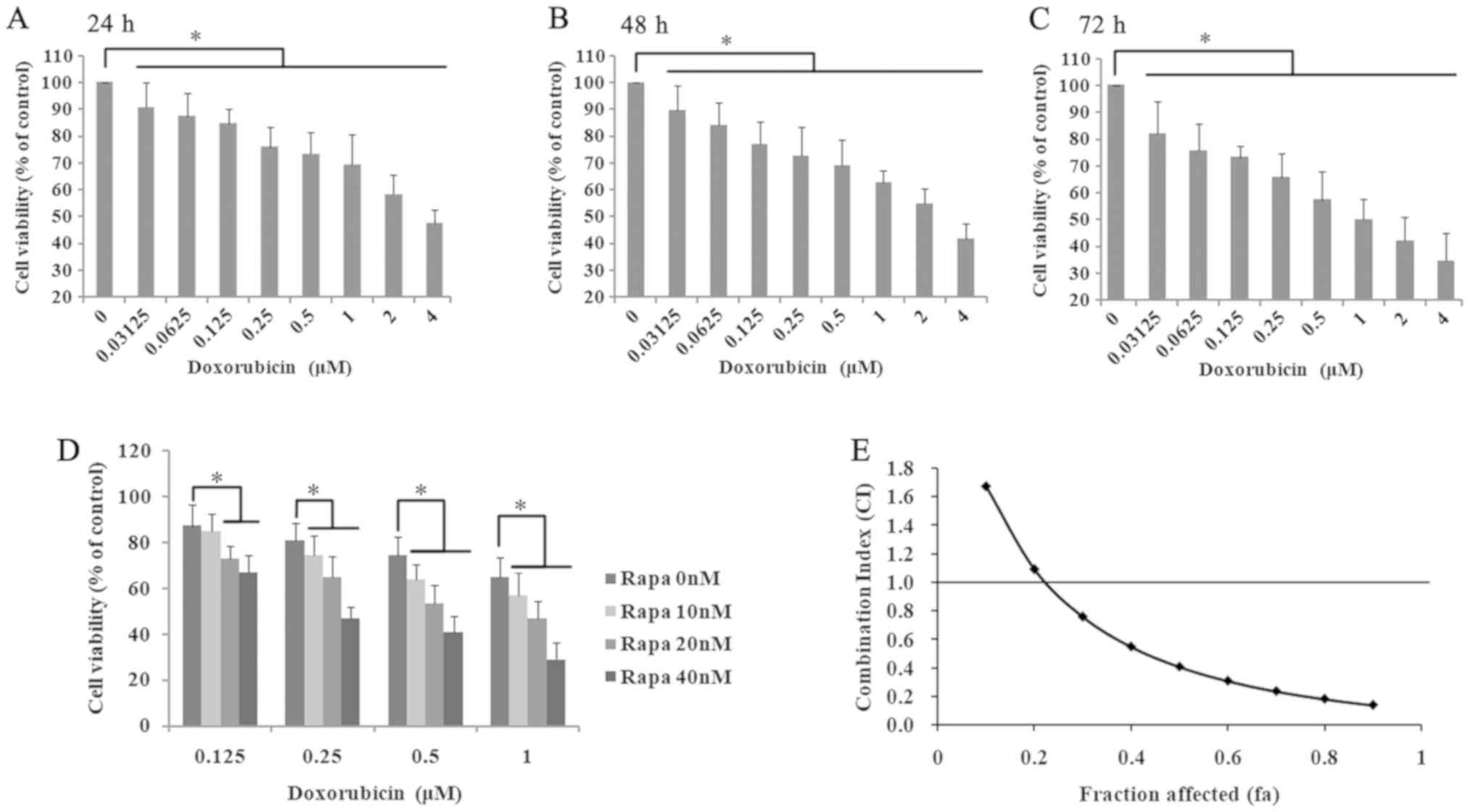
The cytotoxic effect of doxorubicin on K562 cells
was measured by the CCK-8 assay after treating cells with
increasing concentrations for 24, 48 and 72 h (Fig. 1). As shown in Fig. 1A, when the cells were treated with
doxorubicin for 24 h, the cell viability was significantly
decreased in a dose-dependent manner (0.03125–4 µM; r=−0.946;
P<0.05). The IC50 of doxorubicin treated for 24 h was
3.47±0.57 µM. In addition, doxorubicin treatment for 48 and 72 h
also significantly inhibited cell proliferation (48h: r=−0.958,
P<0.05; Fig. 1B; 72 h: r=−0.968;
P<0.05; Fig. 1C).
In the authors' previously published paper (12), K562 cells were incubated with various
concentrations of rapamycin (5–1,000 nM) and the IC50 of
rapamycin treatment for 24 h was 174.94±14.59 nM. To further
understand the possible synergistic mechanism of rapamycin and
doxorubicin, the cells were incubated with 0.125, 0.25, 0.5 and 1.0
µM doxorubicin, combined with 10, 20 and 40 nM rapamycin. The cell
survival rates of K562 cells were significantly decreased in the
two-drug treatment group compared with the matched single-drug
group and the control group (P<0.05; Fig. 1D). Of note, the cell viability in the
0.25 µM doxorubicin and 10 nM rapamycin-treated group was 74.4%,
and the cell inhibition rate was ~25%. In addition, the CI of
rapamycin and doxorubicin was analyzed using the Chou-Talalay
method (30). As shown in Fig. 1D and E, when the rapamycin
concentration was >10 nM and the doxorubicin concentration was
>0.2 µM, the fraction affected (at growth inhibition rates of
25%) is higher than 0.25 and the CI was <1. According to the
definition of the CI (27,28), when CI is <1, the combination of
rapamycin and doxorubicin had a synergistic inhibitory effect on
the growth of K562 cells. Among these groups, the cell survival
rate of K562 cells treated with 20 nM rapamycin and 0.5 µM
doxorubicin was 53.4±8.20%. Therefore, 20 nM rapamycin and 0.5 µM
doxorubicin was chosen for the following experiments.
Rapamycin could enhance the apoptotic
effect of doxorubicin on K562 cells
To assess whether apoptosis contributes to cell
growth inhibition in rapamycin and doxorubicin treatment groups,
the effects of two drugs on K562 cell apoptosis were studied. The
apoptotic rates following treatment with rapamycin, doxorubicin and
a combination of both were significantly increased compared with in
the solvent control group (P<0.05; Fig. 2A). The apoptotic cells, I n
particular, were more frequently detected in the rapamycin +
doxorubicin treatment group (25.50±1.25%) compared with rapamycin
(12.23±1.37%) and doxorubicin (14.87±1.34%) treatment groups
(P<0.05; Fig. 2A; Table II).
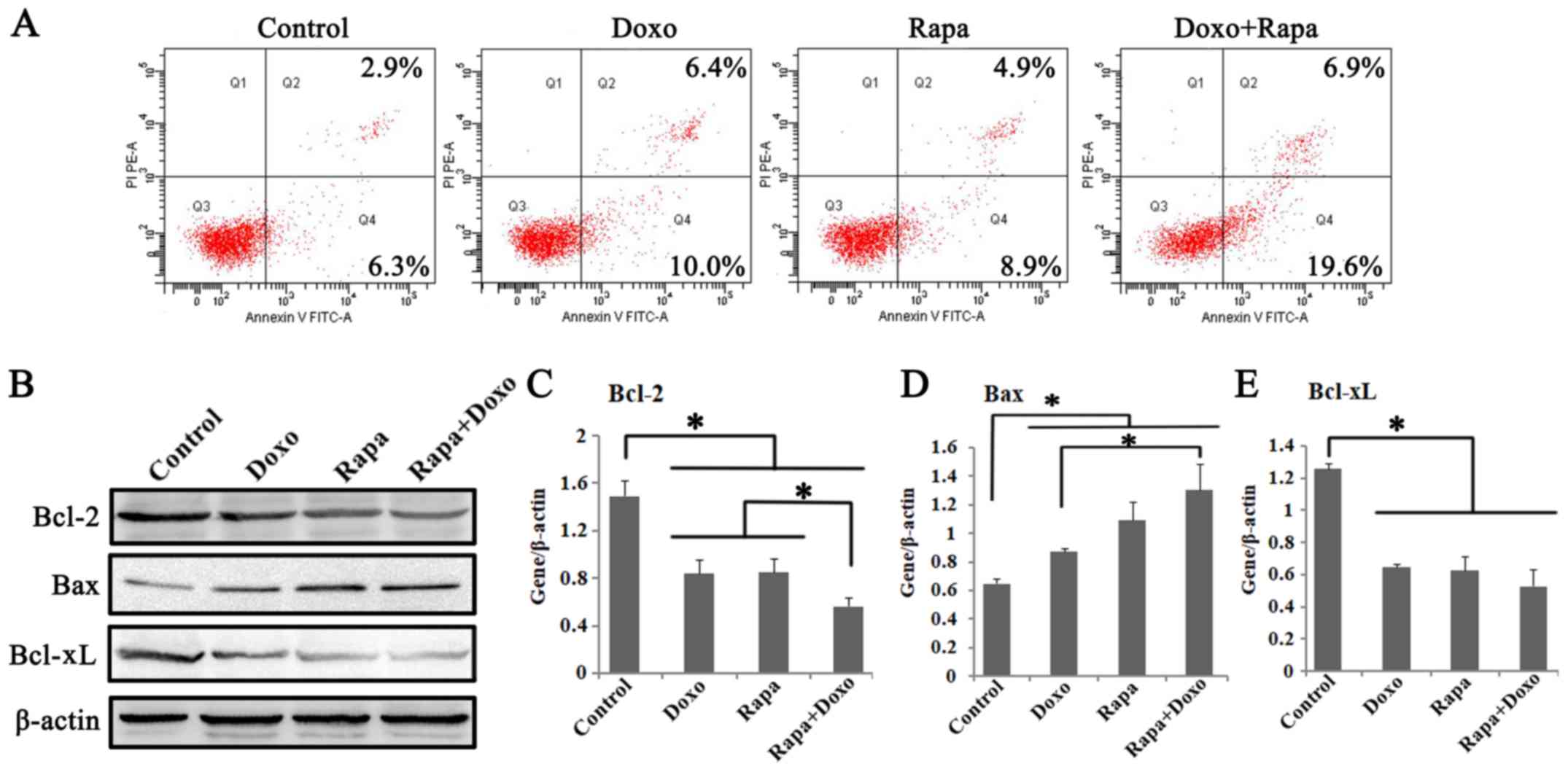 | Figure 2.Rapa and Doxo induced cell apoptosis
of K562 cells. Cells were incubated with Doxo (0.5 µM) and Rapa (20
nM) for 24 h. (A) Apoptosis of K562 cells was determined by Annexin
V/PI apoptosis detection kit. Rapa and Doxo combination
significantly increased apoptotic cells, compared with the matched
single-drug group. (B) Western blotting showed the expressions of
Bcl-2, Bax and Bcl-xL in Rapa, Doxo and the combination treatment
group. The protein expression of (C) Bcl-2, (D) Bax and (E) Bcl-xL
in K562 cells. Rapa and Doxo significantly decreased expression of
(C) Bcl-2 and (E) Bcl-xL, and increase the expression of (D) Bax,
respectively. *P<0.05. Data were presented as the mean ±
standard deviation of triplicates. Rapa, rapamycin; Doxo,
doxorubicin; FITC, fluorescein isothiocyanate; PI, propidium
iodide. |
| Table II.Effect of rapamycin and doxorubicin
on apoptosis in K562 cells. |
Table II.
Effect of rapamycin and doxorubicin
on apoptosis in K562 cells.
|
|
| Apoptosis (%) |
|---|
|
|
|
|
|---|
| Group | Early | Later | Total |
|---|
| Control | 5.63±0.61 | 2.83±0.50 | 8.47±0.95 |
| Doxo |
9.13±0.90a |
5.73±0.65a |
14.87±1.34a |
| Rapa |
8.30±0.66a | 3.93±0.87 |
12.23±1.37a |
| Doxo+Rapa |
18.10±1.37a,b |
7.40±0.62a,b |
25.50±1.25a,b |
To further investigate the apoptotic pathway
activated by rapamycin and doxorubicin, the effects of both drugs
on the expression of proteins that are pivotal for apoptosis,
including Bcl-2, Bax, and Bcl-xL, were studied. As shown in
Fig. 2B, the significant
downregulation of Bcl-2 and Bcl-xL and significant upregulation of
Bax were observed in rapamycin, doxorubicin, and the combination
treatment group (P<0.05; Fig.
2C-E). Of note, compared with the single-treatment group, a
significantly decreased Bcl-2 and increased Bax expression were
observed in the combination treatment cells (P<0.05). It
indicated that a decrease in the Bcl-2/Bax ratio might be involved
in the apoptotic pathways induced by rapamycin and doxorubicin in
K562 cells. The above findings confirmed that rapamycin could
enhance the apoptotic effect of doxorubicin on K562 cells.
Inactivation of the mTOR/p70S6K
pathway in rapamycin- and doxorubicin-treated K562 cells
The effects of rapamycin and doxorubicin on the
mTOR/p70s6k pathway in K562 cells were investigated by western blot
analysis. As shown in Fig. 3A,
although the total mTOR expression showed slight changes in K562
cells, the phosphorylation level of mTOR (Ser-2448) treated with
rapamycin, doxorubicin and a combination of both was significantly
decreased compared with in the solvent-treated control group
(P<0.05; Fig. 3A). The expression
of p70S6K was also investigated. Rapamycin, doxorubicin and a
combination of both induced a significant decrease in the total and
the phosphorylation level of p70S6K (Thr-389) in K562 cells
(P<0.05; Fig. 3B). In addition,
p70S6K and its phosphorylated form were significantly decreased in
cells treated with both drugs compared with those treated with
rapamycin or doxorubicin alone (P<0.05; Fig. 3B).
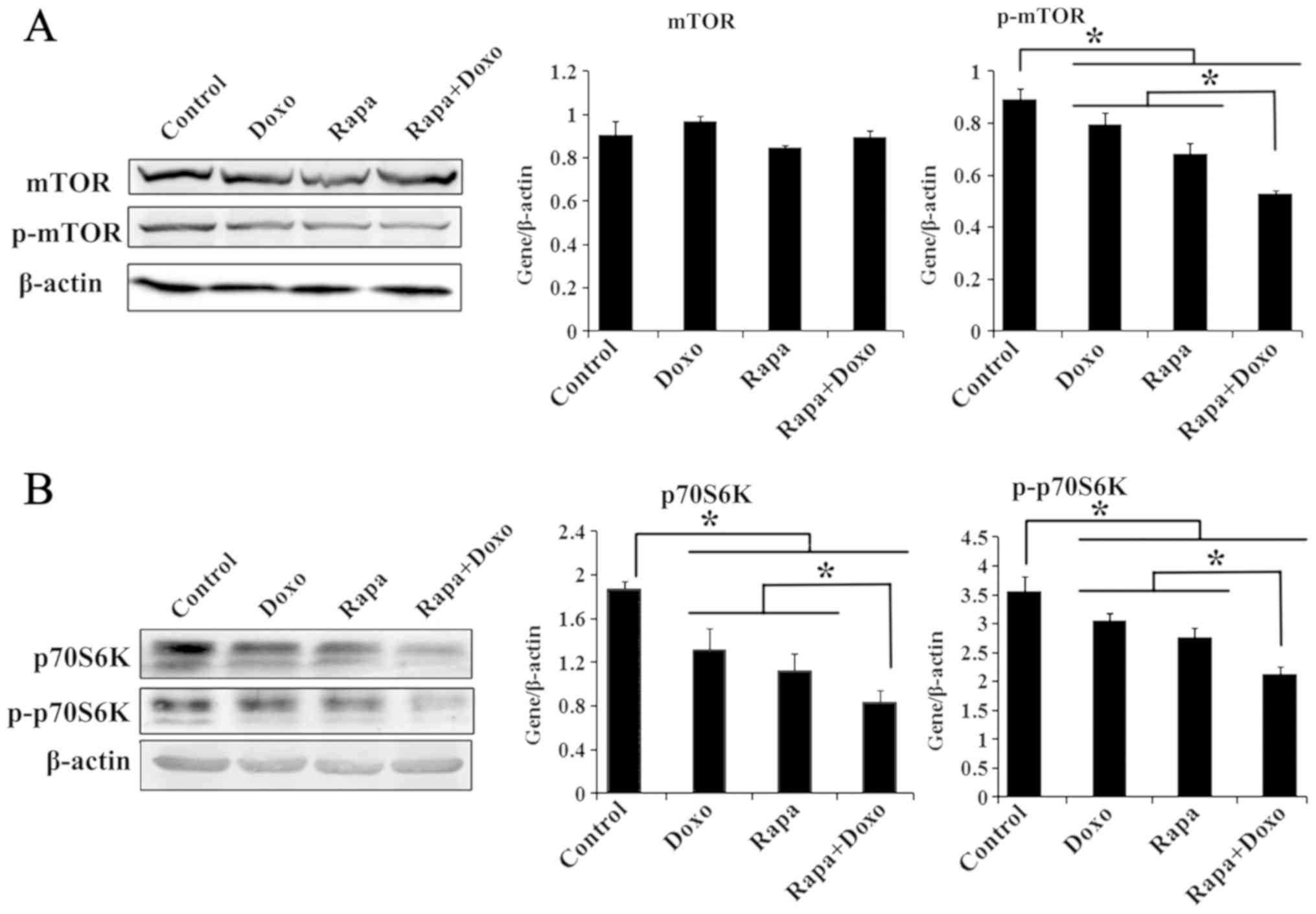 | Figure 3.Rapa and Doxo inhibit expression of
mTOR/p70S6K signaling. Cells were cultured with Doxo (0.5 µM), Rapa
(20 nM) or combination treatment. Total protein was extracted,
followed by immunoblotting with mTOR, p-mTOR (Ser-2448), p70S6K,
p-p70S6K (Thr-389) and β-actin antibodies. Experiments were
repeated 3 times and data were presented as the mean ± standard
deviation of triplicates. (A) mTOR phosphorylation (Ser-2448) was
significantly decreased compared with the solvent control group and
the matched single-drug group. *P<0.05. (B) Rapa, Doxo, alone or
combination treatment decreased the expression of p70S6K and its
phosphorylation (Thr-389) *P<0.05. Rapa, rapamycin; Doxo,
doxorubicin; p-mTOR, phosphorylated-mammalian target of rapamycin;
p70S6K, ribosomal protein S6 kinase. |
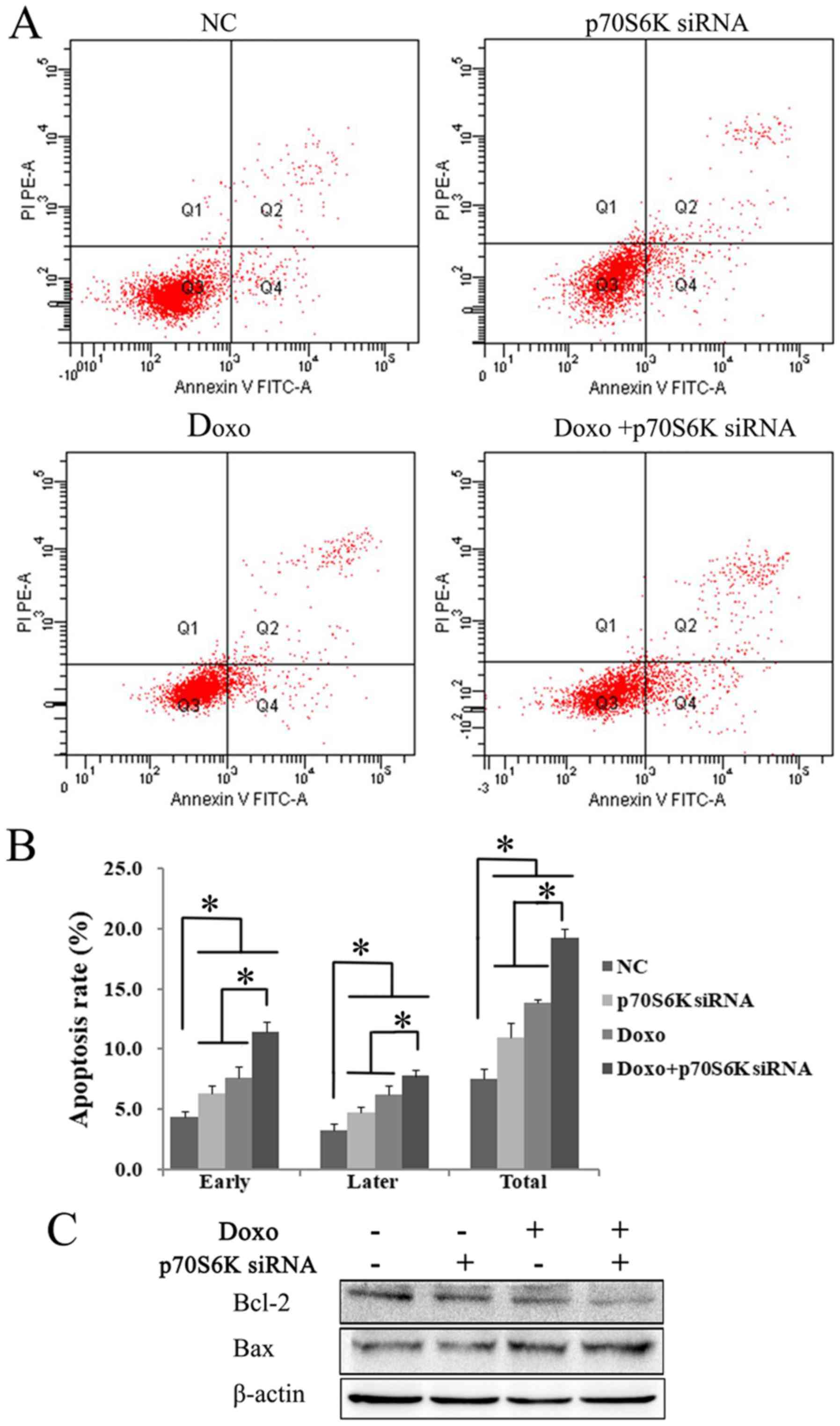
p70S6K siRNA decreases the expression
of p70S6K in K562 cells
To understand the role of p70S6K in the rapamycin
pathway, K562 cells were transfected with targeting siRNA to block
p70S6K. The successful knockdown of p70S6K was confirmed by RT-qPCR
and western blot analysis. Compared with the control siRNA group,
the mRNA expression of p70S6K was significantly decreased to ~1/5
in p70S6K targeting siRNA cells (P<0.05; Fig. 4A). Furthermore, western blot analysis
results showed that the p70S6K expression was markedly decreased in
the p70S6K siRNA treatment group (Fig.
4B). The present data showed that the p70S6K-targeting siRNA
effectively downregulated the expression of p70S6K.
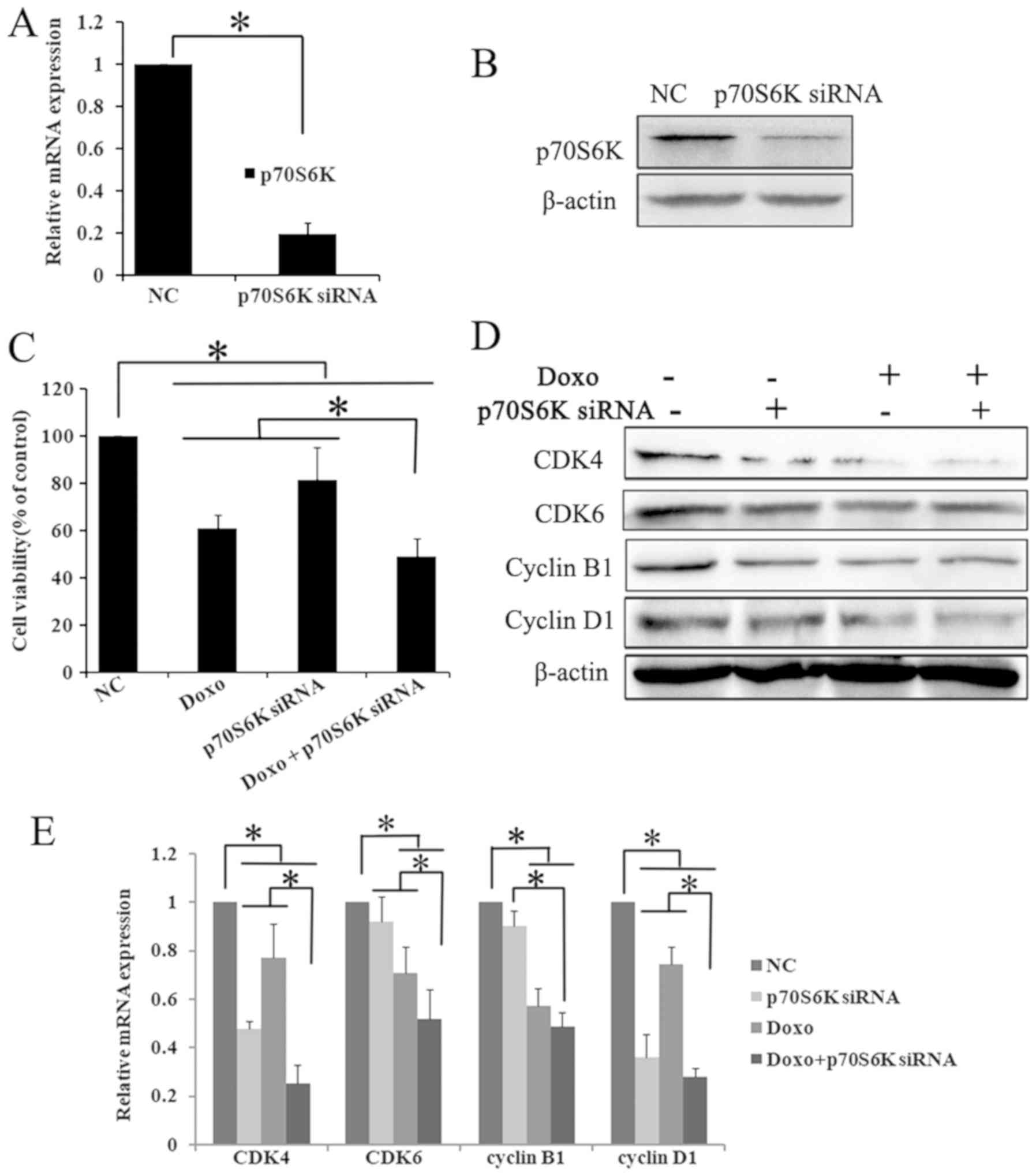 | Figure 4.p70S6K knock down enhances the
anti-proliferative effects of Doxo in K562 cells. K562 cells were
cultured and transfected with p70S6K targeting siRNA for 48 h.
Successful knockdown of p70S6K was confirmed by (A) RT-qPCR and (B)
western blot assays. The results showed that targeting siRNA
significantly decreased the expression of p70S6K, compared with the
control siRNA group. *P<0.05. Data were presented as mean ± SD
of triplicates. (C) Cells were cultured with Doxo (0.5 µM) after
p70S6K knockdown, the cell viability was determined by Cell
Counting Kit-8. The exposure of p70S6K knockdown and doxorubicin
alone or combination significantly inhibited cell viability.
*P<0.05. The values are represented as the mean ± SD. The
expression of cell cycle molecules were detected using (D) western
blotting and (E) RT-qPCR. The expression of CDK4, CDK6, cyclin B1
and cyclin D1 were significantly decreased in p70S6K knockdown and
Doxo treatment group compared withthe single treatment group and
the control group. *P<0.05. Data were presented as mean ± SD of
triplicates. CDK, cyclin dependent kinases; SD, standard deviation;
RT-q, reverse transcription-quantitative; si, small interfering;
Doxo, doxorubicin; p70S6K, ribosomal protein S6 kinase. |
p70S6K downregulation enhances the
inhibitory effect of doxorubicin on cell proliferation
To investigate the role of p70S6K in the inhibitory
effect of rapamycin and doxorubicin on cell proliferation, cell
viability was further detected following treatment with p70S6K
siRNA and doxorubicin. The CCK-8 assay results showed that
p70S6K-targeting siRNA and doxorubicin treatment significantly
decreased cell proliferation compared with the p70S6K siRNA,
doxorubicin and control siRNA groups (P<0.05; Fig. 4C).
Certain critical proteins involved in cell
proliferation were also investigated by western blot analysis and
RT-qPCR. The knockdown of p70S6K and doxorubicin treatment
significantly decreased the mRNA and protein expression of CDK4,
CDK6, cyclin D1 and cyclin B1 (P<0.05; Fig. 4D and E). These results may suggest
that rapamycin could enhance the inhibitory effect of doxorubicin
on cell proliferation by downregulating the mTOR/p70S6K pathway in
K562 cells.
p70S6K downregulation enhances the
doxorubicin-induced K562 cell apoptosis
To study whether the cytotoxic effects of treatment
with p70S6K siRNA and doxorubicin are associated with enhanced
apoptosis, cell apoptosis was examined by FCM. K562 cells were
treated with p70S6K siRNA, doxorubicin alone and a combination of
both. The results showed that the knockdown of p70S6K and
doxorubicin treatment significantly increased the early, late and
total apoptotic rates, as compared with the control group
(P<0.05; Fig. 5A and B). Compared
with single treatment, the exposure of cells to p70S6K siRNA and
doxorubicin significantly increased apoptosis (P<0.05; Fig. 5A and B). In addition, western blot
analysis results showed a decreased expression of Bcl-2 and
increased expression of Bax in K562 cells treated with p70S6K siRNA
and doxorubicin (Fig. 5C). These
data suggested that blocking p70S6K signaling could enhance
doxorubicin-induced K562 cell apoptosis.
Discussion
Despite the fact that Bcr/Abl kinase inhibitors have
significantly improved life expectancy in the majority of patients
in the chronic phase of CML, certain patients progressing to the
blast phase will develop Bcr/Abl-independent mechanisms of
resistance. This patient population requires chemotherapeutics,
including doxorubicin and cytarabine, or needs an alternative drug
target that can be inhibited with a novel compound. Certain studies
have shown that mTOR signaling is frequently activated in CML
(8–12). In addition, Bcr/Abl kinase could
regulate PI3K activity through its regulatory subunit and mTOR was
subsequently activated by the phosphorylation of AKT in CML cell
lines (15). It was found in the
authors' previous studies that rapamycin markedly inhibited cell
growth and enhanced the antitumor effects of celecoxib on CML cells
by downregulating the mTOR pathway (12,17). It
is well known that mTOR exists in two multiprotein complexes,
mTORC1 and mTORC2. The main function of mTORC1 which phosphorylates
p70S6K and 4E-BP1, is sensitive to nutrients and rapamycin. In
contrast to mTORC1, mTORC2 which is insensitive to rapamycin, could
phosphorylate AKT at Ser473 and forms a feedback loop (31). Certain studies have demonstrated that
an mTOR inhibitor, combined additively or synergistically with
several chemotherapies, such aspaclitaxel, doxorubicin and
carboplatin, could inhibit cancer cells proliferation in
vitro (32–34). Collectively, targeting the mTOR
pathway may be a new therapeutic approach. However, the mechanisms
through which rapamycin may have synergistic effects with
doxorubicin on leukemia cells remain virtually unknown.
To further explore the possible synergistic
mechanisms of rapamycin and doxorubicin on cell growth, K562 cells
were cultured with a combination of various concentrations.
According to the authors' published paper (12), the IC50 of rapamycin
treatment for 24 h is 174.94±14.59 nM. It was reported that the
distribution volume of doxorubicin varies markedly in different
patients or for different cell lines (23–26).
Therefore, to investigate effects of doxorubicin on cell
proliferation, K562 cells were cultured with various concentrations
from 0.03125–4 µM. The results showed that doxorubicin decreased
K562 cells in a dose-dependent manner and The IC50 of
doxorubicin treatment for 24 h was 3.47±0.57 µM. The cell survival
rates of K562 cells were markedly lower in two-drug treatment group
than in the matched single-drug group. Meanwhile, when the
rapamycin concentration was >10 nM, the doxorubicin
concentration was >0.2 µM, and the growth inhibition rates is
higher than 25%, CI was less than 1 (CI<1). Co-incubation
markedly increased the response of K562 cells to doxorubicin,
indicating that rapamycin synergistically enhance the
anti-proliferative effects of doxorubicin in K562 cells.
In addition, cell apoptosis was measured in
rapamycin- and doxorubicin-treated K562 cells. The results showed
that individual treatment can induce cell apoptosis and regulate
the expression of Bcl-2, Bax and Bcl-xL. As compared with
single-drug treatment, rapamycin combined with doxorubicin
increased cell apoptosis. It has been demonstrated that the Bcl-2
family members regulate the cell apoptotic pathway. In particular,
increasing Bax expression and/or decreasing Bcl-2 expression may
induce apoptosis (35). The present
results showed that both drugs induced Bcl-2 downregulation and Bax
upregulation, resulting in a decrease in the Bcl-2/Bax ratio. It
was also found that rapamycin and doxorubicin can decrease the
phosphorylation of mTOR and p70S6K. A recent study found combined
treatment with rapamycin and melatonin to be associated with
increased apoptosis and mitophagy in head and neck cancers
(33). These findings suggested that
rapamycin enhances the apoptotic effect of doxorubicin on K562
cells through the mTOR/p70S6K pathway.
As is known, 4EBP1 and p70S6K are the two most
important downstream proteins of the mTOR pathway. The
phosphorylation of 4E-BP1 (Ser37; Thr46; Ser65; Thr70) regulates
cap-dependent transcription and translation of a great number of
proteins. On the other hand, activated p70S6K could increase the
translation of 5′-terminal oligopyrimidine tract mRNAs, regulates
protein synthesis and plays a critical role in controlling cell
growth (36). Extensive evidence has
revealed that the overexpression and phosphorylation of p70S6K
promotes cell proliferation, angiogenesis and suppression of
apoptosis in vitro (37,38),
whereas, the p70S6K specific inhibitor (PF-4708671) has inhibitory
effects on non-small cell lung cancer in vitro and in
vivo (39). High p70S6K
phosphorylation has also been reported to be an independent
biomarker of poor prognosis in right-sided colorectal cancer
(20). Similar observations of the
p-p70S6K prognostic function have been made for lung, esophagealand
ovarian cancer, and hepatocellular carcinoma (40–42).
Studies havepreviously demonstrated that
overexpression of p70S6K in tumor tissues contributes to
chemotherapy resistance (43). High
levels of p-p70S6K were involved in selumetinib resistance, and the
inhibition of p70S6K could reverse this chemotherapeutic resistance
in colorectal cancer (44). The
authors have previously found that the inhibition of p70S6K by
rapamycin was additionally associated with an increased number of
G0/G1 cells and apoptotic cells in leukemia,
while rapamycin combined with celecoxib enhanced this effect
(12,17). A more recent study showed p70S6K1
suppressed autophagy through inhibiting AMPK and JNK in a
TAK1-dependent manner (45).
In the present study, a p70S6K-targeting siRNA was
used as an inhibitor to knockdown p70S6K and more attention was
paid to investigate its mechanism. It was found that p70S6K
knockdown and doxorubicin treatment decreased cell proliferation,
compared with the single treatment. The p70S6K siRNA and
doxorubicin clearly decreased the expression of CDK4, CDK6, cyclin
D1 and cyclin B1, as compared with the control and single-treatment
groups. These molecules are key elements in the cell cycle and
serve an essential role in the progression of various types of
cancer. Among these, cyclin D1 binding to CDK4 is triggered at the
early stage of the G1 phase. The inhibition of cyclin D1
or CDK4 partly restores G1/S arrest and sensitizes cells
to doxorubicin-mediated cell apoptosis, significantly overcoming
doxorubicin resistance (46,47). The authors have previously
demonstrated that rapamycin arrested K562 cells at the
G0/G1 phase through mTOR and its downstream
molecules (12,15). The present study showed that the
downregulation of p70S6K could inhibit CDK4 and cyclin D1
expression. In addition, compared with single treatment, the
knockdown of p70S6K and doxorubicin treatment significantly
increased cell apoptosis. Furthermore, western blot analysis
results showed that p70S6K siRNA and doxorubicin regulate the
expression of Bcl-2 and Bax in K562 cells. It suggested that
rapamycin synergistically enhanced the doxorubicin-induced
inhibition of cell proliferation and apoptosis through the
mTOR/p70S6K pathway in K562 cells.
In conclusion, these findings showed that rapamycin
and doxorubicin synergistically inhibited cell growth, induced
apoptosis, and decreased the expression of mTOR/p70S6K. In
addition, p70S6K blocking by siRNA, combined with doxorubicin,
could inhibit cell proliferation and induce apoptosis in K562
cells. The authors' preliminary results suggested that rapamycin
may enhance the antitumor effects of doxorubicin on K562 cells by
downregulating mTOR/p70S6K signaling. Targeting the mTOR pathway
may be a more suitable therapeutic approach for leukemia.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Hebei Province (grant no. H2017307024) and
the Project of Health Department of Hebei Province (grant no.
20170030).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and LX designed the study, analyzed the data and
wrote the manuscript. WL and QW performed the experiments and
prepared the figures. HH designed the study, prepared the figures
and analyzed the data. JL and LX performed critical revision of the
manuscript and supervised the study. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Hebei General Hospital (Shijiazhuang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rowley JD: Ph1-positive leukaemia,
including chronic myelogenous leukaemia. Clin Haematol. 9:55–86.
1980.PubMed/NCBI
|
|
2
|
Groffen J, Stephenson JR, Heisterkamp N,
de Klein A, Bartram CR and Grosveld G: Philadelphia chromosomal
breakpoints are clustered within a limited region, bcr, on
chromosome 22. Cell. 36:93–98. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gorre ME, Mohammed M, Ellwood K, Hsu N,
Paquette R, Rao PN and Sawyers CL: Clinical resistance to STI-571
cancer therapy caused by BCR-ABL gene mutation or amplification.
Science. 293:876–880. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Okabe S, Tauchi T, Tanaka Y, Kitahara T,
Kimura S, Maekawa T and Ohyashiki K: Efficacy of the dual PI3K and
mTOR inhibitor NVP-BEZ235 in combination with nilotinib against
BCR-ABL-positive leukemia cells involves the ABL kinase domain
mutation. Cancer Biol Ther. 15:207–215. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun Z, Li Q, Zhang S, Chen J, Huang L, Ren
J, Chang Y, Liang Y and Wu G: NVP-BEZ235 overcomes
gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR
phosphorylation. Onco Targets Ther. 8:269–277. 2015.PubMed/NCBI
|
|
6
|
Xu J, Pham CG, Albanese SK, Dong Y, Oyama
T, Lee CH, Rodrik-Outmezguine V, Yao Z, Han S, Chen D, et al:
Mechanistically distinct cancer-associated mTOR activation clusters
predict sensitivity to rapamycin. J Clin Invest. 126:3526–3540.
2016. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morran DC, Wu J, Jamieson NB, Mrowinska A,
Kalna G, Karim SA, Au AY, Scarlett CJ, Chang DK, Pajak MZ, et al:
Targeting mTOR dependency in pancreatic cancer. Gut. 63:1481–1489.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mohi MG, Boulton C, Gu TL, Sternberg DW,
Neuberg D, Griffin JD, Gilliland DG and Neel BG: Combination of
rapamycin and protein tyrosine kinase (PTK) inhibitors for the
treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci
USA. 101:3130–3135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parmar S, Smith J, Sassano A, Uddin S,
Katsoulidis E, Majchrzak B, Kambhampati S, Eklund EA, Tallman MS,
Fish EN and Platanias LC: Differential regulation of the p70 S6
kinase pathway by interferon alpha (IFNalpha) and imatinib mesylate
(STI571) in chronic myelogenous leukemia cells. Blood.
106:2436–2443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hirase C, Maeda Y, Takai S and Kanamaru A:
Hypersensitivity of Ph-positive lymphoid cell lines to rapamycin:
Possible clinical application of mTOR inhibitor. Leuk Res.
33:450–459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Batista A, Barata JT, Raderschall E,
Sallan SE, Carlesson N, Nadler LM and Cardoso AA: Targeting of
active mTOR inhibits primary leukemia T cells and synergizes with
cytotoxic drugs and signaling inhibitors. Exp Hematol.
39:457–472.e3. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Xue L, Hao H, Han Y, Yang J and Luo
J: Rapamycin provides a therapeutic option through inhibition of
mTOR signaling in chronic myelogenous leukemia. Oncol Rep.
27:461–466. 2012.PubMed/NCBI
|
|
13
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: Effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo N, Azadniv M, Coppage M, Nemer M,
Mendler J, Becker M and Liesveld J: Effects of neddylation and mTOR
inhibition in acute myelogenous leukemia. Transl Oncol. 12:602–613.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ly C, Arechiga AF, Melo JV, Walsh CM and
Ong ST: Bcr-Abl kinase modulates the translation regulators
ribosomal protein S6 and 4E-BP1 in chronic myelogenous leukemia
cells via the mammalian target of rapamycin. Cancer Res.
63:5716–5722. 2003.PubMed/NCBI
|
|
16
|
Xin P, Li C, Zheng Y, Peng Q, Xiao H,
Huang Y and Zhu X: Efficacy of the dual PI3K and mTOR inhibitor
NVP-BEZ235 in combination with imatinib mesylate against chronic
myelogenous leukemia cell lines. Drug Des Devel Ther. 11:1115–1126.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Xue L, Hao H, Li R and Luo J:
Rapamycin combined with celecoxib enhanced antitumor effects of
mono treatment on chronic myelogenous leukemia cells through
downregulating mTOR pathway. Tumour Biol. 35:6467–6474. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Harada H, Andersen JS, Mann M, Terada N
and Korsmeyer SJ: p70S6 kinase signals cell survival as well as
growth, inactivating the pro-apoptotic molecule BAD. Proc Natl Acad
Sci USA. 98:9666–9670. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie Y, Naizabekov S, Chen Z and Tokay T:
Power of PTEN/AKT: Molecular switch between tumor suppressors and
oncogenes. Oncol Lett. 12:375–378. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vicary GW and Roman J: Targeting the
mammalian target of rapamycin in lung cancer. Am J Med Sci.
352:507–516. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kurgan N, Tsakiridis E, Kouvelioti R,
Moore J, Klentrou P and Tsiani E: Inhibition of human lung cancer
cell proliferation and survival by post-exercise serum is
associated with the inhibition of Akt, mTOR, p70 S6K, and Erk1/2.
Cancers (Basel). 9:E462017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wiesweg M, Reis H, Köster T, Goetz M, Worm
K, Herold T, Paul A, Dechêne A, Schumacher B, Markus P, et al:
Phosphorylation of p70 ribosomal protein S6 kinase β-1 is an
independent prognostic parameter in metastatic colorectal cancer.
Clin Colorectal Cancer. 17:e331–e352. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morjani H, Pignon B, Millot JM, Debal V,
Lamiable D, Potron G, Etienne JC and Manfait M: Intranuclear
concentration measurements of doxorubicin in living leucocytes from
patients treated for a lympho-proliferative disorder. Leuk Res.
16:647–653. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Speth PA, Linssen PC, Termond EF, Boezeman
JB, Wessels HM and Haanen C: In vivo and in vitro pharmacokinetic
differences between four structurally closely related
anthracyclines in hematopoietic cell subtypes in humans. Drug Metab
Dispos. 17:98–105. 1989.PubMed/NCBI
|
|
25
|
Skorkina MY, Shamray EA, Salo VA,
Buchelnikov AS and Evstigneev MP: Study of the properties of
doxorubicin-resistant cells affected by acute leucosis. J Bioenerg
Biomembr. 50:53–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chekin F, Myshin V, Ye R, Melinte S, Singh
SK, Kurungot S, Boukherroub R and Szunerits S: Graphene-modified
electrodes for sensing doxorubicin hydrochloride in human plasma.
Anal Bioanal Chem. 411:1509–1516. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin ZJ: Addition in drug combination
(author's transl). Zhongguo Yao Li Xue Bao. 1:70–76. 1980.(In
Chinese). PubMed/NCBI
|
|
28
|
Ding Y, Niu W, Zhang T, Wang J, Cao J,
Chen H, Wang R and An H: Levistolide A synergistically enhances
doxorubicin-induced apoptosis of k562/dox cells by decreasing MDR1
expression through the ubiquitin pathway. Oncol Rep. 41:1198–1208.
2019.PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chou TC: Theoretical basis, experimental
design, and computerized simulation of synergism and antagonism in
drug combination studies. Pharmacol Rev. 58:621–681. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yuan HX and Guan KL: The SIN1-PH domain
connects mTORC2 to PI3K. Cancer Discov. 5:1127–1129. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
MondesireW H, Jian W, Zhang H, Ensor J,
Hung MC, Mills GB and Meric-Bernstam F: Targeting mammalian target
of rapamycin synergistically enhances chemotherapy-induced
cytotoxicity in breast cancer cells. Clin Cancer Res. 10:7031–7042.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shen YQ, Guerra-Librero A, Fernandez-Gil
BI, Florido J, García-López S, Martinez-Ruiz L, Mendivil-Perez M,
Soto-Mercado V, Acuña-Castroviejo D, Ortega-Arellano H, et al:
Combination of melatonin and rapamycin for head and neck cancer
therapy: Suppression of AKT/mTOR pathway activation, and activation
of mitophagy and apoptosis via mitochondrial function regulation. J
Pineal Res. 64:2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hsu PY, Wu VS, Kanaya N, Petrossian K, Hsu
HK, Nguyen D, Schmolze D, Kai M, Liu CY, Lu H, et al: Dual mTOR
kinase inhibitor MLN0128 sensitizes HR+/HER2+
breast cancer patient-derived xenografts to trastuzumab or
fulvestrant. Clin Cancer Res. 24:395–406. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zheng JH, Viacava Follis A, Kriwacki RW
and Moldoveanu T: Discoveries and controversies in BCL-2
protein-mediated apoptosis. FEBS J. 283:2690–2700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma XM and Blenis J: Molecular mechanisms
of mTOR-mediated translational control. Nat Rev Mol Cell Biol.
10:307–318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bian CX, Shi Z, Meng Q, Jiang Y, Liu LZ
and Jiang BH: P70S6K 1 regulation of angiogenesis through VEGF and
HIF-1alpha expression. Biochem Biophys Res Commun. 398:395–399.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamnik RL, Digilova A, Davis DC, Brodt ZN,
Murphy CJ and Holz MK: S6 kinase 1 regulates estrogen receptor
alpha in control of breast cancer cell proliferation. J Biol Chem.
284:6361–6369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qiu ZX, Sun RF, Mo XM and Li WM: The
p70S6K specific inhibitor PF-4708671 impedes non-small cell lung
cancer growth. PLoS One. 11:e01471852016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Baba HA, Wohlschlaeger J, Cicinnati VR,
Hilgard P, Lang H, Sotiropoulos GC, Takeda A, Beckebaum S and
Schmitz KJ: Phosphorylation of p70S6 kinase predicts overall
survival in patients with clear margin-resected hepatocellular
carcinoma. Liver Int. 29:399–405. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li SH, Chen CH, Lu HI, Huang WT, Tien WY,
Lan YC, Lee CC, Chen YH, Huang HY, Chang AY and Lin WC:
Phosphorylated p70S6K expression is an independent prognosticator
for patients with esophageal squamous cell carcinoma. Surgery.
157:570–580. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
No JH, Jeon YT, Park IA, Kim YB, Kim JW,
Park NH, Kang SB, Han JY, Lim JM and Song YS: Activation of mTOR
signaling pathway associated with adverse prognostic factors of
epithelial ovarian cancer. Gynecol Oncol. 121:8–12. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoshida S, Matsumoto K, Arao T, Taniguchi
H, Goto I, Hanafusa T, Nishio K and Yamada Y: Gene amplification of
ribosomal protein S6 kinase-1 and −2 in gastric cancer. Anticancer
Res. 33:469–475. 2013.PubMed/NCBI
|
|
44
|
Grasso S, Tristante E, Saceda M, Carbonell
P, Mayor-López L, Carballo-Santana M, Carrasco-García E,
Rocamora-Reverte L, García-Morales P, Carballo F, et al: Resistance
to Selumetinib (AZD6244) in colorectal cancer cell lines is
mediated by p70S6K and RPS6 activation. Neoplasia. 16:845–860.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xu X, Sun J, Song R, Doscas ME, Williamson
AJ, Zhou J, Sun J, Jiao X, Liu X and Li Y: Inhibition of p70 S6
kinase (S6K1) activity by A77 1726, the active metabolite of
leflunomide, induces autophagy through TAK1-mediated AMPK and JNK
activation. Oncotarget. 8:30438–30454. 2017.PubMed/NCBI
|
|
46
|
Ji ZP, Qiang L and Zhang JL: Transcription
activated p73-modulated cyclin D1 expression leads to doxorubicin
resistance in gastric cancer. Exp Ther Med. 15:1831–1838.
2018.PubMed/NCBI
|
|
47
|
Gogolin S, Ehemann V, Becker G, Brueckner
LM, Dreidax D, Bannert S, Nolte I, Savelyeva L, Bell E and
Westermann F: CDK4 inhibition restores G(1)-S arrest in
MYCN-amplified neuroblastoma cells in the context of
doxorubicin-induced DNA damage. Cell Cycle. 12:1091–1104. 2013.
View Article : Google Scholar : PubMed/NCBI
|