Introduction
As a biologically aggressive subtype of malignant
glioma, glioblastoma multiforme (GBM) is the most common and lethal
brain tumour in adults (1). GBM
diffusely infiltrates the brain at an early time point, and its
complexity and distinct pathophysiology are facilitated and
dictated by the unique brain milieu and cellular interactions
(2,3). In total, >80% of GBM cases are
primary tumours, which typically respond poorly to current
therapeutic approaches (4). Although
previous studies detected a number of events associated with
initiation or progression (5,6),
identification of the key molecular targets underlying the
regulatory mechanisms of GBM remains to be elucidated.
At present, the wealth of molecular information
generated by massively parallel sequencing technologies provides a
great opportunity for discovering novel biomarkers in different
types of cancer (7,8). Transcriptome analysis involving
protein-coding genes and noncoding microRNAs (miRNAs/miRs) has
proven to be a valuable first step for studying the genetic
characteristics of various complex human diseases, especially in
malignant gliomas (9,10). For example, Lai et al
(11) found that miRNA-210 could
serve as a potentially non-invasive biomarker for the diagnosis and
prognosis of GBM. An increased expression level of the
protein-coding gene PLAUR was determined to be a valuable predictor
of the mesenchymal GBM subtype (12). However, it is unlikely that a single
dysfunctional gene would lead to abnormal phenotypes. Multiple
aberrant gene-gene interactions may have co-operative effects in
the development and progression of complex diseases such as cancer
(13,14).
Understanding the molecular events associated with
activation of the risk pathways in patients with GBM may facilitate
the development and clinical testing of potential therapeutic
targets.
In the present study, differentially expressed genes
and miRNAs were identified by analysing a large protein-coding gene
and miRNA expression dataset comprised of normal and tumour tissues
from GBM samples obtained from The Cancer Genome Atlas (TCGA). A
GBM-specific regulatory network involving transcription factors
(TFs) and miRNAs was built. The GBM-specific regulatory network
showed a scale-free network with a small-world property. The key
molecules in the regulatory network were significantly associated
with immune-related functions. Linear risk pathways of GBM were
identified through systems-level analyses of the regulatory
network. The identified molecular risk pathways may be a potential
resource for understanding the pathogenesis and aetiology
underlying GBM.
Materials and methods
Acquiring known GBM-related genes
The GeneCards database provides a comprehensive map
of manually curated human disease genes (15). In total, 32 GBM-associated genes were
obtained by searching the GeneCards database according to all
descriptions and aliases of GBM, including ‘GBM’, ‘glioblastoma
multiforme’, ‘malignant brain tumour’, ‘gliomatosis’ and
‘high-grade glioma’. In addition, 41 experimentally validated
GBM-related miRNAs from the miR2Disease database were retrieved
(16), a manually curated database
of miRNA deregulation in various human diseases.
Identification of differentially
expressed genes and miRNAs
The mRNA and miRNA expression profiles for 378
patients with GBM were downloaded from TCGA database (https://portal.gdc.cancer.gov/). The dataset
contained 378 tumour samples and 10 patient-matched normal samples.
The genes were mapped to the Entrez Gene IDs for mRNA expression
data using biomaRt software version 2.40.0 (17). In total, 11,273 genes and 470 miRNAs
were included in the subsequent analysis. A Student's t-test was
used to identify significantly differentially expressed genes and
miRNAs from the GBM expression profiles. All P-values were adjusted
using Bonferroni correction to account for multiple comparisons.
Only genes and miRNAs with an adjusted P-value <0.01 and a fold
change >1.2 were considered statistically significant.
Extraction of the GBM-specific
regulatory network
A TF and miRNA regulatory network was constructed by
integrating the molecular interaction associations between genes
and miRNAs from four databases [miRTarBase (18); The Transcription Factor Database
(19); miRecords (20); and TarBase (21)]. To ensure the reliability of the
relationships between the curated regulations, each individual
interaction presented is supported by the literature.
Although differential expression analysis is often
used to screen disease candidate genes, previous studies have shown
that a number of disease-associated genes do not always exhibit
differential expression patterns in microarray experiments
(14,22). Therefore, a candidate risk regulatory
network was constructed by connecting all of the risk-associated
differentially expressed nodes with their immediate neighbouring
non-differentially expressed nodes.
Mining for GBM-specific risk
regulatory linear pathways
Linear risk pathways potentially associated with GBM
were obtained according to the following criteria: i) The beginning
of a risk pathway was defined as a gene or miRNA with a 0-indegree
(it could not be regulated by other genes or miRNAs); and ii) the
end of a risk pathway was defined as a gene or miRNA with a
0-outdegree (it could not regulate other genes or miRNAs). In the
GBM-specific regulatory network, all linear regulatory pathways
were defined as pathways from the beginning node to the end node
and were detected using R (23).
Functional enrichment analysis
The R package ‘clusterProfiler’ (24) was used to examine the significantly
enriched gene ontology (GO) biological functions (25) of the differentially expressed genes.
The adjusted P-values were calculated using the Benjamini and
Hochberg method.
Results
Differential expression analysis of
mRNAs and miRNAs
To detect potential key molecules underlying the
pathology of GBM, the mRNA and miRNA gene profiles of patients with
GBM from TCGA database were downloaded and differential expression
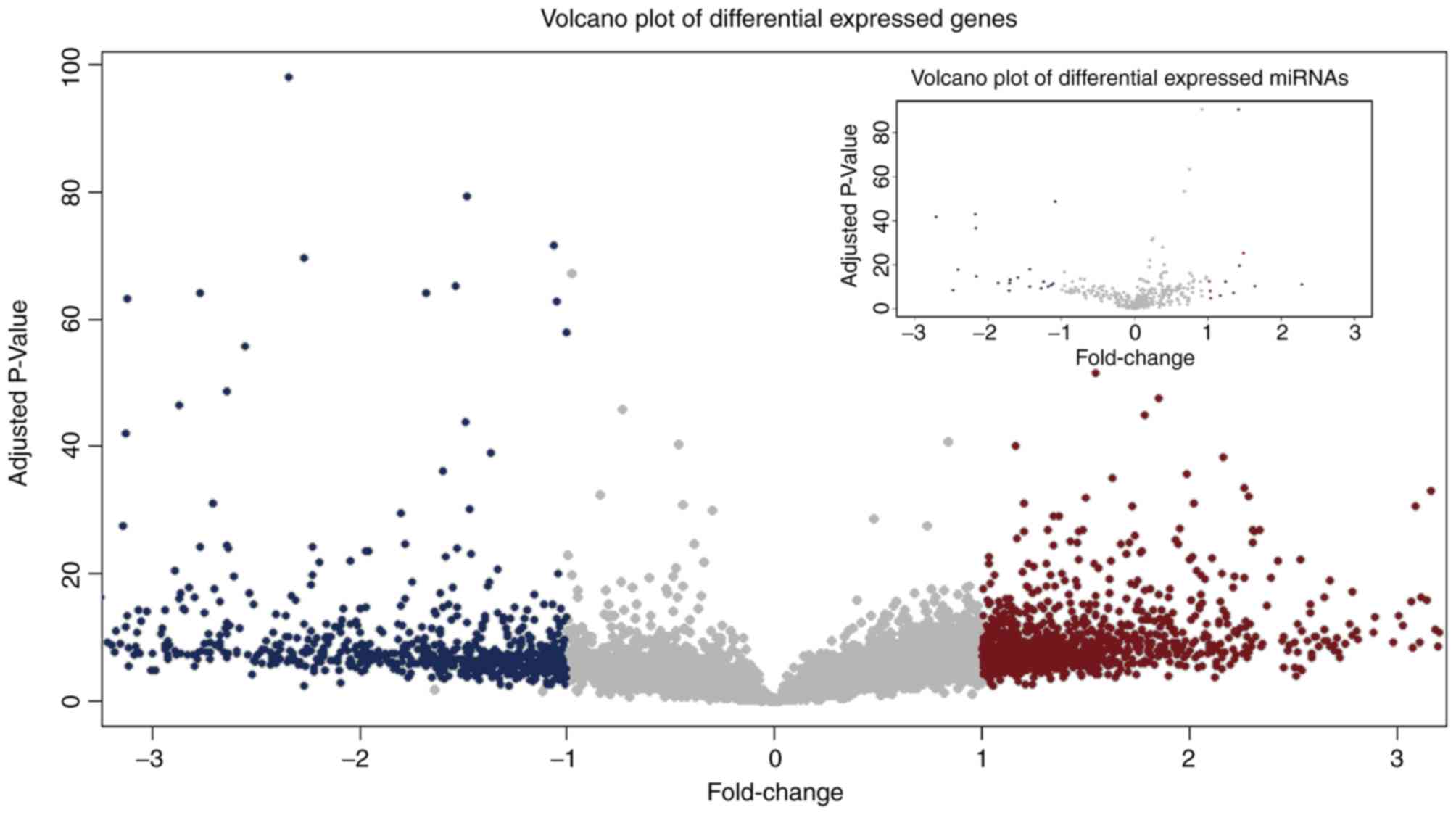
analysis was performed. In total, 1,827 and 30 significantly
differentially expressed genes and miRNAs were detected,
respectively. The number of upregulated genes, (1,085) was greater
than the number of downregulated genes (742). Conversely, the
number of upregulated miRNAs (11)
was smaller than the number of downregulated miRNAs, 19 (Fig. 1). These results suggested that a
number of candidate molecules may contribute to the carcinogenesis
of GBM, and that miRNAs may serve a negative regulatory role.
Systematic characterization of a
regulatory network specific to GBM
To examine the systems-level characteristics of the
‘true’ biological processes, including interactions and other
combined activities associated with GBM, a comprehensive regulatory
network among TFs, miRNAs and genes was built by combining four
manually curated databases that focused on both transcriptional and
post-transcriptional regulation. The background regulatory network
was composed of 6,036 regulatory relationships. Of the background
regulatory relationships, 1,827 differentially expressed genes and
30 differentially expressed miRNAs were considered as the risk
seeds and mapped into the regulatory network to obtain a
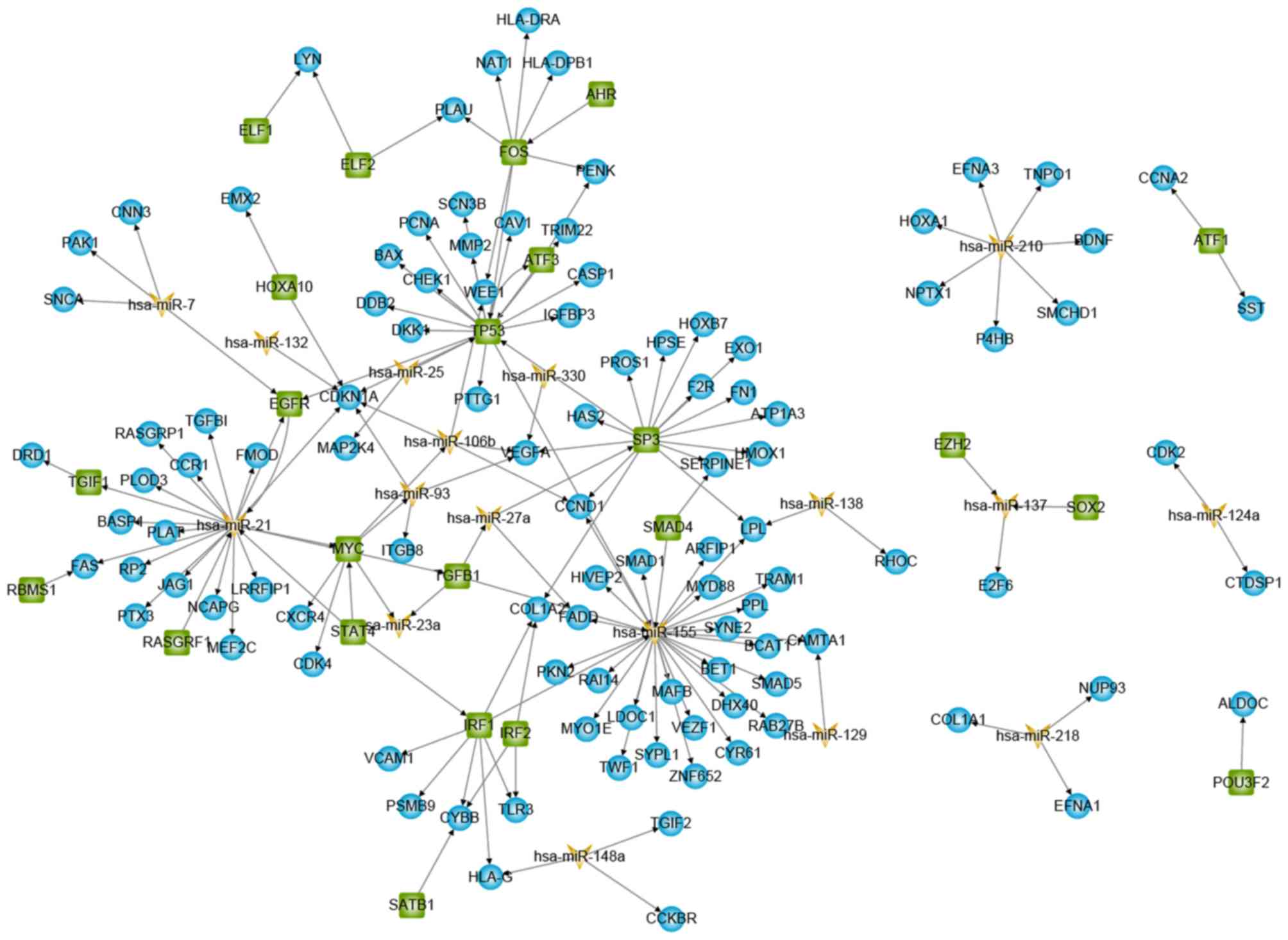
GBM-specific sub-network (Fig. 2).
Given that networks provide knowledge that can be used to infer the
information flow between regulators and target genes, a
GBM-specific sub-network consisting of differentially expressed
genes/miRNAs and their immediate neighbouring nodes from the
TF-miRNA-gene regulatory network was further extracted. The
GBM-specific regulatory network contained 160 interactions
involving 23 TFs, 17 miRNAs and 105 target genes.
The network showed dense local interconnectivity and
complied with a power-law distribution suggesting that the
GBM-specific sub-network was scale-free, similar to most biological
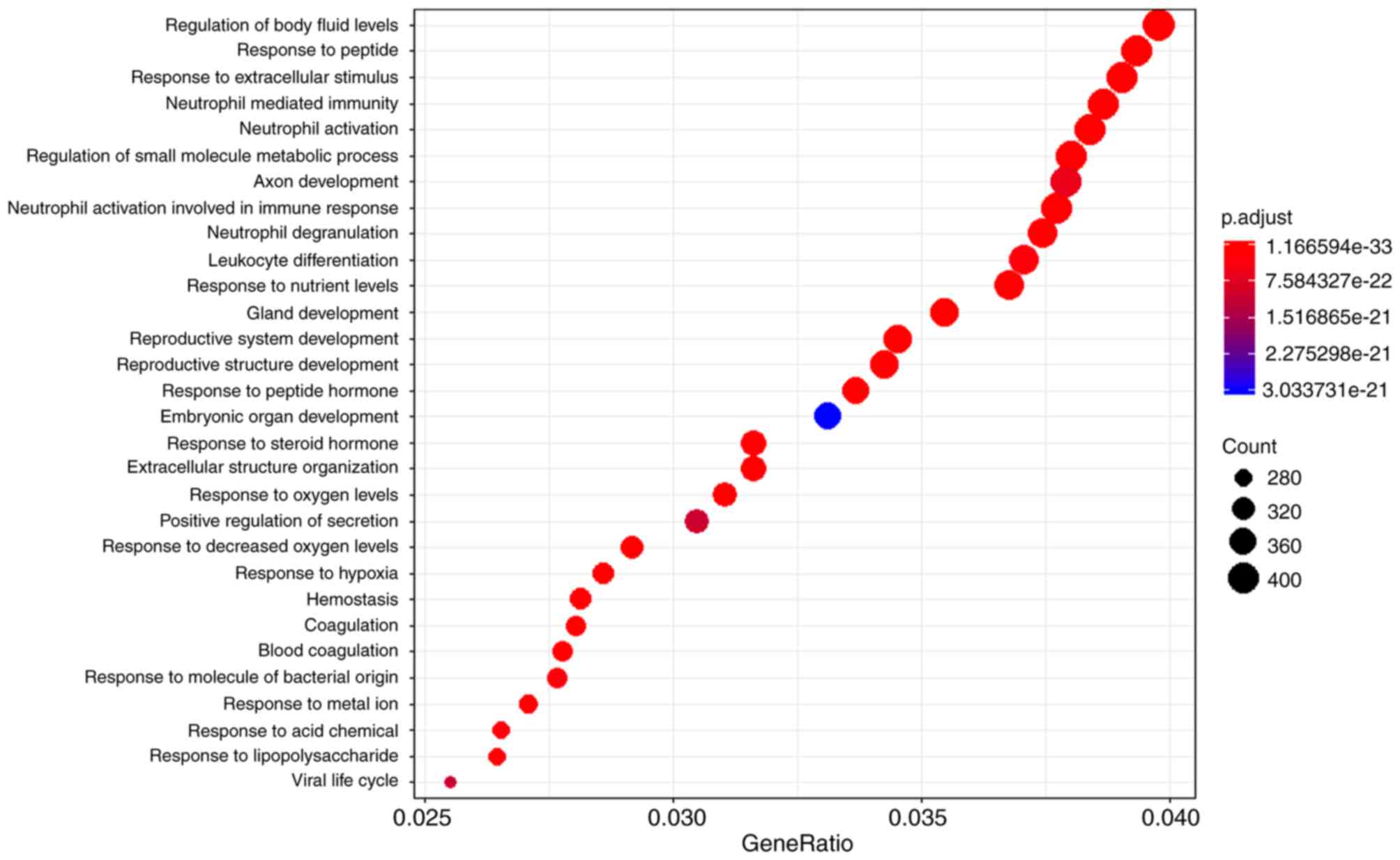
networks (26) (Fig. 3). The biological functions of the
GBM-specific network were investigated. Functional enrichment
analysis was performed, and the top 30 significantly enriched Gene
Ontology functions are presented in Fig.
3. Notably, the results demonstrated a number of immune
response-associated functions, including ‘neutrophil-mediated
immunity’, ‘neutrophil activation involved in immune response’ and
‘leukocyte differentiation’. Numerous studies have reported
profound and generalised immunosuppression of GBM tumours,
particularly within the context of cell-mediated immunity (27,28). In
addition, a number of other types of functions were found, such as
‘axon development’, ‘haemostasis’ and ‘response to peptide’, which
might be unique or important to the brain. Together, these results
support the possibility that the GBM-specific sub-network may be a
potential resource for GBM-related research.
GBM-specific risk pathways
A biological interaction network contains a large
amount of information, and its complex structure may hinder the
deduction of new insights for interested risk pathways. The risk
pathways obtained from the regulatory network can shed light on the
molecular mechanisms of GBM. Detecting disease-associated pathways
in a clear and simple manner is an important goal. In the present
study, 615 linear risk pathways from the GBM-specific regulatory
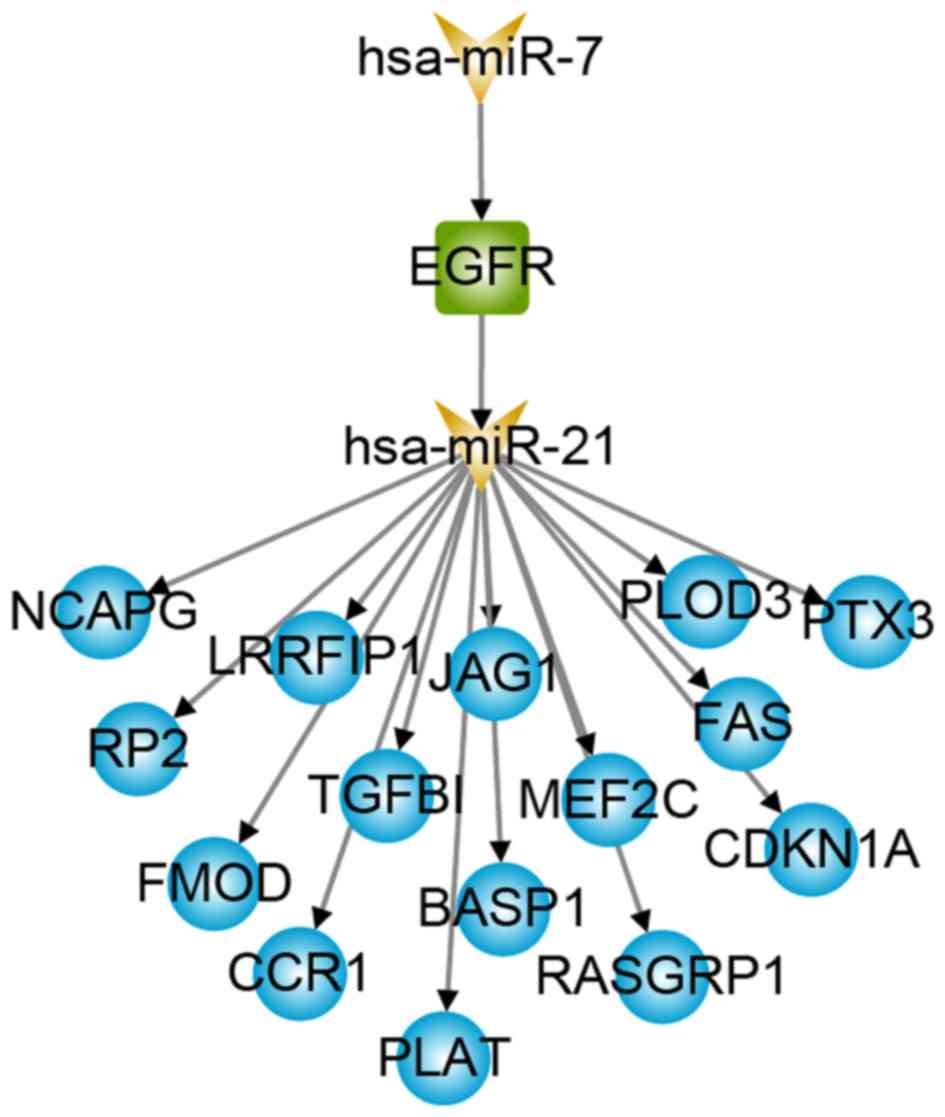
network were identified. Notably, a TF named EGFR was not only a
known GBM disease gene, but also a differentially expressed gene
(29). A total of 15-risk pathways
were connected by EGFR and its up-stream regulator hsa-miR-7
(Fig. 4), which is a miRNA that has
been reported as a prospective risk factor for GBM (30,31).
Discussion
Glioblastoma is an astrocyte-derived tumour with a
propensity for malignancy and is caused by an abnormal disorder of
multiple interacting genes rather than a single gene acting in
isolation (32). Activation of
molecular signalling cascades have been shown in GBM tumourigenesis
(33). Identification of a cascade
regulatory pathway, which usually has a clear and simple linear
structure (34), may help reveal
molecular mechanisms of the disease. A network-based approach to
analyse risk regulatory pathways in GBM was used in the present
study. Noncoding RNAs are emerging as key regulators of diverse
biological processes and are important in cancer pathogenesis
(35,36). Therefore, noncoding RNAs involved in
pathway regulation were also considered.
Through differential expression analysis, a total of
1,827 genes and 30 miRNAs were identified in normal and tumour
tissues from GBM samples. When these candidate risk factors were
taken as seed nodes, a GBM-specific regulatory network involving
TFs, miRNAs and target genes was identified. Functional analysis
showed that the GBM-specific network was significantly associated
with several important functions of the immune response, axon
development, haemostasis and response to peptides. From these, 15
reliable risk pathways specific to GBM were identified, which may
be a useful resource for increasing our fundamental understanding
of the molecular role and regulation of GBM. These results showed
that the 15 GBM-associated risk pathways were connected by a TF
named EGFR that was both a known GBM gene and a differentially
expressed gene. Numerous studies have highlighted the vital role
played by EGFR during the emergence and development of GBM. Shao
et al (37) showed that ~80%
of patients with GBM presented with EGFR genomic amplification or
overexpression. Mischel et al (38) reported that EGFR-overexpressing GBMs
promoted GBM cell proliferation, survival and angiogenesis. Wheeler
et al (39) showed that EGFR
inhibitors may be a novel therapeutic strategy for treating
patients with GBM. In the present study, EGFR indirectly regulates
15 GBM-related genes (Fig. 4) and
forms a cascade regulatory pathway with hsa-miR-7 and hsa-miR-21,
participating in the occurrence and development of GBM. Regarding
the downstream target genes, FAS-induced expression of chemokines
in human glioma cells (40) and
TGFβ1 gene expression are potential signatures for the mesenchymal
high-grade glioma subtype (41).
Therefore, the risk regulatory pathways miR-7, EGFR, miR-21 and
FAS, and miR-7, EGFR, miR-21 and TGFB1 may cause disease by
affecting the abnormal function of glioma cells.
In conclusion, accumulating evidence indicates that
a single mutated or aberrantly expressed gene is not sufficient to
result in an abnormal phenotype and that complex diseases are
caused by the accumulative and co-operative effects of multiple
dysregulated genes and gene interactions (42,43).
Risk regulatory pathways that consist of interacting genes with a
linear chain structure can help reveal the molecular mechanisms of
diseases. Through integration of transcriptional and
post-transcriptional regulation, risk regulatory pathways in GBM
that might serve as potential targets for clinical treatment were
identified.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the author on reasonable request.
Authors' contributions
JL, YJX, YW, and YY collected and analyzed all data.
CZ, YX and JXW performed statistical analysis. ZYL, JL and JXW
contributed to the concept and design of the study and drafted the
manuscript. JL, YW, and YY revised the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choe G, Horvath S, Cloughesy TF, Crosby K,
Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL and Mischel PS:
Analysis of the phosphatidylinositol 3′-kinase signaling pathway in
glioblastoma patients in vivo. Cancer Res. 63:2742–2746.
2003.PubMed/NCBI
|
|
3
|
Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer
A, Llobet-Navas D, Rajbhandari P, Bansal M, Guarnieri P, Silva J
and Califano A: An extensive microRNA-mediated network of RNA-RNA
interactions regulates established oncogenic pathways in
glioblastoma. Cell. 147:370–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Davis ME: Glioblastoma: Overview of
disease and treatment. Clin J Oncol Nurs. 20:S2–S8. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwartzentruber J, Korshunov A, Liu XY,
Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA,
Tonjes M, et al: Driver mutations in histone H3.3 and chromatin
remodelling genes in paediatric glioblastoma. Nature. 482:226–231.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Frattini V, Trifonov V, Chan JM, Castano
A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al: The
integrated landscape of driver genomic alterations in glioblastoma.
Nat Genet. 45:1141–1149. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang Y, Li X, Zhou D, Zhi H, Wang P, Gao
Y, Guo M, Yue M, Wang Y, Shen W, et al: Inferences of individual
drug responses across diverse cancer types using a novel competing
endogenous RNA network. Mol Oncol. 12:1429–1446. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu F, Quan F, Xu J, Zhang Y, Xie Y, Zhang
J, Lan Y, Yuan H, Zhang H, Cheng S, et al: Breast cancer prognosis
signature: Linking risk stratification to disease subtypes. Brief
Bioinform. Sep 3–2018.(Epub ahead of print). View Article : Google Scholar
|
|
9
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Best MG, Sol N, Kooi I, Tannous J,
Westerman BA, Rustenburg F, Schellen P, Verschueren H, Post E,
Koster J, et al: RNA-Seq of tumor-educated platelets enables
blood-based pan-cancer, multiclass, and molecular pathway cancer
diagnostics. Cancer Cell. 28:666–676. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lai NS, Wu DG, Fang XG, Lin YC, Chen SS,
Li ZB and Xu SS: Serum microRNA-210 as a potential noninvasive
biomarker for the diagnosis and prognosis of glioma. Br J Cancer.
112:1241–1246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gilder AS, Natali L, Van Dyk DM, Zalfa C,
Banki MA, Pizzo DP, Wang H, Klemke RL, Mantuano E and Gonias SL:
The urokinase receptor induces a mesenchymal gene expression
signature in glioblastoma cells and promotes tumor cell survival in
neurospheres. Sci Rep. 8:29822018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Liu D, Wang L, Wang S, Yu X, Dai
E, Liu X, Luo S and Jiang W: Integrated systems approach identifies
risk regulatory pathways and key regulators in coronary artery
disease. J Mol Med (Berl). 93:1381–1390. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang W, Zhang Y, Meng F, Lian B, Chen X,
Yu X, Dai E, Wang S, Liu X, Li X, et al: Identification of active
transcription factor and miRNA regulatory pathways in Alzheimer's
disease. Bioinformatics. 29:2596–2602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stelzer G, Rosen N, Plaschkes I, Zimmerman
S, Twik M, Fishilevich S, Stein TI, Nudel R, Lieder I, Mazor Y, et
al: The genecards suite: From gene data mining to disease genome
sequence analyses. Curr Protoc Bioinformatics. 54:1.30.1–1.30.33.
2016.
|
|
16
|
Jiang Q, Wang Y, Hao Y, Juan L, Teng M,
Zhang X, Li M, Wang G and Liu Y: miR2Disease: A manually curated
database for microRNA deregulation in human disease. Nucleic Acids
Res 37 (Database Issu). D98–D104. 2009. View Article : Google Scholar
|
|
17
|
Durinck S, Spellman PT, Birney E and Huber
W: Mapping identifiers for the integration of genomic datasets with
the R/Bioconductor package biomaRt. Nat Protoc. 4:1184–1191. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu SD, Lin FM, Wu WY, Liang C, Huang WC,
Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, et al: miRTarBase: A
database curates experimentally validated microRNA-target
interactions. Nucleic Acids Res. 39:(Database Issue). D163–D169.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wingender E, Chen X, Hehl R, Karas H,
Liebich I, Matys V, Meinhardt T, Pruss M, Reuter I and Schacherer
F: TRANSFAC: An integrated system for gene expression regulation.
Nucleic Acids Res. 28:316–319. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xiao F, Zuo Z, Cai G, Kang S, Gao X and Li
T: miRecords: An integrated resource for microRNA-target
interactions. Nucleic Acids Res. 37:(Database Issue). D105–D110.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sethupathy P, Corda B and Hatzigeorgiou
AG: TarBase: A comprehensive database of experimentally supported
animal microRNA targets. RNA. 12:192–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao J, Yang TH, Huang Y and Holme P:
Ranking candidate disease genes from gene expression and protein
interaction: A Katz-centrality based approach. PLoS One.
6:e243062011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
R Core Team: R, . A language and
environment for statistical computing. R Foundation for Statistical
Computing; Vienna: 2012, http://www.R-project.org/
|
|
24
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
The Gene Ontology Consortium: The gene
ontology resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Girvan M and Newman ME: Community
structure in social and biological networks. Proc Natl Acad Sci
USA. 99:7821–7826. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Osuka S and Van Meir EG: Cancer therapy:
Neutrophils traffic in cancer nanodrugs. Nat Nanotechnol.
12:616–618. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Junttila MR and de Sauvage FJ: Influence
of tumour micro-environment heterogeneity on therapeutic response.
Nature. 501:346–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chakravarty D, Pedraza AM, Cotari J, Liu
AH, Punko D, Kokroo A, Huse JT, Altan-Bonnet G and Brennan CW: EGFR
and PDGFRA co-expression and heterodimerization in glioblastoma
tumor sphere lines. Sci Rep. 7:90432017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Y, Vogel G, Yu Z and Richard S: The
QKI-5 and QKI-6 RNA binding proteins regulate the expression of
microRNA 7 in glial cells. Mol Cell Biol. 33:1233–1243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang B, Sun F, Dong N, Sun Z, Diao Y,
Zheng C, Sun J, Yang Y and Jiang D: MicroRNA-7 directly targets
insulin-like growth factor 1 receptor to inhibit cellular growth
and glucose metabolism in gliomas. Diagn Pathol. 9:2112014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Patel VN, Gokulrangan G, Chowdhury SA,
Chen Y, Sloan AE, Koyuturk M, Barnholtz-Sloan J and Chance MR:
Network signatures of survival in glioblastoma multiforme. PLoS
Comput Biol. 9:e10032372013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jhanwar-Uniyal M, Amin AG, Cooper JB, Das
K, Schmidt MH and Murali R: Discrete signaling mechanisms of mTORC1
and mTORC2: Connected yet apart in cellular and molecular aspects.
Adv Biol Regul. 64:39–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Beguerisse-Díaz M, Desikan R and Barahona
M: Linear models of activation cascades: Analytical solutions and
coarse-graining of delayed signal transduction. J R Soc Interface.
13(pii): 201604092016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pekarsky Y and Croce CM: Noncoding RNA
genes in cancer pathogenesis. Adv Biol Regul. 71:219–223. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu F, Zhang G, Shi A, Hu J, Li F, Zhang X,
Zhang Y, Huang J, Xiao Y, Li X and Cheng S: LnChrom: A resource of
experimentally validated lncRNA-chromatin interactions in human and
mouse. Database (Oxford). 2018:2018. View Article : Google Scholar
|
|
37
|
Shao H, Chung J, Balaj L, Charest A,
Bigner DD, Carter BS, Hochberg FH, Breakefield XO, Weissleder R and
Lee H: Protein typing of circulating microvesicles allows real-time
monitoring of glioblastoma therapy. Nat Med. 18:1835–1840. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mischel PS, Shai R, Shi T, Horvath S, Lu
KV, Choe G, Seligson D, Kremen TJ, Palotie A, Liau LM, et al:
Identification of molecular subtypes of glioblastoma by gene
expression profiling. Oncogene. 22:2361–2373. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors-impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Choi C, Xu X, Oh JW, Lee SJ, Gillespie GY,
Park H, Jo H and Benveniste EN: Fas-induced expression of
chemokines in human glioma cells: involvement of extracellular
signal-regulated kinase 1/2 and p38 mitogen-activated protein
kinase. Cancer Res. 61:3084–3091. 2001.PubMed/NCBI
|
|
41
|
Pan YB, Zhang CH, Wang SQ, Ai PH, Chen K,
Zhu L, Sun ZL and Feng DF: Transforming growth factor beta induced
(TGFBI) is a potential signature gene for mesenchymal subtype
high-grade glioma. J Neurooncol. 137:395–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cordell HJ: Detecting gene-gene
interactions that underlie human diseases. Nat Rev Genet.
10:392–404. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cole BS, Hall MA, Urbanowicz RJ,
Gilbert-Diamond D and Moore JH: Analysis of gene-gene interactions.
Curr Protoc Hum Genet. 95:1.14.1–1.14.10. 2017. View Article : Google Scholar
|